Glycan Mimetics from Natural Products: New Therapeutic Opportunities for Neurodegenerative Disease

Abstract
:1. Introduction
2. Glycan Biosynthesis and Function
2.1. N-linked Glycosylation
2.2. O-linked Glycosylation
2.3. Attachment of Glycans to Lipids
3. Glycans in Neurodegenerative Diseases
3.1. Glycans and Alzheimer’s Disease
3.2. Glycans in Parkinson’s Disease
3.3. Glycans in Huntington’s Disease
3.4. Glycans in Multiple Sclerosis
3.5. Glycans and Amyotrophic Lateral Sclerosis
4. Glycan-Based Therapies for Neurodegenerative Disease
4.1. Glycosylation Modulators
4.2. Glycan Mimetics from Natural Products
4.2.1. Human Natural Killer-1 (HNK-1) Mimicking Natural Compound
4.2.2. Lewis X (LeX) Mimicking Natural Compounds
4.2.3. L1CAM Mimicking Natural Compound
4.2.4. Polysialic Acid (PSA) Mimicking Natural Compounds
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AGEs | Advanced glycation end-product |
| AICD | Amyloid precursor protein intracellular domain |
| ALS | Amyotrophic lateral sclerosis |
| ANP | Atrial natriuretic peptide |
| APP | Amyloid precursor protein |
| Asn | Asparagine |
| Aβ | Amyloid beta |
| BACE-1 | β-Site APP-cleaving enzyme 1 |
| CAG | Glutamine-coding |
| CCI4 | Carbon tetrachloride |
| CMD | Congenital muscular dystrophy |
| CML | N-carboxymethyl-lysine |
| CN | Caudate nucleus |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CSF | Cerebrospinal fluid |
| CTF | C-terminal fragments |
| CTLA-4 | Cytotoxic T lymphocyte antigen 4 |
| dolichol-P | Dolichol-phosphate |
| DON | 6-Diazo-5-oxo-L-norleucine |
| ECM | Extracellular matrix |
| ELISA | Enzyme-linked immunosorbent assay |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| ErkFDA | Extracellular signal-regulated kinasesFood and Drug Administration |
| GlcNAc | N-acetylglucosamine |
| GlcNAc-1-P | N-acetylglucosamine-1-phosphate |
| GlcNAc6ST1 | A carbohydrate sulfotransferase |
| GM1 | Monosialotetrahexosylganglioside |
| GM2 | Monosialic ganglioside |
| GM3 | Monosialodihexosylganglioside |
| GSLs | Glycosphingolipids |
| HD | Huntington’s disease |
| HMGB1 | High mobility group box 1 protein |
| HNK-1 | Human natural killer-1 glycan antigen |
| HSAS | X-linked hydrocephalus with stenosis of the aqueduct of sylvius |
| I-R | Ischemia–reperfusion |
| IgG | Immunoglobulin G |
| IL-6 | Interleukin 6 |
| L-DOPA | Levodopa |
| L1CAM | Neural cell adhesion molecule L1 |
| LacCer | Lactosylceramide |
| Lewy bodies | α-Synuclein aggregates |
| LeX | LewisX |
| LIGA-20 | A blood–brain barrier-permeable GM1 analogue |
| MALDI-TOF | Matrix-assisted Laser Desorption/Ionization |
| MASA syndrome | A rare X-linked recessive neurological disorder on the L1 disorder spectrum |
| MEB | Muscle–eye–brain disease |
| mHtt | Mutant huntingtin |
| MS | Multiple Sclerosis |
| NCAM | Neural cell adhesion molecule |
| NeuGc | N-glycolylneuraminic acid |
| NF-kβ | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| NSPC | Neural stem and progenitor cells |
| NTD | Neural tube defect |
| O-GlcNAc | O-linked β-N-acetylglucosamine |
| OGA | O-GlcNAcase |
| OGT | O-GlcNAc transferase |
| P0 | Myelin protein zero |
| p3 peptide | 3-kDa fragments of amino-terminal truncated Aβ peptides |
| PD | Parkinson’s disease |
| PHFs | Paired helical filaments |
| PNS | Peripheral nervous system |
| pRb | Etinoblastoma protein |
| PS | Presenilin |
| PSA | Polysialic acid |
| PST | ST8Sia IV |
| PTM | Post-translational modification |
| RAGE | Receptor for advanced glycation end-products |
| ROS | Reactive oxygen species |
| Ser | Serine |
| Sirt1 | NAD-dependent deacetylase sirtuin-1 |
| sLeX | Sialyl LewisX |
| SOD1 | Superoxide dismutase 1 |
| sRAGE | Soluble receptor for advanced glycation end-products |
| ST | Sialyltransferase |
| STAT3 | Signal transducer and activator of transcription 3 |
| STX | ST8Sia II |
| Thr | Threonine |
| TNF-α | Tumor necrosis factor alpha |
| UA | Ursolic acid |
| UDP | Uridine diphosphate |
| α-Sp22 | A glycosylated form of α-synuclein |
References
- Freeze, H.H.; Kinoshita, T.; Varki, A. Glycans in Acquired Human Diseases. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 583–595. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, P.H. Monosaccharide Diversity. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Schnaar, R.L., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 19–30. [Google Scholar] [CrossRef]
- Ryu, K.; Lin, S.; Shao, J.; Song, J.; Chen, M.; Wang, W.; Li, H.; Yi, W.; Wang, P.G. Synthesis of complex carbohydrates and glyconjugates: Enzymatic synthesis of globotetraose using alpha-1,3-N-acetylgalactosaminyltransferase LgtD from Haemophilus infuenzae strain Rd. Methods Mol. Biol. 2005, 310, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic acids in the brain: Gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rini, J.M.; Esko, J.D. Glycosyltransferases and Glycan-Processing Enzymes. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 65–75. [Google Scholar] [CrossRef]
