
A Spotlight on Viruses—Application of Click Chemistry to Visualize Virus-Cell Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Minimally Invasive Labeling of Viral Proteins Using Click Chemistry
2.1. Metabolic Labeling of Viral Components
2.1.1. Glycans
2.1.2. Lipids
2.1.3. Proteins
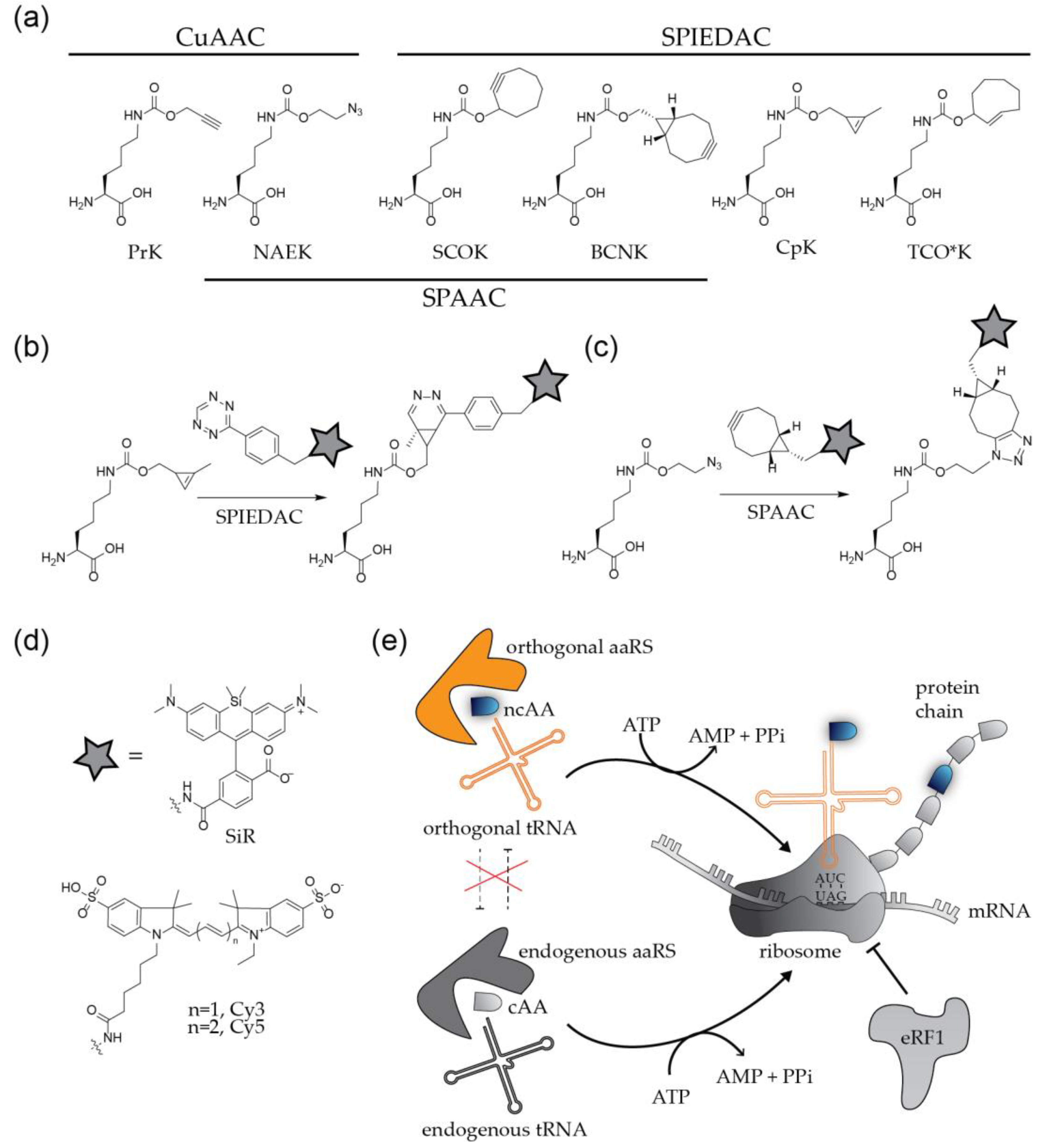
2.2. Labeling of Viral Proteins Using Genetic Code Expansion and Click Chemistry
2.3. Application of Genetic Code Expansion and Click Labeling in Virology
2.3.1. Non-Enveloped Viruses
2.3.2. Enveloped Viruses
Metabolic Labeling of Glycosylated Viral Proteins
Labeling of Viral Proteins by Amber Suppression
2.4. Virus-Specific Challenges and Limitations of GCE/Click Labeling and Possible Solutions
2.4.1. Choice of Position
2.4.2. Detection Sensitivity
2.4.3. Incomplete Suppression
2.4.4. Choice of Cell Line
2.4.5. Low Yield of Engineered Virus
2.4.6. Unwanted Amber Suppression and Click Labeling
2.4.7. Choice of ncAA and Fluorophore
2.4.8. Effect of Virus Replication on GCE
3. Click Labeling of Nucleic Acids Allows Detailed Analyses of Viral Replication
3.1. Viral Replication Sites
3.2. Site-Specific Nucleic Acid Labeling
3.3. Stochastic Nucleic Acid Labeling
3.4. Sensitivity of Viral Genome Detection
4. Insights into Virus Biology Obtained Using Click Labeling of Viral Nucleic Acids
4.1. DNA Viruses
4.1.1. Recent Insights into Herpes Simplex Virus Biology are Fueled by Click Chemistry
4.1.2. Adenovirus Replication: Click Labeling Revealed a New Virus Induced Nuclear Substructure
4.1.3. Human Papillomavirus Protects Its Genome by Special Transport Vesicles that are Delivered into the Nucleus During Mitosis
4.1.4. Visualization of Vaccinia Virus DNA in Viral Replication Factories and Incoming Virions
4.2. Retroviruses
4.2.1. Using Click-Labeled Viral RNA to Monitor HIV-1 Uncoating
4.2.2. Detection of HIV-1 Reverse Transcription Complexes by EdU/Click Labeling
4.3. RNA Viruses
4.4. Future Challenges and Opportunities
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, I.H.; Burckhardt, C.J.; Yakimovich, A.; Greber, U.F. Imaging, Tracking and Computational Analyses of Virus Entry and Egress with the Cytoskeleton. Viruses 2018, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Witte, R.; Andriasyan, V.; Georgi, F.; Yakimovich, A.; Greber, U.F. Concepts in Light Microscopy of Viruses. Viruses 2018, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Hanne, J.; Zila, V.; Heilemann, M.; Muller, B.; Krausslich, H.G. Super-resolved insights into human immunodeficiency virus biology. FEBS Lett. 2016, 590, 1858–1876. [Google Scholar] [CrossRef]
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P.C. Super-resolution microscopy demystified. Nat. Cell Biol. 2019, 21, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Beerli, C.; Pereira, P.M.; Scherer, K.M.; Samolej, J.; Bleck, C.K.; Mercer, J.; Henriques, R. VirusMapper: Open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci. Rep. 2016, 6, 29132. [Google Scholar] [CrossRef] [PubMed]
- Sbalzarini, I.F. Seeing Is Believing: Quantifying Is Convincing: Computational Image Analysis in Biology. Adv. Anat. Embryol. Cell Biol. 2016, 219, 1–39. [Google Scholar]
