Covalent Inhibition of the Histamine H3 Receptor

 , , ,
, , ,  ,
,  ,
,
Abstract
1. Introduction
2. Results
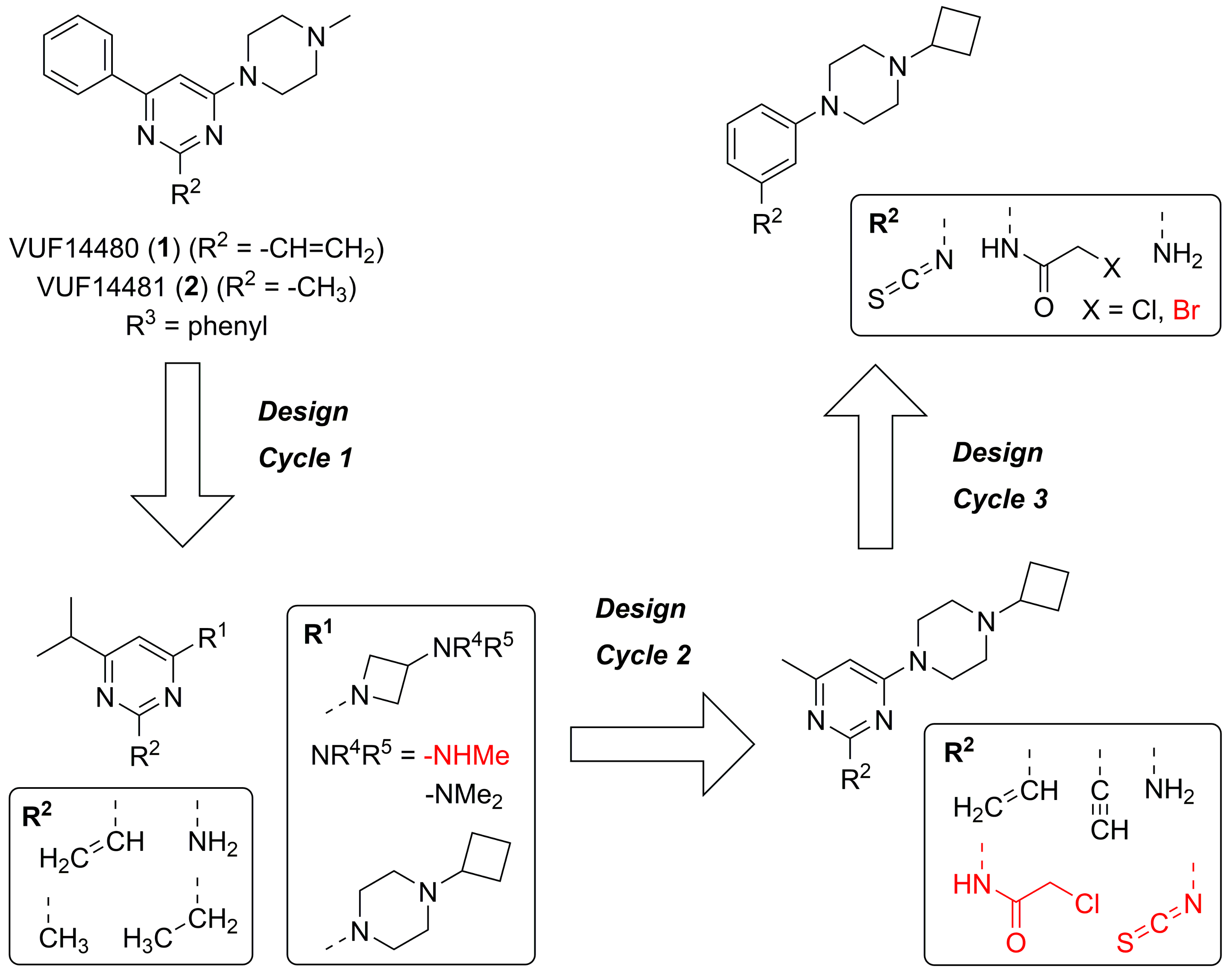
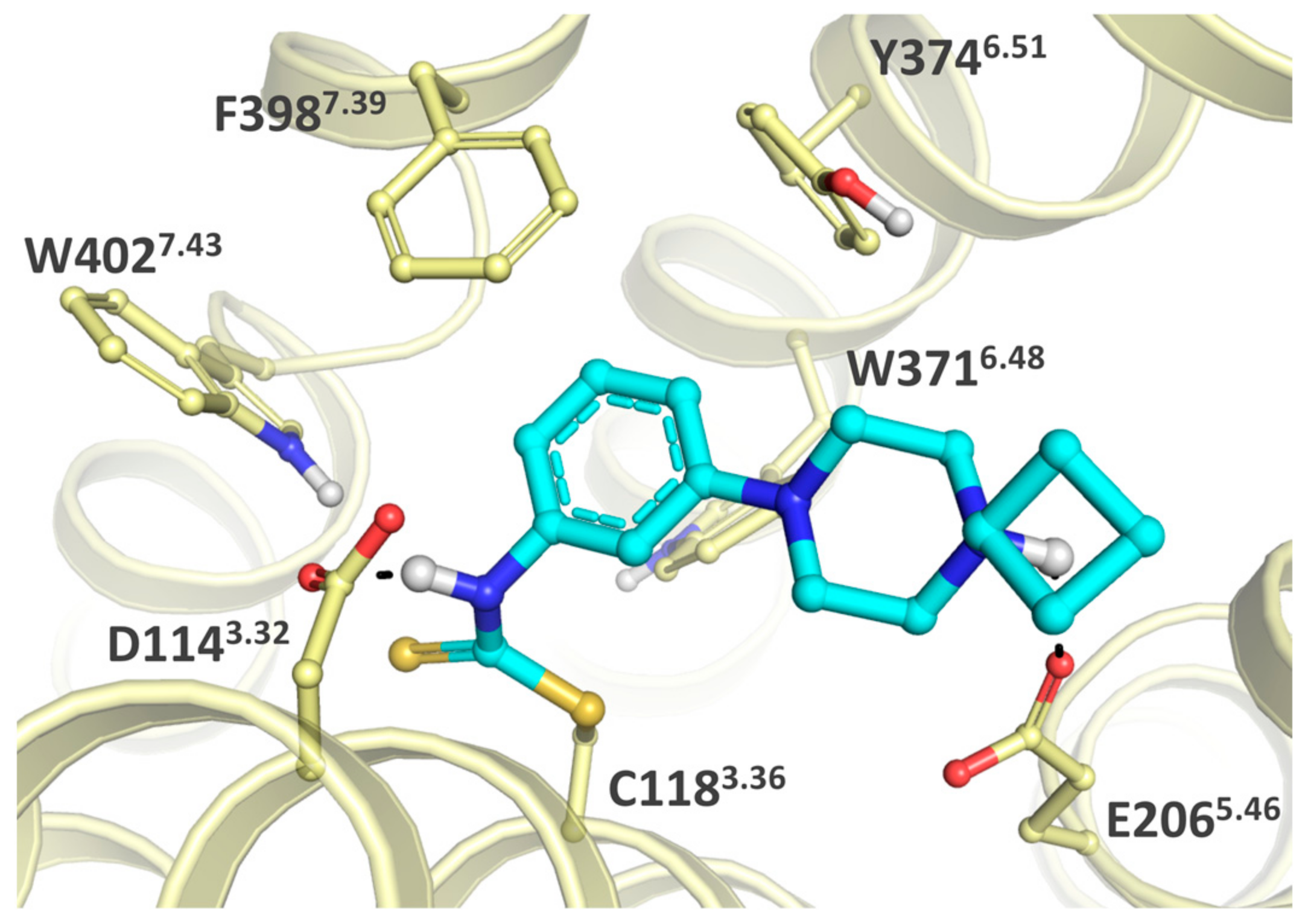
2.1. Design
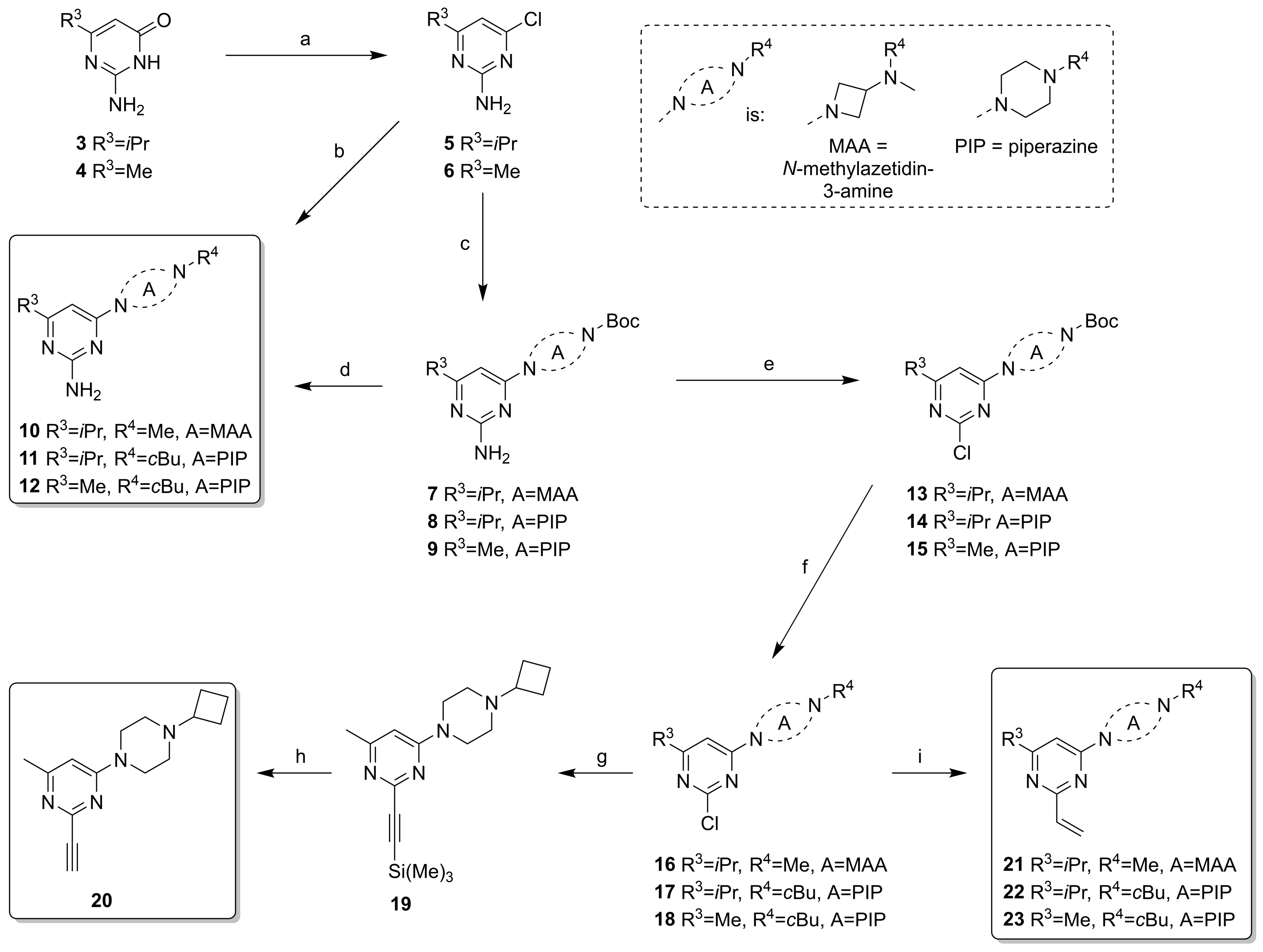
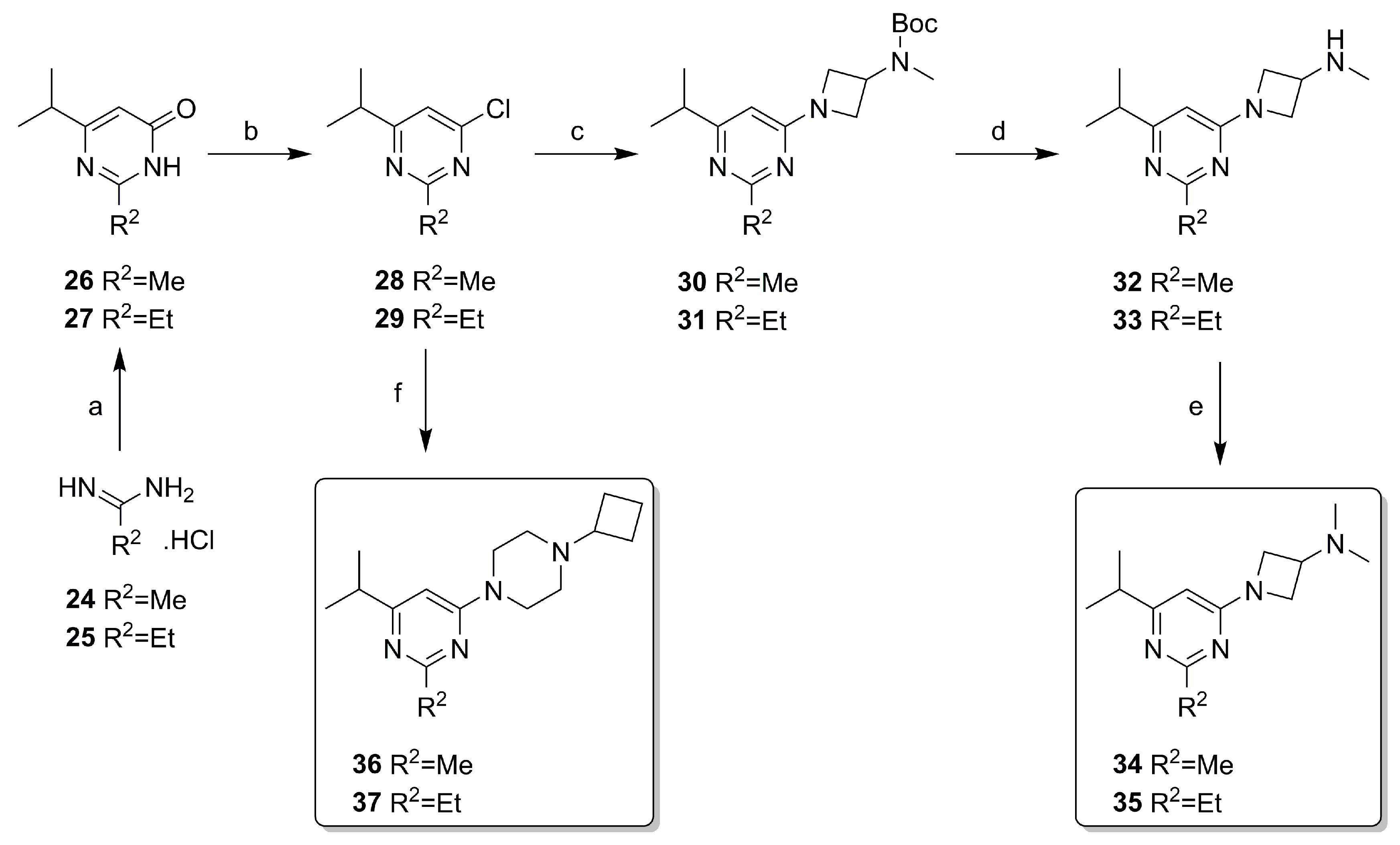
2.2. Synthesis
2.3. Structure-Activity Relationship
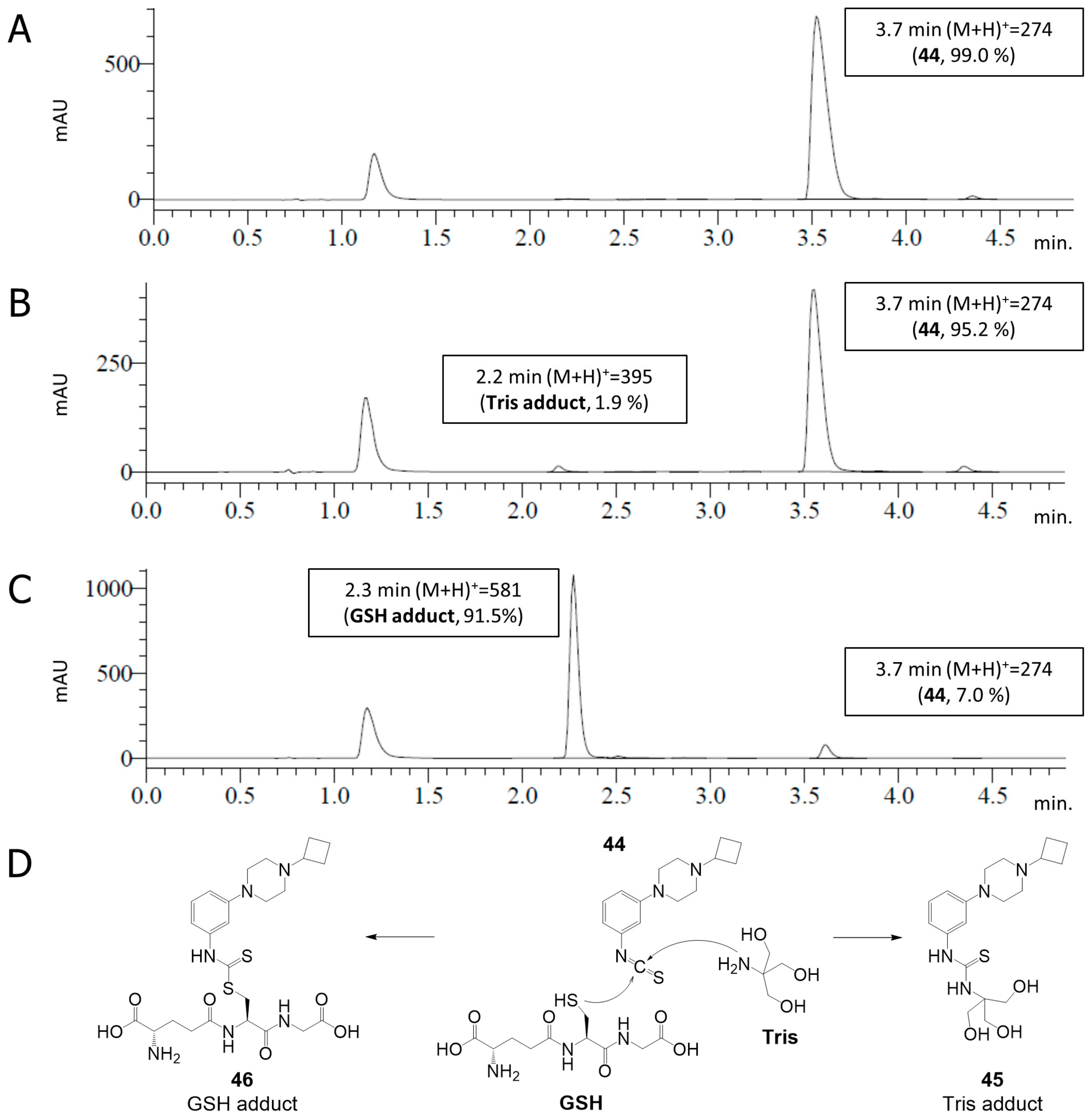
2.4. Covalent Binding to Glutathione and Nonapeptide
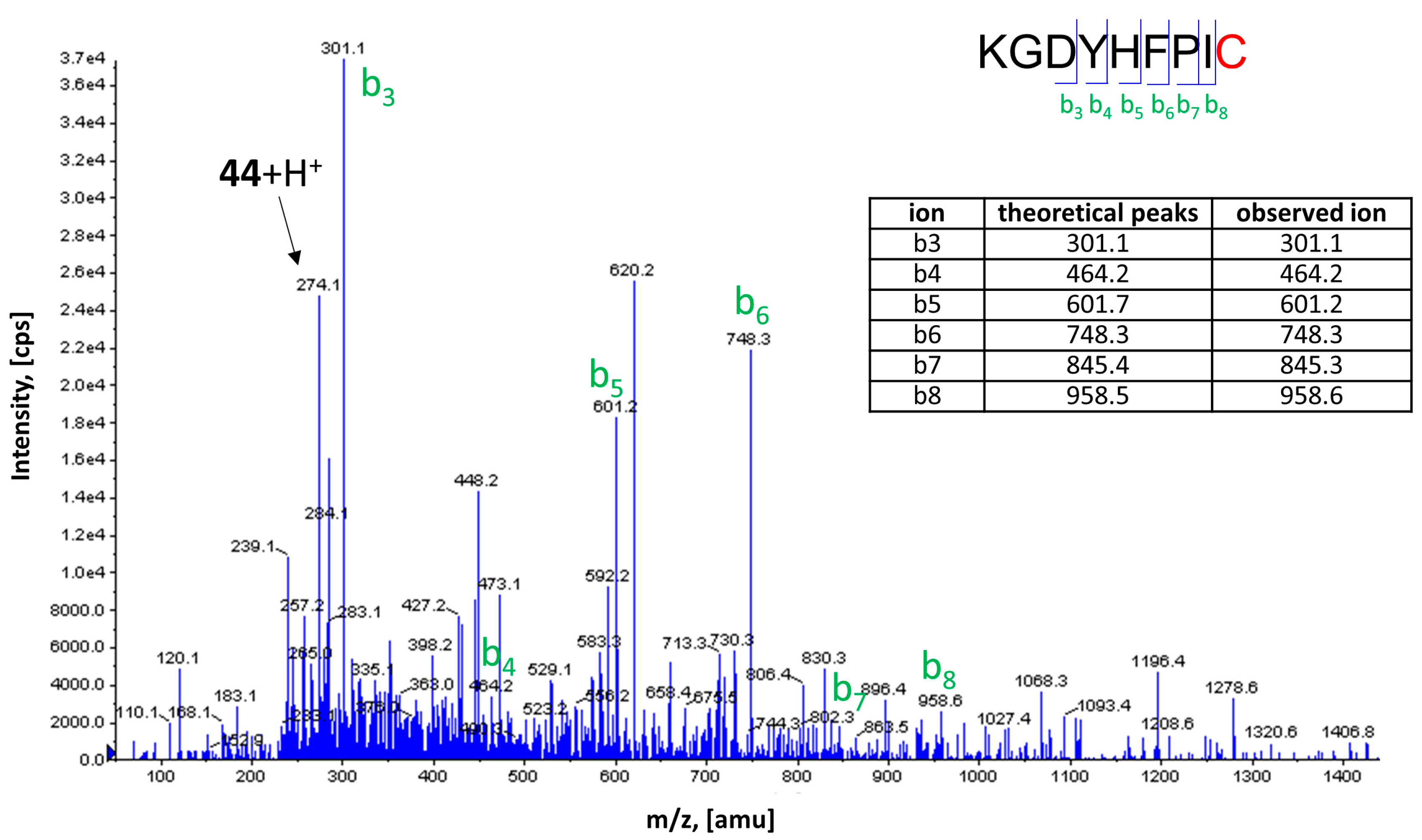
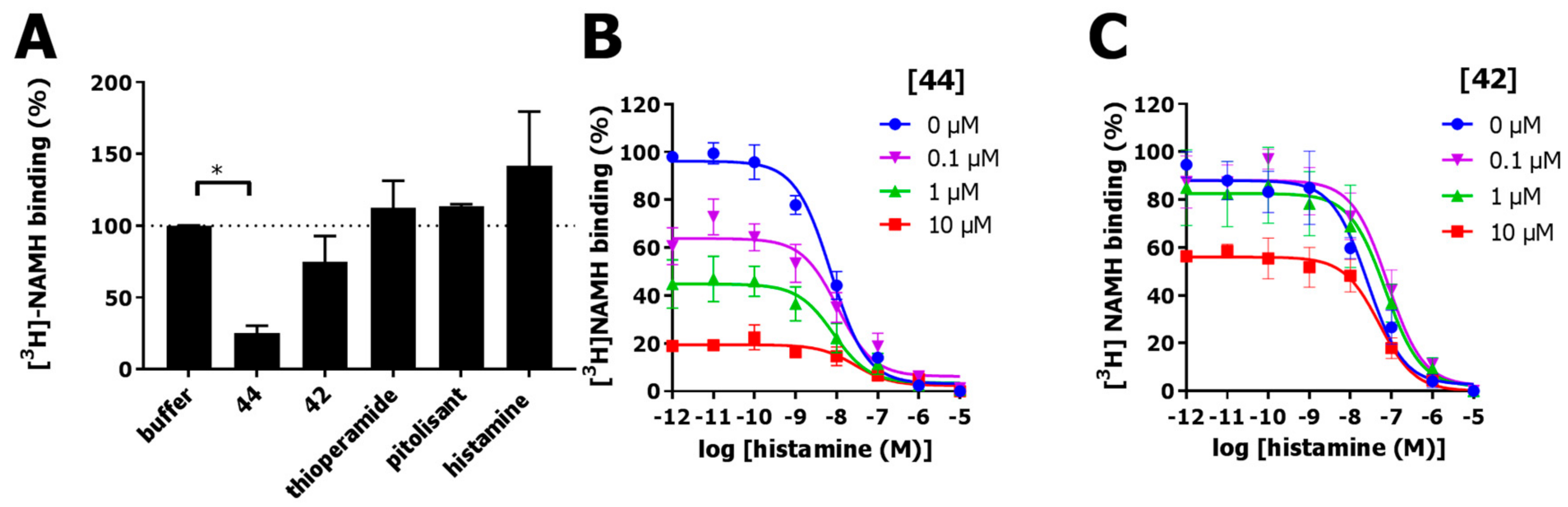
2.5. Covalent Labeling of the H3R by 44
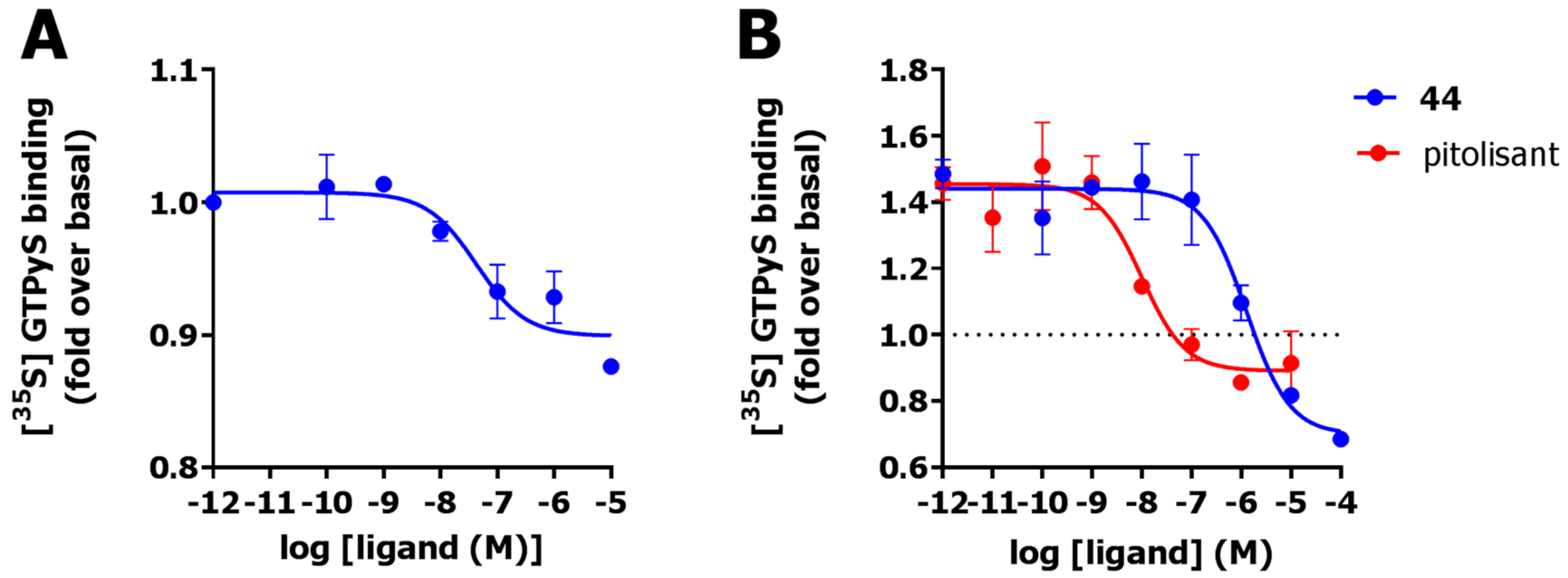
2.6. Functional Characterization