- Bojarova, P.; Kren, V. Glycosidases: A key to tailored carbohydrates. Trends Biotechnol. 2009, 27, 199–209. [Google Scholar] [CrossRef]
- Nilsson, J.; Halim, A.; Grahn, A.; Larson, G. Targeting the glycoproteome. Glycoconj. J. 2013, 30, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.B.; Loganathan, D.; Merchant, Z.M.; Linhardt, R.J. Carbohydrate analysis of glycoproteins. A review. Appl. Biochem. Biotechnol. 1990, 23, 53–80. [Google Scholar] [CrossRef]
- Sandhoff, K.; Kolter, T. Glycolipids of the cell surface—Biochemistry of their decomposition. Naturwissenschaften 1995, 82, 403–413. [Google Scholar]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomin, V.H.; Mulloy, B. Glycosaminoglycans and Proteoglycans. Pharmaceuticals 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, C.M.; Hart, G.W. Nucleocytoplasmic Glycosylation. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 223–238. [Google Scholar] [CrossRef]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chui, D.; Sellakumar, G.; Green, R.; Sutton-Smith, M.; McQuistan, T.; Marek, K.; Morris, H.; Dell, A.; Marth, J. Genetic remodeling of protein glycosylation in vivo induces autoimmune disease. Proc. Natl. Acad. Sci. USA 2001, 98, 1142–1147. [Google Scholar] [CrossRef] [Green Version]
- Abou-Abbass, H.; Abou-El-Hassan, H.; Bahmad, H.; Zibara, K.; Zebian, A.; Youssef, R.; Ismail, J.; Zhu, R.; Zhou, S.; Dong, X.; et al. Glycosylation and other PTMs alterations in neurodegenerative diseases: Current status and future role in neurotrauma. Electrophoresis 2016, 37, 1549–1561. [Google Scholar] [CrossRef]
- Hwang, H.; Zhang, J.; Chung, K.A.; Leverenz, J.B.; Zabetian, C.P.; Peskind, E.R.; Jankovic, J.; Su, Z.; Hancock, A.M.; Pan, C.; et al. Glycoproteomics in neurodegenerative diseases. Mass Spectrom. Rev. 2010, 29, 79–125. [Google Scholar] [CrossRef] [Green Version]
- Colley, K.J.; Varki, A.; Kinoshita, T. Cellular Organization of Glycosylation. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 41–49. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Nagata, K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 2013, 24, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Nakatsukasa, K.; Brodsky, J.L. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 2008, 9, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell. Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Coker, L.H.; Wagenknecht, L.E. Advanced glycation end products, diabetes, and the brain. Neurology 2011, 77, 1326–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabaton, M.; Perry, G.; Smith, M.; Vitek, M.; Angelini, G.; Dapino, D.; Garibaldi, S.; Zaccheo, D.; Odetti, P. Is amyloid beta-protein glycated in Alzheimer’s disease? Neuroreport 1997, 8, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Schmidt, A.M.; Brett, J.; Godman, G.; Zou, Y.S.; Scott, C.W.; Caputo, C.; Frappier, T.; Smith, M.A.; et al. Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc. Natl. Acad. Sci. USA 1994, 91, 7787–7791. [Google Scholar] [CrossRef] [Green Version]
- Konig, A.; Vicente Miranda, H.; Outeiro, T.F. Alpha-Synuclein Glycation and the Action of Anti-Diabetic Agents in Parkinson’s Disease. J. Parkinsons. Dis. 2018, 8, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Salahuddin, P.; Rabbani, G.; Khan, R.H. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, W.; Lin, P.; Wu, B.; Fu, Z.G.; Shen, H.M.; Jing, L.; Liu, Z.Y.; Zhou, Y.; Meng, Y.; et al. N-linked glycosylation at Asn152 on CD147 affects protein folding and stability: Promoting tumour metastasis in hepatocellular carcinoma. Sci. Rep. 2016, 6, 35210. [Google Scholar] [CrossRef] [Green Version]
- Wildt, S.; Gerngross, T.U. The humanization of N-glycosylation pathways in yeast. Nat. Rev. Microbiol. 2005, 3, 119–128. [Google Scholar] [CrossRef]
- Spiro, R.G. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002, 12, 43R–56R. [Google Scholar] [CrossRef]
- Stanley, P.; Schachter, H.; Taniguchi, N. N-Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Freeze, H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006, 7, 537–551. [Google Scholar] [CrossRef]
- Stanley, P.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 99–111. [Google Scholar] [CrossRef]
- Peixoto, A.; Relvas-Santos, M.; Azevedo, R.; Santos, L.L.; Ferreira, J.A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.S.; Ma, J.; Hart, G.W. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 6050–6055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachara, N.; Akimoto, Y.; Hart, G.W. The O-GlcNAc Modification. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 239–251. [Google Scholar] [CrossRef]
- Ernst, J.F.; Prill, S.K. O-glycosylation. Med. Mycol. 2001, 39 (Suppl. 1), 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.; Li, D.; Chen, K. Structures and functions of invertebrate glycosylation. Open. Biol. 2019, 9, 180232. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Sasmal, A.; Zhang, Y.; Gao, T.; Liu, C.C.; Khan, N.; Varki, A.; Wang, F.; Cao, H. Chemoenzymatic Assembly of Mammalian O-Mannose Glycans. Angew. Chem. Int. Ed. Engl. 2018, 57, 9003–9007. [Google Scholar] [CrossRef] [PubMed]