- Shaner, N.C.; Patterson, G.H.; Davidson, M.W. Advances in fluorescent protein technology. J. Cell Sci. 2007, 120 Pt 24, 4247–4260. [Google Scholar] [CrossRef]
- Sakin, V.; Paci, G.; Lemke, E.A.; Muller, B. Labeling of virus components for advanced, quantitative imaging analyses. FEBS Lett. 2016, 590, 1896–1914. [Google Scholar] [CrossRef]
- Gautier, A.; Tebo, A.G. Fluorogenic Protein-Based Strategies for Detection, Actuation, and Sensing. Bioessays 2018, 40, e1800118. [Google Scholar] [CrossRef]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef]
- Lang, K.; Davis, L.; Chin, J.W. Genetic encoding of unnatural amino acids for labeling proteins. Methods Mol. Biol. 2015, 217–228. [Google Scholar]
- Nikic, I.; Lemke, E.A. Genetic code expansion enabled site-specific dual-color protein labeling: Superresolution microscopy and beyond. Curr. Opin. Chem. Biol. 2015, 28, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.S.; Ostapchuk, P.; Hearing, P.; Carrico, I. Chemoselective attachment of small molecule effector functionality to human adenoviruses facilitates gene delivery to cancer cells. J. Am. Chem. Soc. 2010, 132, 13615–13617. [Google Scholar] [CrossRef] [PubMed]
- Erickson, S.B.; Mukherjee, R.; Kelemen, R.E.; Wrobel, C.J.; Cao, X.; Chatterjee, A. Precise Photoremovable Perturbation of a Virus-Host Interaction. Angew. Chem. 2017, 56, 4234–4237. [Google Scholar] [CrossRef]
- Fratini, M.; Wiegand, T.; Funaya, C.; Jiang, Z.; Shah, P.N.M.; Spatz, J.P.; Cavalcanti-Adam, E.A.; Boulant, S. Surface Immobilization of Viruses and Nanoparticles Elucidates Early Events in Clathrin-Mediated Endocytosis. ACS Infect. Dis. 2018, 4, 1585–1600. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Xu, H.; Zhou, X.; Zhang, Z.; Tian, Z.; Wang, Y.; Wu, Y.; Zhang, B.; Niu, Z.; Zhang, C.; et al. Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 2016, 354, 1170–1173. [Google Scholar] [CrossRef]
- Kelemen, R.E.; Erickson, S.B.; Chatterjee, A. Synthesis at the interface of virology and genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 164–171. [Google Scholar] [CrossRef]
- Ratnatilaka Na Bhuket, P.; Luckanagul, J.A.; Rojsitthisak, P.; Wang, Q. Chemical modification of enveloped viruses for biomedical applications. Integr. Biol. 2018, 10, 666–679. [Google Scholar] [CrossRef]
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar]
- Bumpus, T.W.; Baskin, J.M. Greasing the Wheels of Lipid Biology with Chemical Tools. Trends Biochem. Sci. 2018, 43, 970–983. [Google Scholar] [CrossRef]
- Huang, L.L.; Lu, G.H.; Hao, J.; Wang, H.; Yin, D.L.; Xie, H.Y. Enveloped virus labeling via both intrinsic biosynthesis and metabolic incorporation of phospholipids in host cells. Anal. Chem. 2013, 85, 5263–5270. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.S.; Ostapchuk, P.; Hearing, P.; Carrico, I.S. Unnatural amino acid incorporation onto adenoviral (Ad) coat proteins facilitates chemoselective modification and retargeting of Ad type 5 vectors. J. Virol. 2011, 85, 7546–7554. [Google Scholar] [CrossRef]
- Atkins, J.F.; Gesteland, R. Biochemistry. The 22nd amino acid. Science 2002, 296, 1409–1410. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Greber, U.F. Principles of Virus Uncoating: Cues and the Snooker Ball. Traffic 2016, 17, 569–592. [Google Scholar] [CrossRef] [PubMed]