3. Discussion
4. Materials and Methods
4.1. Pharmacology
4.1.1. Materials
4.1.2. Cell Culture and Transfection
4.1.3. Preparation of Cell Homogenates
4.1.4. Radioligand Displacement Assays
4.1.5. Receptor Recovery Assay
4.1.6. [35S]GTPγs Assay
4.1.7. Chemical Stability and Reactivity of 44
4.1.8. Nonapeptide Assay
4.1.9. Data Analysis
4.2. Modelling
4.3. Chemistry
4.3.1. General Information
4.3.2. Synthesis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| [3H]NAMH | [3H]N-α-methylhistamine |
| ANOVA | analysis of variance |
| DCM | Dichloromethane |
| DIPEA | N,N-Diisopropylethylamine |
| DME | 1,2-Dimethoxyethane |
| DMSO | dimethyl sulfoxide |
| GPCR | G protein-coupled receptor |
| GSH | glutathion |
| GTPγS | guanosine 5′-O-[gamma-thio]triphosphate |
| LSD | least significant difference |
| MeCN | Acetonitrile |
| MIDA | N-methyliminodiacetic acid |
| Mp | Melting point |
| NP | nonapeptide (KGDYHFPIC) |
| SAR | structure-activity relationship |
| satd. aq. | saturated aqueous |
| SD | standard deviation |
| SEM | standard error of mean |
| rt | room temperature |
| TCI | targeted covalent inhibitors |
| TEA | Triethylamine |
| Tris | 2-Amino-2-(hydroxymethyl)propane-1,3-diol |
| μW | microwave reaction |
| wt | wild type |