- Breloy, I. O-glycomics: Profiling and structural analysis of mucin-type O-linked glycans. Methods Mol. Biol. 2012, 842, 165–177. [Google Scholar] [PubMed]
- Freeze, H.H.; Haltiwanger, R.S. Other Classes of ER/Golgi-derived Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Martin, P.T. The dystroglycanopathies: The new disorders of O-linked glycosylation. Semin. Pediatr. Neurol. 2005, 12, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.T. Congenital muscular dystrophies involving the O-mannose pathway. Curr. Mol. Med. 2007, 7, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.W.; Sorum, A.W.; Hsieh-Wilson, L.C. Deciphering the Functions of O-GlcNAc Glycosylation in the Brain: The Role of Site-Specific Quantitative O-GlcNAcomics. Biochemistry 2018, 57, 4010–4018. [Google Scholar] [CrossRef] [Green Version]
- Lagerlof, O. O-GlcNAc cycling in the developing, adult and geriatric brain. J. Bioenerg. Biomembr. 2018, 50, 241–261. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. alpha-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akan, I.; Olivier-Van Stichelen, S.; Bond, M.R.; Hanover, J.A. Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration. J. Neurochem. 2018, 144, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmano, K.; Rowan, A.; Guillermo, R.; Guan, J.; McJarrow, P. The role of gangliosides in neurodevelopment. Nutrients 2015, 7, 3891–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo. Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.K.; Tsai, Y.T.; Ariga, T. Functional roles of gangliosides in neurodevelopment: An overview of recent advances. Neurochem. Res. 2012, 37, 1230–1244. [Google Scholar] [CrossRef] [Green Version]
- Benady, A.; Freidin, D.; Pick, C.G.; Rubovitch, V. GM1 ganglioside prevents axonal regeneration inhibition and cognitive deficits in a mouse model of traumatic brain injury. Sci. Rep. 2018, 8, 13340. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.; Ghanimi Fard, M.; Everest-Dass, A.; Packer, N.H.; Parker, L.M. Understanding cellular glycan surfaces in the central nervous system. Biochem. Soc. Trans. 2019, 47, 89–100. [Google Scholar] [CrossRef]
- Scott, H.; Panin, V.M. N-glycosylation in regulation of the nervous system. Adv. Neurobiol. 2014, 9, 367–394. [Google Scholar]
- Morita, I.; Kakuda, S.; Takeuchi, Y.; Kawasaki, T.; Oka, S. HNK-1 (human natural killer-1) glyco-epitope is essential for normal spine morphogenesis in developing hippocampal neurons. Neuroscience 2009, 164, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Prineas, J.W.; Kwon, E.E.; Goldenberg, P.Z.; Ilyas, A.A.; Quarles, R.H.; Benjamins, J.A.; Sprinkle, T.J. Multiple sclerosis. Oligodendrocyte proliferation and differentiation in fresh lesions. Lab. Investig. 1989, 61, 489–503. [Google Scholar] [PubMed]
- Bonfanti, L.; Theodosis, D.T. Polysialic acid and activity-dependent synapse remodeling. Cell Adh. Migr. 2009, 3, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, L.B.; Winslow, A.R.; Strasser, S.D. Systems biology of neurodegenerative diseases. Integr. Biol. (Camb) 2015, 7, 758–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate beta-Amyloid Aggregation in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Camara, H.; De-Souza, E.A. beta-Amyloid Accumulation Slows Earlier than Expected in Preclinical Alzheimer’s Disease Patients. J. Neurosci. 2018, 38, 9123–9125. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Bharadwaj, P.R.; Dubey, A.K.; Masters, C.L.; Martins, R.N.; Macreadie, I.G. Abeta aggregation and possible implications in Alzheimer’s disease pathogenesis. J. Cell Mol. Med. 2009, 13, 412–421. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M. Molecular mechanism of the intramembrane cleavage of the beta-carboxyl terminal fragment of amyloid precursor protein by gamma-secretase. Front. Physiol. 2014, 5, 463. [Google Scholar] [CrossRef] [Green Version]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromolecular. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Emendato, A.; Milordini, G.; Zacco, E.; Sicorello, A.; Dal Piaz, F.; Guerrini, R.; Thorogate, R.; Picone, D.; Pastore, A. Glycation affects fibril formation of Abeta peptides. J. Biol. Chem. 2018, 293, 13100–13111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Kozutsumi, Y.; Saito, Y.; Taniguchi, N.; Murayama, S.; Spitalnik, S.L.; Endo, T. Protective effect of N-glycan bisecting GlcNAc residues on beta-amyloid production in Alzheimer’s disease. Glycobiology 2010, 20, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, T.M.; Gillian, A.M.; O’Mahony, D.; Coughlan, C.M.; Dennihan, A.; Breen, K.C. A decrease in serum sialyltransferase levels in Alzheimer’s disease. Neurobiol. Aging 1994, 15, 99–102. [Google Scholar] [CrossRef]