- Lux, K.; Goerlitz, N.; Schlemminger, S.; Perabo, L.; Goldnau, D.; Endell, J.; Leike, K.; Kofler, D.M.; Finke, S.; Hallek, M.; et al. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 2005, 79, 11776–11787. [Google Scholar] [CrossRef]
- Joo, K.I.; Fang, Y.; Liu, Y.; Xiao, L.; Gu, Z.; Tai, A.; Lee, C.L.; Tang, Y.; Wang, P. Enhanced real-time monitoring of adeno-associated virus trafficking by virus-quantum dot conjugates. ACS Nano 2011, 5, 3523–3535. [Google Scholar] [CrossRef]
- Kelemen, R.E.; Mukherjee, R.; Cao, X.; Erickson, S.B.; Zheng, Y.; Chatterjee, A. A Precise Chemical Strategy to Alter the Receptor Specificity of the Adeno-Associated Virus. Angew. Chem. 2016, 55, 10645–10649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhou, X.; Yao, T.; Tian, Z.; Zhou, D. Precision Fluorescent Labeling of an Adeno-Associated Virus Vector to Monitor the Viral Infection Pathway. Biotechnol. J. 2018, 13, e1700374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Cai, L.; Adogla, E.A.; Guan, H.; Lin, Y.; Wang, Q. Labeling of Enveloped Virus via Metabolic Incorporation of Azido Sugars. Bioconjug. Chem. 2015, 26, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Oum, Y.H.; Desai, T.M.; Marin, M.; Melikyan, G.B. Click labeling of unnatural sugars metabolically incorporated into viral envelope glycoproteins enables visualization of single particle fusion. J. Virol. Methods 2016, 233, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Nikic, I.; Plass, T.; Schraidt, O.; Szymanski, J.; Briggs, J.A.; Schultz, C.; Lemke, E.A. Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew. Chem. 2014, 53, 2245–2249. [Google Scholar] [CrossRef]
- Sakin, V.; Hanne, J.; Dunder, J.; Anders-Osswein, M.; Laketa, V.; Nikic, I.; Krausslich, H.G.; Lemke, E.A.; Muller, B. A Versatile Tool for Live-Cell Imaging and Super-Resolution Nanoscopy Studies of HIV-1 Env Distribution and Mobility. Cell Chem. Biol. 2017, 24, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Govindan, R.; Nikic-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 2018, 174, 926–937. [Google Scholar] [CrossRef]
- Lin, S.; Yan, H.; Li, L.; Yang, M.; Peng, B.; Chen, S.; Li, W.; Chen, P.R. Site-specific engineering of chemical functionalities on the surface of live hepatitis D virus. Angew. Chem. 2013, 52, 13970–13974. [Google Scholar] [CrossRef] [PubMed]
- Pott, M.; Schmidt, M.J.; Summerer, D. Evolved sequence contexts for highly efficient amber suppression with noncanonical amino acids. ACS Chem. Biol. 2014, 9, 2815–2822. [Google Scholar] [CrossRef]
- Rihn, S.J.; Wilson, S.J.; Loman, N.J.; Alim, M.; Bakker, S.E.; Bhella, D.; Gifford, R.J.; Rixon, F.J.; Bieniasz, P.D. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 2013, 9, e1003461. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lewis, T.L., Jr.; Igo, P.; Polleux, F.; Chatterjee, A. Virus-Enabled Optimization and Delivery of the Genetic Machinery for Efficient Unnatural Amino Acid Mutagenesis in Mammalian Cells and Tissues. ACS Synth. Biol. 2017, 6, 13–18. [Google Scholar] [CrossRef]
- Nikic, I.; Estrada Girona, G.; Kang, J.H.; Paci, G.; Mikhaleva, S.; Koehler, C.; Shymanska, N.V.; Ventura Santos, C.; Spitz, D.; Lemke, E.A. Debugging Eukaryotic Genetic Code Expansion for Site-Specific Click-PAINT Super-Resolution Microscopy. Angew. Chem. 2016, 55, 16172–16176. [Google Scholar] [CrossRef]