References
- Arrang, J.M.; Garbarg, M.; Schwartz, J.C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 1983, 302, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.S.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2014, 67, 601–655. [Google Scholar] [CrossRef] [PubMed]
- Mocking, T.A.M.; Bosma, R.; Rahman, S.N.; Verweij, E.W.E.; McNaught-Flores, D.A.; Vischer, H.F.; Leurs, R. Molecular Aspects of Histamine Receptors. In Histamine Receptors: Preclinical and Clinical Aspects; Blandina, P., Passani, M.B., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–49. ISBN 978-3-319-40308-3. [Google Scholar]
- Schlicker, E.; Kathmann, M. Role of the Histamine H3 Receptor in the Central Nervous System. Handb. Exp. Pharmacol. 2017, 241, 277–299. [Google Scholar]
- Lovenberg, T.W.; Roland, B.L.; Wilson, S.J.; Jiang, X.; Pyati, J.; Huvar, A.; Jackson, M.R.; Erlander, M.G. Cloning and Functional Expression of the Human Histamine H3 Receptor. Mol. Pharmacol. 2018, 55, 1101–1107. [Google Scholar] [CrossRef]
- Celanire, S.; Wijtmans, M.; Talaga, P.; Leurs, R.; de Esch, I.J.P. Keynote review: Histamine H3 receptor antagonists reach out for the clinic. Drug Discov. Today 2005, 10, 1613–1627. [Google Scholar] [CrossRef]