- Guevara, J.; Espinosa, B.; Zenteno, E.; Vazguez, L.; Luna, J.; Perry, G.; Mena, R. Altered glycosylation pattern of proteins in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 57, 905–914. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Pahlsson, P.; Shakin-Eshleman, S.H.; Spitalnik, S.L. N-linked glycosylation of beta-amyloid precursor protein. Biochem. Biophys. Res. Commun. 1992, 189, 1667–1673. [Google Scholar] [CrossRef]
- Tsatsanis, A.; Dickens, S.; Kwok, J.C.F.; Wong, B.X.; Duce, J.A. Post Translational Modulation of beta-Amyloid Precursor Protein Trafficking to the Cell Surface Alters Neuronal Iron Homeostasis. Neurochem. Res. 2019, 44, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, X.; Li, G.; Zhang, Y.; Wu, Y.; Song, W. Modifications and Trafficking of APP in the Pathogenesis of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef]
- Perdivara, I.; Petrovich, R.; Allinquant, B.; Deterding, L.J.; Tomer, K.B.; Przybylski, M. Elucidation of O-glycosylation structures of the beta-amyloid precursor protein by liquid chromatography-mass spectrometry using electron transfer dissociation and collision induced dissociation. J. Proteome. Res. 2009, 8, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.C.; Cheng, K.M.; Ma, Y.L.; Hsu, W.L.; Chen, Y.C.; Fuh, J.L.; Lee, W.J.; Chao, C.C.; Lee, E.H.Y. Galectin-3 promotes Abeta oligomerization and Abeta toxicity in a mouse model of Alzheimer’s disease. Cell Death Differ. 2019. [Google Scholar] [CrossRef]
- Malito, E.; Hulse, R.E.; Tang, W.J. Amyloid beta-degrading cryptidases: Insulin degrading enzyme, presequence peptidase, and neprilysin. Cell Mol. Life. Sci. 2008, 65, 2574–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, L.H.; Madsen, T.D.; Goth, C.K.; Clausen, H.; Chen, Y.; Dzhoyashvili, N.; Iyer, S.R.; Sangaralingham, S.J.; Burnett, J.C., Jr.; Rehfeld, J.F.; et al. Discovery of O-glycans on atrial natriuretic peptide (ANP) that affect both its proteolytic degradation and potency at its cognate receptor. J. Biol. Chem. 2019, 294, 12567–12578. [Google Scholar] [CrossRef] [Green Version]
- Halim, A.; Brinkmalm, G.; Ruetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizuka, Y.; Kitazume, S.; Fujinawa, R.; Saito, T.; Iwata, N.; Saido, T.C.; Nakano, M.; Yamaguchi, Y.; Hashimoto, Y.; Staufenbiel, M.; et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol. Med. 2015, 7, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Naito, Y.; Grundke-Iqbal, I.; Iqbal, K.; Endo, T. Analysis of N-glycans of pathological tau: Possible occurrence of aberrant processing of tau in Alzheimer’s disease. FEBS Lett. 2001, 496, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Regan, P.; McClean, P.L.; Smyth, T.; Doherty, M. Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines 2019, 6, 92. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Glycosylation of microtubule-associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat. Med. 1996, 2, 871–875. [Google Scholar] [CrossRef]
- Bourre, G.; Cantrelle, F.X.; Kamah, A.; Chambraud, B.; Landrieu, I.; Smet-Nocca, C. Direct Crosstalk Between O-GlcNAcylation and Phosphorylation of Tau Protein Investigated by NMR Spectroscopy. Front. Endocrinol. (Lausanne) 2018, 9, 595. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [Green Version]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin, cognition, and dementia. Eur. J. Pharmacol. 2013, 719, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accardi, G.; Caruso, C.; Colonna-Romano, G.; Camarda, C.; Monastero, R.; Candore, G. Can Alzheimer disease be a form of type 3 diabetes? Rejuvenation. Res. 2012, 15, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Ko, H.A.; Chu, K.H.; Shieh, T.M.; Chi, T.C.; Chen, H.I.; Chang, W.C.; Chang, S.S. The Possible Mechanism of Advanced Glycation End Products (AGEs) for Alzheimer’s Disease. PLoS ONE 2015, 10, e0143345. [Google Scholar] [CrossRef]
- Chen, J.H.; Lin, X.; Bu, C.; Zhang, X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr. Metab. (Lond.) 2018, 15, 72. [Google Scholar] [CrossRef]
- Ozansoy, M.; Basak, A.N. The central theme of Parkinson’s disease: Alpha-synuclein. Mol. Neurobiol. 2013, 47, 460–465. [Google Scholar] [CrossRef]
- Lehtonen, S.; Sonninen, T.M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of Cellular Proteostasis in Parkinson’s Disease. Front. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef] [Green Version]
- Videira, P.A.Q.; Castro-Caldas, M. Linking Glycation and Glycosylation With Inflammation and Mitochondrial Dysfunction in Parkinson’s Disease. Front. Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef]
- Zhang, G.; Xia, Y.; Wan, F.; Ma, K.; Guo, X.; Kou, L.; Yin, S.; Han, C.; Liu, L.; Huang, J.; et al. New Perspectives on Roles of Alpha-Synuclein in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 370. [Google Scholar] [CrossRef] [Green Version]