- Plass, T.; Milles, S.; Koehler, C.; Szymanski, J.; Mueller, R.; Wiessler, M.; Schultz, C.; Lemke, E.A. Amino acids for Diels-Alder reactions in living cells. Angew. Chem. 2012, 51, 4166–4170. [Google Scholar] [CrossRef] [PubMed]
- Schmied, W.H.; Elsasser, S.J.; Uttamapinant, C.; Chin, J.W. Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. J. Am. Chem. Soc. 2014, 136, 15577–15583. [Google Scholar] [CrossRef]
- Elsasser, S.J.; Ernst, R.J.; Walker, O.S.; Chin, J.W. Genetic code expansion in stable cell lines enables encoded chromatin modification. Nat. Methods 2016, 13, 158–164. [Google Scholar] [CrossRef]
- Kita, A.; Hino, N.; Higashi, S.; Hirota, K.; Narumi, R.; Adachi, J.; Takafuji, K.; Ishimoto, K.; Okada, Y.; Sakamoto, K.; et al. Adenovirus vector-based incorporation of a photo-cross-linkable amino acid into proteins in human primary cells and cancerous cell lines. Sci. Rep. 2016, 6, 36946. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Xiang, Z.; Miller, B.; Louie, G.; Wang, W.; Noel, J.P.; Gage, F.H.; Wang, L. Genetically encoding unnatural amino acids in neural stem cells and optically reporting voltage-sensitive domain changes in differentiated neurons. Stem Cells 2011, 29, 1231–1240. [Google Scholar] [CrossRef]
- Ernst, R.J.; Krogager, T.P.; Maywood, E.S.; Zanchi, R.; Beranek, V.; Elliott, T.S.; Barry, N.P.; Hastings, M.H.; Chin, J.W. Genetic code expansion in the mouse brain. Nat. Chem. Biol. 2016, 12, 776–778. [Google Scholar] [CrossRef]
- Chatterjee, A.; Xiao, H.; Bollong, M.; Ai, H.W.; Schultz, P.G. Efficient viral delivery system for unnatural amino acid mutagenesis in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 11803–11808. [Google Scholar] [CrossRef]
- Wan, W.; Huang, Y.; Wang, Z.; Russell, W.K.; Pai, P.J.; Russell, D.H.; Liu, W.R. A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew. Chem. 2010, 49, 3211–3214. [Google Scholar] [CrossRef]
- Kozma, E.; Nikic, I.; Varga, B.R.; Aramburu, I.V.; Kang, J.H.; Fackler, O.T.; Lemke, E.A.; Kele, P. Hydrophilic trans-Cyclooctenylated Noncanonical Amino Acids for Fast Intracellular Protein Labeling. Chembiochem 2016, 17, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Lukinavicius, G.; Umezawa, K.; Olivier, N.; Honigmann, A.; Yang, G.; Plass, T.; Mueller, V.; Reymond, L.; Correa, I.R., Jr.; Luo, Z.G.; et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell 2005, 97, 147–172. [Google Scholar] [CrossRef]
- Schmid, M.; Speiseder, T.; Dobner, T.; Gonzalez, R.A. DNA virus replication compartments. J. Virol. 2014, 88, 1404–1420. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Randall, G. (+) RNA virus replication compartments: A safe home for (most) viral replication. Curr. Opin. Microbiol. 2016, 32, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudriere-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Malyshev, D.A.; Dhami, K.; Lavergne, T.; Chen, T.; Dai, N.; Foster, J.M.; Correa, I.R., Jr.; Romesberg, F.E. A semi-synthetic organism with an expanded genetic alphabet. Nature 2014, 509, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lamb, B.M.; Feldman, A.W.; Zhou, A.X.; Lavergne, T.; Li, L.; Romesberg, F.E. A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl. Acad. Sci. USA 2017, 114, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ptacin, J.L.; Fischer, E.C.; Aerni, H.R.; Caffaro, C.E.; San Jose, K.; Feldman, A.W.; Turner, C.R.; Romesberg, F.E. A semi-synthetic organism that stores and retrieves increased genetic information. Nature 2017, 551, 644–647. [Google Scholar] [CrossRef]