- Lebois, E.P.; Jones, C.K.; Lindsley, C.W. The evolution of histamine H3 antagonists/inverse agonists. Curr. Top. Med. Chem. 2011, 11, 648–660. [Google Scholar] [CrossRef]
- Kuhne, S.; Wijtmans, M.; Lim, H.D.; Leurs, R.; de Esch, I.J.P. Several down, a few to go: Histamine H3 receptor ligands making the final push towards the market? Expert Opin. Investig. Drugs 2011, 20, 1629–1648. [Google Scholar] [CrossRef]
- Sadek, B.; Łażewska, D.; Hagenow, S.; Kieć-Kononowicz, K.; Stark, H. Histamine H3R Antagonists: From Scaffold Hopping to Clinical Candidates. In Histamine Receptors: Preclinical and Clinical Aspects; Blandina, P., Passani, M.B., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 109–155. ISBN 978-3-319-40308-3. [Google Scholar]
- Kollb-Sielecka, M.; Demolis, P.; Emmerich, J.; Markey, G.; Salmonson, T.; Haas, M. The European Medicines Agency review of pitolisant for treatment of narcolepsy: Summary of the scientific assessment by the Committee for Medicinal Products for Human Use. Sleep Med. 2018, 33, 125–129. [Google Scholar] [CrossRef]
- Harmony Biosciences LLC. Available online: https://www.harmonybiosciences.com/newsroom/harmony-biosciences-announces-fda-approval-of-wakix-r-pitolisant-a-first-in/ (accessed on 15 August 2019).