- Birol, M.; Wojcik, S.P.; Miranker, A.D.; Rhoades, E. Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated alpha-Synuclein. PLoS Biol. 2019, 17, e3000318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.S. Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson’s disease. PLoS ONE 2018, 13, e0199189. [Google Scholar] [CrossRef] [PubMed]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Cisbani, G.; Cicchetti, F. An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis. 2012, 3, e382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, A.; Larsen, K.E.; Behr, G.G.; Romero, N.; Przedborski, S.; Brundin, P.; Sulzer, D. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum. Mol. Genet. 2001, 10, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.A.; Denny, C.A.; Kass, K.E.; Gilmartin, T.; Head, S.R.; Sutcliffe, J.G.; Seyfried, T.N.; Thomas, E.A. Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol. Dis. 2007, 27, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gizaw, S.T.; Koda, T.; Amano, M.; Kamimura, K.; Ohashi, T.; Hinou, H.; Nishimura, S. A comprehensive glycome profiling of Huntington’s disease transgenic mice. Biochim. Biophys. Acta 2015, 1850, 1704–1718. [Google Scholar] [CrossRef] [Green Version]
- Goh, J.B.; Ng, S.K. Impact of host cell line choice on glycan profile. Crit. Rev. Biotechnol. 2018, 38, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Singh, P.K.; Parihar, R.; Dwivedi, V.; Lakhotia, S.C.; Ganesh, S. Decreased O-linked GlcNAcylation protects from cytotoxicity mediated by huntingtin exon1 protein fragment. J. Biol. Chem. 2014, 289, 13543–13553. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Nicholson, L.F. Expression of the receptor for advanced glycation end products in Huntington’s disease caudate nucleus. Brain Res. 2004, 1018, 10–17. [Google Scholar] [CrossRef]
- Hong, Y.; Tang, H.R.; Ma, M.; Chen, N.; Xie, X.; He, L. Multiple sclerosis and stroke: A systematic review and meta-analysis. BMC Neurol. 2019, 19, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mkhikian, H.; Grigorian, A.; Li, C.F.; Chen, H.L.; Newton, B.; Zhou, R.W.; Beeton, C.; Torossian, S.; Tatarian, G.G.; Lee, S.U.; et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat. Commun. 2011, 2, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.U.; Grigorian, A.; Pawling, J.; Chen, I.J.; Gao, G.; Mozaffar, T.; McKerlie, C.; Demetriou, M. N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J. Biol. Chem. 2007, 282, 33725–33734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, Z.; Ostrow, P.; Vaughan, M.; Chichelli, T.; Munschauer, F. AGE-RAGE in multiple sclerosis brain. Immunol. Investig. 2011, 40, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Juranek, J.K.; Daffu, G.K.; Geddis, M.S.; Li, H.; Rosario, R.; Kaplan, B.J.; Kelly, L.; Schmidt, A.M. Soluble RAGE Treatment Delays Progression of Amyotrophic Lateral Sclerosis in SOD1 Mice. Front. Cell. Neurosci. 2016, 10, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edri-Brami, M.; Rosental, B.; Hayoun, D.; Welt, M.; Rosen, H.; Wirguin, I.; Nefussy, B.; Drory, V.E.; Porgador, A.; Lichtenstein, R.G. Glycans in sera of amyotrophic lateral sclerosis patients and their role in killing neuronal cells. PLoS ONE 2012, 7, e35772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Takeda-Uchimura, Y.; Foyez, T.; Ohtake-Niimi, S.; Narentuya; Akatsu, H.; Nishitsuji, K.; Michikawa, M.; Wyss-Coray, T.; Kadomatsu, K.; et al. Deficiency of a sulfotransferase for sialic acid-modified glycans mitigates Alzheimer’s pathology. Proc. Natl. Acad. Sci. USA 2017, 114, E2947–E2954. [Google Scholar] [CrossRef] [Green Version]
- Drannik, A.; Martin, J.; Peterson, R.; Ma, X.; Jiang, F.; Turnbull, J. Cerebrospinal fluid from patients with amyotrophic lateral sclerosis inhibits sonic hedgehog function. PLoS ONE 2017, 12, e0171668. [Google Scholar] [CrossRef]
- Esko, J.D.; Bertozzi, C.; Schnaar, R.L. Chemical Tools for Inhibiting Glycosylation. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 701–712. [Google Scholar] [CrossRef]
- Cole, N.B.; Ellenberg, J.; Song, J.; DiEuliis, D.; Lippincott-Schwartz, J. Retrograde transport of Golgi-localized proteins to the ER. J. Cell Biol. 1998, 140, 1–15. [Google Scholar] [CrossRef]
- Fishman, P.H.; Curran, P.K. Brefeldin A inhibits protein synthesis in cultured cells. FEBS Lett. 1992, 314, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Duan, F.; Jia, D.; Zhao, J.; Wu, W.; Min, L.; Song, S.; Wu, H.; Wang, L.; Wang, H.; Ruan, Y.; et al. Loss of GFAT1 promotes epithelial-to-mesenchymal transition and predicts unfavorable prognosis in gastric cancer. Oncotarget 2016, 7, 38427–38439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manivannan, S.; Baxter, V.K.; Schultz, K.L.; Slusher, B.S.; Griffin, D.E. Protective Effects of Glutamine Antagonist 6-Diazo-5-Oxo-l-Norleucine in Mice with Alphavirus Encephalomyelitis. J. Virol. 2016, 90, 9251–9262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, Y.; Jiang, L.; Zhang, J.; Gao, J.; Shen, Z.; Zheng, Y.; Deng, T.; Yan, H.; Li, W.; et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014, 10, 1801–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantagrel, V.; Lefeber, D.J. From glycosylation disorders to dolichol biosynthesis defects: A new class of metabolic diseases. J. Inherit. Metab. Dis. 2011, 34, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Gloster, T.M.; Davies, G.J. Glycosidase inhibition: Assessing mimicry of the transition state. Org. Biomol. Chem. 2010, 8, 305–320. [Google Scholar] [CrossRef] [Green Version]