- de Bruyn Kops, A.; Knipe, D.M. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 1988, 55, 857–868. [Google Scholar] [CrossRef]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef]
- Qu, D.; Wang, G.; Wang, Z.; Zhou, L.; Chi, W.; Cong, S.; Ren, X.; Liang, P.; Zhang, B. 5-Ethynyl-2′-deoxycytidine as a new agent for DNA labeling: Detection of proliferating cells. Anal. Biochem. 2011, 417, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Neef, A.B.; Samain, F.; Luedtke, N.W. Metabolic labeling of DNA by purine analogues in vivo. Chembiochem 2012, 13, 1750–1753. [Google Scholar] [CrossRef]
- Jao, C.Y.; Salic, A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 15779–15784. [Google Scholar] [CrossRef] [PubMed]
- Neef, A.B.; Luedtke, N.W. Dynamic metabolic labeling of DNA in vivo with arabinosyl nucleosides. Proc. Natl. Acad. Sci. USA 2011, 108, 20404–20409. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.; Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 2013, 14, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Hagemeijer, M.C.; Vonk, A.M.; Monastyrska, I.; Rottier, P.J.; de Haan, C.A. Visualizing coronavirus RNA synthesis in time by using click chemistry. J. Virol. 2012, 86, 5808–5816. [Google Scholar] [CrossRef]
- Bejarano, D.A.; Puertas, M.C.; Borner, K.; Martinez-Picado, J.; Muller, B.; Krausslich, H.G. Detailed Characterization of Early HIV-1 Replication Dynamics in Primary Human Macrophages. Viruses 2018, 10, 620. [Google Scholar] [CrossRef]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Muller, B.; Krausslich, H.G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 2014, 3, e04114. [Google Scholar] [CrossRef]
- Stultz, R.D.; Cenker, J.J.; McDonald, D. Imaging HIV-1 Genomic DNA from Entry through Productive Infection. J. Virol. 2017, 91, e00034-17. [Google Scholar] [CrossRef]
- Dembowski, J.A.; DeLuca, N.A. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 2015, 11, e1004939. [Google Scholar] [CrossRef]
- Dembowski, J.A.; Dremel, S.E.; DeLuca, N.A. Replication-Coupled Recruitment of Viral and Cellular Factors to Herpes Simplex Virus Type 1 Replication Forks for the Maintenance and Expression of Viral Genomes. PLoS Pathog. 2017, 13, e1006166. [Google Scholar] [CrossRef] [PubMed]
- Sekine, E.; Schmidt, N.; Gaboriau, D.; O’Hare, P. Spatiotemporal dynamics of HSV genome nuclear entry and compaction state transitions using bioorthogonal chemistry and super-resolution microscopy. PLoS Pathog. 2017, 13, e1006721. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Robinson, D.R.; Hisaoka, M.; Ueshima, S.; Okuwaki, M.; Nagata, K.; Wodrich, H. Tracking adenovirus genomes identifies morphologically distinct late DNA replication compartments. Traffic 2016, 17, 1168–1180. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Luszczek, W.; Keiffer, T.R.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M.J. Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proc. Natl. Acad. Sci. USA 2016, 113, 6289–6294. [Google Scholar] [CrossRef]