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. Adv. Neurobiol. 2014, 9, 47–70. [Google Scholar]
- Ratner, N.; Elbein, A.; Bunge, M.B.; Porter, S.; Bunge, R.P.; Glaser, L. Specific asparagine-linked oligosaccharides are not required for certain neuron-neuron and neuron-Schwann cell interactions. J. Cell Biol. 1986, 103, 159–170. [Google Scholar] [CrossRef]
- Bird, M.M. The effect of castanospermine on embryonic mouse cerebellar neurons in culture. J. Electron. Microsc. (Tokyo) 1999, 48, 261–266. [Google Scholar] [CrossRef]
- Tulsiani, D.R.; Touster, O. Swainsonine, a potent mannosidase inhibitor, elevates rat liver and brain lysosomal alpha-D-mannosidase, decreases Golgi alpha-D-mannosidase II, and increases the plasma levels of several acid hydrolases. Arch. Biochem. Biophys. 1983, 224, 594–600. [Google Scholar] [CrossRef]
- Mohla, S.; Humphries, M.J.; White, S.L.; Matsumoto, K.; Newton, S.A.; Sampson, C.C.; Bowen, D.; Olden, K. Swainsonine: A new antineoplastic immunomodulator. J. Natl. Med. Assoc. 1989, 81, 1049–1056. [Google Scholar] [PubMed]
- Butters, T.D.; Alonzi, D.S.; Kukushkin, N.V.; Ren, Y.; Bleriot, Y. Novel mannosidase inhibitors probe glycoprotein degradation pathways in cells. Glycoconj. J. 2009, 26, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Yale, A.R.; Nourse, J.L.; Lee, K.R.; Ahmed, S.N.; Arulmoli, J.; Jiang, A.Y.L.; McDonnell, L.P.; Botten, G.A.; Lee, A.P.; Monuki, E.S.; et al. Cell Surface N-Glycans Influence Electrophysiological Properties and Fate Potential of Neural Stem Cells. Stem Cell Rep. 2018, 11, 869–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.Y.; Wang, J.; Ma, J.; Zhang, Y.Q. Interference effect of oral administration of mulberry branch bark powder on the incidence of type II diabetes in mice induced by streptozotocin. Food Nutr. Res. 2016, 60, 31606. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Sulfated Glycans and Related Digestive Enzymes in the Zika Virus Infectivity: Potential Mechanisms of Virus-Host Interaction and Perspectives in Drug Discovery. Interdiscip. Perspect. Infect. Dis. 2017, 2017, 4894598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, N.B.; Wang, X.; Song, L.; Butts, B.D.; Grotz, D.; Hargreaves, R.; Fred Hess, J.; Hong, K.K.; Huang, C.R.; Hyde, L.; et al. Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol. Neurodegener. 2017, 12, 39. [Google Scholar] [CrossRef]
- Nicolas, C.; Martin, O.R. Glycoside Mimics from Glycosylamines: Recent Progress. Molecules 2018, 23, 1612. [Google Scholar] [CrossRef] [Green Version]
- Hudak, J.E.; Bertozzi, C.R. Glycotherapy: New advances inspire a reemergence of glycans in medicine. Chem. Biol. 2014, 21, 16–37. [Google Scholar] [CrossRef] [Green Version]
- Hevey, R. Strategies for the Development of Glycomimetic Drug Candidates. Pharmaceuticals 2019, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- Sahu, S.; Li, R.; Kadeyala, P.K.; Liu, S.; Schachner, M. The human natural killer-1 (HNK-1) glycan mimetic ursolic acid promotes functional recovery after spinal cord injury in mouse. J. Nutr. Biochem. 2018, 55, 219–228. [Google Scholar] [CrossRef]
- Theis, T.; Johal, A.S.; Kabat, M.; Basak, S.; Schachner, M. Enhanced Neuronal Survival and Neurite Outgrowth Triggered by Novel Small Organic Compounds Mimicking the LewisX Glycan. Mol. Neurobiol. 2018, 55, 8203–8215. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hu, C.; Jiang, Q.; Pan, H.; Shen, H.; Schachner, M. Trimebutine, a small molecule mimetic agonist of adhesion molecule L1, contributes to functional recovery after spinal cord injury in mice. Dis. Models Mech. 2017, 10, 1117–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, V.; Lutz, D.; Kataria, H.; Kaur, G.; Schachner, M.; Loers, G. The polysialic acid mimetics 5-nonyloxytryptamine and vinorelbine facilitate nervous system repair. Sci. Rep. 2016, 6, 26927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morise, J.; Takematsu, H.; Oka, S. The role of human natural killer-1 (HNK-1) carbohydrate in neuronal plasticity and disease. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2455–2461. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Lok, J.; Washida, K.; Arai, K. Mechanisms of Axonal Damage and Repair after Central Nervous System Injury. Transl. Stroke Res. 2017, 8, 14–21. [Google Scholar] [CrossRef]
- Yoshihara, T.; Sugihara, K.; Kizuka, Y.; Oka, S.; Asano, M. Learning/memory impairment and reduced expression of the HNK-1 carbohydrate in beta4-galactosyltransferase-II-deficient mice. J. Biol. Chem. 2009, 284, 12550–12561. [Google Scholar] [CrossRef] [Green Version]
- Senn, C.; Kutsche, M.; Saghatelyan, A.; Bosl, M.R.; Lohler, J.; Bartsch, U.; Morellini, F.; Schachner, M. Mice deficient for the HNK-1 sulfotransferase show alterations in synaptic efficacy and spatial learning and memory. Mol. Cell. Neurosci. 2002, 20, 712–729. [Google Scholar] [CrossRef]