- Alandijany, T.; Roberts, A.P.E.; Conn, K.L.; Loney, C.; McFarlane, S.; Orr, A.; Boutell, C. Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection. PLoS Pathog. 2018, 14, e1006769. [Google Scholar]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Hennig, T.; Serwa, R.A.; Marchetti, M.; O’Hare, P. Remote Activation of Host Cell DNA Synthesis in Uninfected Cells Signaled by Infected Cells in Advance of Virus Transmission. J. Virol. 2015, 89, 11107–11115. [Google Scholar] [CrossRef]
- Neef, A.B.; Pernot, L.; Schreier, V.N.; Scapozza, L.; Luedtke, N.W. A Bioorthogonal Chemical Reporter of Viral Infection. Angew. Chem. 2015, 54, 7911–7914. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Sapp, M. Human Papillomavirus Entry: Hiding in a Bubble. J. Virol. 2016, 90, 8032–8035. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, C.; Samperio Ventayol, P.; Vogeley, C.; Schelhaas, M. Kallikrein-8 Proteolytically Processes Human Papillomaviruses in the Extracellular Space To Facilitate Entry into Host Cells. J. Virol. 2015, 89, 7038–7052. [Google Scholar] [CrossRef]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kuhbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Massimi, P.; Bergant, M.; Banks, L. Human Papillomavirus Infectious Entry and Trafficking Is a Rapid Process. J. Virol. 2015, 89, 8727–8732. [Google Scholar] [CrossRef]
- Yakimovich, A.; Huttunen, M.; Zehnder, B.; Coulter, L.J.; Gould, V.; Schneider, C.; Kopf, M.; McInnes, C.J.; Greber, U.F.; Mercer, J. Inhibition of Poxvirus Gene Expression and Genome Replication by Bisbenzimide Derivatives. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Kilcher, S.; Schmidt, F.I.; Schneider, C.; Kopf, M.; Helenius, A.; Mercer, J. siRNA screen of early poxvirus genes identifies the AAA+ ATPase D5 as the virus genome-uncoating factor. Cell Host Microbe 2014, 15, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef]
- Francis, A.C.; Melikyan, G.B. Live-Cell Imaging of Early Steps of Single HIV-1 Infection. Viruses 2018, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Borner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Muller, B.; Krausslich, H.G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the Nuclear Pore Complex. eLife 2019, 8, e41800. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Franks, T.; Gibson, G.; Huber, K.; Rahm, N.; Strambio De Castillia, C.; Luban, J.; Aiken, C.; Watkins, S.; Sluis-Cremer, N.; et al. Evidence for biphasic uncoating during HIV-1 infection from a novel imaging assay. Retrovirology 2013, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Forster, F.; Ecke, M.; Plitzko, J.M.; Melchior, F.; Gerisch, G.; Baumeister, W.; Medalia, O. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science 2004, 306, 1387–1390. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wilk, T.; Welker, R.; Krausslich, H.G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef]