- Bizzoca, A.; Corsi, P.; Gennarini, G. The mouse F3/contactin glycoprotein: Structural features, functional properties and developmental significance of its regulated expression. Cell Adh. Migr. 2009, 3, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid: An anti- and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42. [Google Scholar] [CrossRef]
- Tsai, S.J.; Yin, M.C. Antioxidative and anti-inflammatory protection of oleanolic acid and ursolic acid in PC12 cells. J. Food. Sci. 2008, 73, H174–H178. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Cui, L.; Wang, L.; Liu, H.; Ji, H.; Du, Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013, 1497, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Q.; Ding, J.; Zhang, L.; Liu, C.M. Ursolic acid protects mouse liver against CCl4-induced oxidative stress and inflammation by the MAPK/NF-kappaB pathway. Environ. Toxicol. Pharmacol. 2014, 37, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F.; Ye, Q.; Liu, C.M.; Shan, Q.; Wang, Y.J. Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-kappaB pathway activation. Cereb. Cortex 2010, 20, 2540–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.Y.; Jeong, W.S.; Jun, M. Protective effects of the key compounds isolated from Corni fructus against beta-amyloid-induced neurotoxicity in PC12 cells. Molecules 2012, 17, 10831–10845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, H.; Saito, T.; Yanagisawa, M.; Yu, R.K.; Kato, K. Lewis X-carrying N-glycans regulate the proliferation of mouse embryonic neural stem cells via the Notch signaling pathway. J. Biol. Chem. 2012, 287, 24356–24364. [Google Scholar] [CrossRef] [Green Version]
- Trinchera, M.; Aronica, A.; Dall’Olio, F. Selectin Ligands Sialyl-Lewis a and Sialyl-Lewis x in Gastrointestinal Cancers. Biology 2017, 6, 16. [Google Scholar] [CrossRef]
- Qian, S.Z.; Wang, Z.G. Gossypol: A potential antifertility agent for males. Annu. Rev. Pharmacol. Toxicol. 1984, 24, 329–360. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Wierda, W.G.; Keating, M.J.; Gandhi, V. Gossypol, a BH3 mimetic, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2008, 112, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- Bagrodia, S.; Derijard, B.; Davis, R.J.; Cerione, R.A. Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 1995, 270, 27995–27998. [Google Scholar]
- Karimi, P.; Kamali, E.; Mousavi, S.M.; Karahmadi, M. Environmental factors influencing the risk of autism. J. Res. Med. Sci. 2017, 22, 27. [Google Scholar]
- van Gool, J.D.; Hirche, H.; Lax, H.; De Schaepdrijver, L. Folic acid and primary prevention of neural tube defects: A review. Reprod. Toxicol. 2018, 80, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Linneberg, C.; Toft, C.L.F.; Kjaer-Sorensen, K.; Laursen, L.S. L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing. Sci. Rep. 2019, 9, 3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocock, R.; Benard, C.Y.; Shapiro, L.; Hobert, O. Functional dissection of the C. elegans cell adhesion molecule SAX-7, a homologue of human L1. Mol. Cell. Neurosci. 2008, 37, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Tagliavacca, L.; Colombo, F.; Racchetti, G.; Meldolesi, J. L1CAM and its cell-surface mutants: New mechanisms and effects relevant to the physiology and pathology of neural cells. J. Neurochem. 2013, 124, 397–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, M.K.; Frotscher, M. Role of L1CAM for axon sprouting and branching. Cell Tissue Res. 2012, 349, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Duan, X.; Yang, G.; Zhang, X.; Deng, L.; Zheng, H.; Deng, C.; Wen, J.; Wang, N.; Peng, C.; et al. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS ONE 2011, 6, e18490. [Google Scholar] [CrossRef]
- Shen, J.L.; Man, K.M.; Huang, P.H.; Chen, W.C.; Chen, D.C.; Cheng, Y.W.; Liu, P.L.; Chou, M.C.; Chen, Y.H. Honokiol and magnolol as multifunctional antioxidative molecules for dermatologic disorders. Molecules 2010, 15, 6452–6465. [Google Scholar] [CrossRef]
- Lin, Y.R.; Chen, H.H.; Ko, C.H.; Chan, M.H. Neuroprotective activity of honokiol and magnolol in cerebellar granule cell damage. Eur. J. Pharmacol. 2006, 537, 64–69. [Google Scholar] [CrossRef]
- Talarek, S.; Listos, J.; Barreca, D.; Tellone, E.; Sureda, A.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Neuroprotective effects of honokiol: From chemistry to medicine. Biofactors 2017, 43, 760–769. [Google Scholar] [CrossRef]
- Woodbury, A.; Yu, S.P.; Wei, L.; Garcia, P. Neuro-modulating effects of honokiol: A review. Front. Neurol. 2013, 4, 130. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Peng, S.; Li, X.; Yao, J.; Xu, J.; Fang, J. Honokiol Alleviates Oxidative Stress-Induced Neurotoxicity via Activation of Nrf2. ACS Chem. Neurosci. 2018, 9, 3108–3116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, X.; Zhu, Y.; Chen, S.; Zhou, D.; Wang, Y. Honokiol inhibits the inflammatory reaction during cerebral ischemia reperfusion by suppressing NF-kappaB activation and cytokine production of glial cells. Neurosci. Lett. 2013, 534, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Hoi, C.P.; Ho, Y.P.; Baum, L.; Chow, A.H. Neuroprotective effect of honokiol and magnolol, compounds from Magnolia officinalis, on beta-amyloid-induced toxicity in PC12 cells. Phytother. Res. 2010, 24, 1538–1542. [Google Scholar] [CrossRef] [PubMed]
- Angata, K.; Huckaby, V.; Ranscht, B.; Terskikh, A.; Marth, J.D.; Fukuda, M. Polysialic acid-directed migration and differentiation of neural precursors are essential for mouse brain development. Mol. Cell. Biol. 2007, 27, 6659–6668. [Google Scholar] [CrossRef] [Green Version]
- Stamatos, N.M.; Zhang, L.; Jokilammi, A.; Finne, J.; Chen, W.H.; El-Maarouf, A.; Cross, A.S.; Hankey, K.G. Changes in polysialic acid expression on myeloid cells during differentiation and recruitment to sites of inflammation: Role in phagocytosis. Glycobiology 2014, 24, 864–879. [Google Scholar] [CrossRef] [Green Version]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Simoens, C.; Lardon, F.; Pauwels, B.; De Pooter, C.M.; Lambrechts, H.A.; Pattyn, G.G.; Breillout, F.; Vermorken, J.B. Comparative study of the radiosensitizing and cell cycle effects of vinflunine and vinorelbine, in vitro. BMC Cancer 2008, 8, 65. [Google Scholar] [CrossRef] [Green Version]

| Diseases | Protein/Gene Products | Known Glycosylation Types | Glycosylation Sites (confirmed) | Functions/Comments | |
|---|---|---|---|---|---|
| Alzheimer’s disease (AD) | APP | N-glycosylated | Asn467 | Asn496 | 1. Defects in N-glycosylation prevent the transportation and secretion of APP 2. O-glycosylated APP decreases Aβ secretion 3. Increase in tyrosine-linked glycan on Aβ fragments has been identified in the CSF samples of AD patients |
| O-GlcNAcylated (APP695) | Thr291 | Thr292 | |||
| Thr576 | |||||
| O-GlcNAcylated (APP770) | Ser597 | Ser606 | |||
| Ser611 | Thr616 | ||||
| Thr634 | Thr635 | ||||
| Ser662 | Ser680 | ||||
| BACE-1 | N-glycosylated | Asn153 | Asn172 | 1. Bisecting GlcNAc modification of BACE-1 increases Aβ production | |
| Asn223 | Asn354 | ||||
| Tau | N-glycosylated | Asn167 | Asn359 | 1. N-glycosylation of Tau appeared to be responsible for the maintenance of the PHFs structure 2. Level of O-GlcNAcylation of Tau is decreased in AD brains | |
| Asn359 | |||||
| O-GlcNAcylated | Ser400 | Thr123 | |||
| Nicastrin | N-glycosylated | 16 potential sites | 1. Defects of O-GlcNAcylation decrease Aβ plaques 2. Function of N-glycosylated remains poorly understood | ||
| O-GlcNAcylated | Ser708 | ||||
| PS | None | ||||
| Parkinson’s disease (PD) | α-synuclein | O-GlcNAcylated | Thr33 | Thr44 | 1. Accumulation of O-linked glycosylation of α-synuclein was found in PD patients |
| Thr54 | Thr59 | ||||
| Thr64 | Thr72 | ||||
| Thr75 | Thr81 | ||||
| Thr87 | |||||
| Huntington’s disease (HD) | huntingtin | O-GlcNAcylated | N/A | 1. O-GlcNAcylation regulates clearance of mHtt 2. O-GlcNAcylation stimulates autophagy and reduces huntingtin aggregation | |
| Multiple Sclerosis (MS) | TNF-α | N/A | N/A | 1. Absence of GlcNAc brancing in neurons induces apoptosis and promotes demyelination 2. N-glycan branching is required to prevent T cell hyperactivity, cytotoxic T lymphocyte antigen 4 (CTLA-4) endocytosis, spontaneous inflammatory demyelination in MS pathology | |
| Amyotrophic Lateral Sclerosis (ALS) | SOD1 | N/A | N/A | 1. CSF IgG N-glycosylation as a potential biomarker for ALS 2. Altered expression of glycoproteins in the sera or CSF were detected in ALS patients | |
| Glycan/Glycoprotein | Natural/Semisynthetic Glycan Mimetics | |
|---|---|---|
| Human natural killer-1 (HNK-1) | 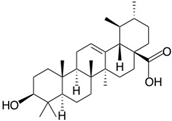 | |
| Ursolic acid | ||
| Lewis X (Lex) | 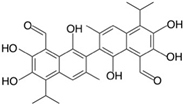 |  |
| Gossypol | Folic acid | |
| Neural cell adhesion molecule L1 (L1CAM) | 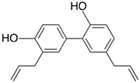 | |
| Honokiol | ||
| Polysialic acid (PSA) |  | |
| Vinorelbine | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Gopal, S.; Pocock, R.; Xiao, Z. Glycan Mimetics from Natural Products: New Therapeutic Opportunities for Neurodegenerative Disease. Molecules 2019, 24, 4604. https://doi.org/10.3390/molecules24244604
Wang W, Gopal S, Pocock R, Xiao Z. Glycan Mimetics from Natural Products: New Therapeutic Opportunities for Neurodegenerative Disease. Molecules. 2019; 24(24):4604. https://doi.org/10.3390/molecules24244604
Chicago/Turabian StyleWang, Wenyue, Sandeep Gopal, Roger Pocock, and Zhicheng Xiao. 2019. "Glycan Mimetics from Natural Products: New Therapeutic Opportunities for Neurodegenerative Disease" Molecules 24, no. 24: 4604. https://doi.org/10.3390/molecules24244604
APA StyleWang, W., Gopal, S., Pocock, R., & Xiao, Z. (2019). Glycan Mimetics from Natural Products: New Therapeutic Opportunities for Neurodegenerative Disease. Molecules, 24(24), 4604. https://doi.org/10.3390/molecules24244604