- Wang, G.Z.; Wang, Y.; Goff, S.P. Histones Are Rapidly Loaded onto Unintegrated Retroviral DNAs Soon after Nuclear Entry. Cell Host Microbe 2016, 20, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Wu, L. SAMHD1 Suppression of Antiviral Immune Responses. Trends Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of ebolavirus replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef]
- Yang, C.W.; Lee, Y.Z.; Hsu, H.Y.; Shih, C.; Chao, Y.S.; Chang, H.Y.; Lee, S.J. Targeting Coronaviral Replication and Cellular JAK2 Mediated Dominant NF-kappaB Activation for Comprehensive and Ultimate Inhibition of Coronaviral Activity. Sci. Rep. 2017, 7, 4105. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Tritsch, S.R.; Kota, K.; Chiang, C.Y.; Dong, L.; Kenny, T.; Brueggemann, E.E.; Ward, M.D.; Cazares, L.H.; Bavari, S. Sphingosine kinase 2 is a chikungunya virus host factor co-localized with the viral replication complex. Emerg. Microbes Infect. 2015, 4, e61. [Google Scholar] [CrossRef]
- Baird, N.L.; York, J.; Nunberg, J.H. Arenavirus infection induces discrete cytosolic structures for RNA replication. J. Virol. 2012, 86, 11301–11310. [Google Scholar] [CrossRef] [PubMed]
- Kalveram, B.; Lihoradova, O.; Indran, S.V.; Head, J.A.; Ikegami, T. Using click chemistry to measure the effect of viral infection on host-cell RNA synthesis. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kalveram, B.; Lihoradova, O.; Ikegami, T. NSs protein of rift valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J. Virol. 2011, 85, 6234–6243. [Google Scholar] [CrossRef]
- Kalveram, B.; Ikegami, T. Toscana virus NSs protein promotes degradation of double-stranded RNA-dependent protein kinase. J. Virol. 2013, 87, 3710–3718. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef]
- Sivaraman, D.; Biswas, P.; Cella, L.N.; Yates, M.V.; Chen, W. Detecting RNA viruses in living mammalian cells by fluorescence microscopy. Trends Biotechnol. 2011, 29, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Quentin-Froignant, C.; Carlon-Andres, I.; Lagadec, F.; Rayne, F.; Ragues, J.; Kehlenbach, R.H.; Zhang, W.; Ehrhardt, A.; Bystricky, K.; et al. In vivo labelling of adenovirus DNA identifies chromatin anchoring and biphasic genome replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Rieder, U.; Luedtke, N.W. Alkene-tetrazine ligation for imaging cellular DNA. Angew. Chem. 2014, 53, 9168–9172. [Google Scholar] [CrossRef]
- Neef, A.B.; Luedtke, N.W. An azide-modified nucleoside for metabolic labeling of DNA. Chembiochem 2014, 15, 789–793. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, T.G.; Sakin, V.; Müller, B. A Spotlight on Viruses—Application of Click Chemistry to Visualize Virus-Cell Interactions. Molecules 2019, 24, 481. https://doi.org/10.3390/molecules24030481
Müller TG, Sakin V, Müller B. A Spotlight on Viruses—Application of Click Chemistry to Visualize Virus-Cell Interactions. Molecules. 2019; 24(3):481. https://doi.org/10.3390/molecules24030481
Chicago/Turabian StyleMüller, Thorsten G., Volkan Sakin, and Barbara Müller. 2019. "A Spotlight on Viruses—Application of Click Chemistry to Visualize Virus-Cell Interactions" Molecules 24, no. 3: 481. https://doi.org/10.3390/molecules24030481
APA StyleMüller, T. G., Sakin, V., & Müller, B. (2019). A Spotlight on Viruses—Application of Click Chemistry to Visualize Virus-Cell Interactions. Molecules, 24(3), 481. https://doi.org/10.3390/molecules24030481