- Zhao, Z.; Bourne, P.E. Progress with covalent small-molecule kinase inhibitors. Drug Discov. Today 2018, 23, 727–735. [Google Scholar] [CrossRef]
- Weichert, D.; Gmeiner, P. Covalent Molecular Probes for Class A G Protein-Coupled Receptors: Advances and Applications. ACS Chem. Biol. 2015, 10, 1376–1386. [Google Scholar] [CrossRef]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Nijmeijer, S.; Engelhardt, H.; Schultes, S.; van de Stolpe, A.C.; Lusink, V.; de Graaf, C.; Wijtmans, M.; Haaksma, E.E.J.; de Esch, I.J.P.; Stachurski, K.; et al. Design and pharmacological characterization of VUF14480, a covalent partial agonist that interacts with cysteine 983.36 of the human histamine H4 receptor. Br. J. Pharmacol. 2013, 170, 89–100. [Google Scholar] [CrossRef]
- Moss, S.M.; Jayasekara, P.S.; Paoletta, S.; Gao, Z.G.; Jacobson, K.A. Structure-based design of reactive nucleosides for site-specific modification of the A2A adenosine receptor. ACS Med. Chem. Lett. 2014, 5, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Weichert, D.; Kruse, A.C.; Manglik, A.; Hiller, C.; Zhang, C.; Hubner, H.; Kobilka, B.K.; Gmeiner, P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA 2014, 111, 10744–10748. [Google Scholar] [CrossRef] [PubMed]
- Janero, D.R.; Yaddanapudi, S.; Zvonok, N.; Subramanian, K.V.; Shukla, V.G.; Stahl, E.; Zhou, L.; Hurst, D.; Wager-Miller, J.; Bohn, L.M.; et al. Molecular-Interaction and Signaling Profiles of AM3677, a Novel Covalent Agonist Selective for the Cannabinoid 1 Receptor. ACS Chem. Neurosci. 2015, 6, 1400–1410. [Google Scholar] [CrossRef]
- Kling, R.C.; Plomer, M.; Lang, C.; Banerjee, A.; Hübner, H.; Gmeiner, P. Development of Covalent Ligand-Receptor Pairs to Study the Binding Properties of Nonpeptidic Neurotensin Receptor 1 Antagonists. ACS Chem. Biol. 2016, 11, 869–875. [Google Scholar] [CrossRef]
- Yang, X.; Dong, G.; Michiels, T.J.M.; Lenselink, E.B.; Heitman, L.; Louvel, J.; Ijzerman, A.P. A covalent antagonist for the human adenosine A2A receptor. Purinergic Signal. 2017, 13, 191–201. [Google Scholar] [CrossRef]
- Soethoudt, M.; Stolze, S.C.; Westphal, M.V.; van Stralen, L.; Martella, A.; van Rooden, E.J.; Guba, W.; Varga, Z.V.; Deng, H.; van Kasteren, S.I.; et al. Selective Photoaffinity Probe That Enables Assessment of Cannabinoid CB 2 Receptor Expression and Ligand Engagement in Human Cells. J. Am. Chem. Soc. 2018, 140, 6067–6075. [Google Scholar] [CrossRef]
- Yang, X.; van Veldhoven, J.P.D.; Offringa, J.; Kuiper, B.J.; Lenselink, E.B.; Heitman, L.H.; van der Es, D.; Ijzerman, A.P. Development of Covalent Ligands for G Protein-Coupled Receptors: A Case for the Human Adenosine A3 Receptor. J. Med. Chem. 2019, 62, 3539–3552. [Google Scholar] [CrossRef]
- Schwalbe, T.; Huebner, H.; Gmeiner, P. Development of covalent antagonists for β1- and β2-adrenergic receptors. Bioorg. Med. Chem. 2019, 27, 2959–2971. [Google Scholar] [CrossRef]
- Lonsdale, R.; Burgess, J.; Colclough, N.; Davies, N.L.; Lenz, E.M.; Orton, A.L.; Ward, R.A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57, 3124–3137. [Google Scholar] [CrossRef] [PubMed]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef]
- Fell, N.; Rodney, G.; Marshall, D.E. Histamine Protein Complexes: Synthesis and Immunologic Investigation: I. Histamine-Azo-Protein. J. Immunol. 1943, 47, 237–249. [Google Scholar]
- Rodney, G.; Fell, N. Histamine-protein complexes: Synthesis and immunological investigation: II. ß-(5-Imidazoyl) ethyl carbamido protein. J. Immunol. 1943, 47, 251–259. [Google Scholar]
- Morse, K.L.; Behan, J.; Laz, T.M.; West, R.E.J.; Greenfeder, S.A.; Anthes, J.C.; Umland, S.; Wan, Y.; Hipkin, R.W.; Gonsiorek, W.; et al. Cloning and characterization of a novel human histamine receptor. J. Pharmacol. Exp. Ther. 2001, 269, 1058–1066. [Google Scholar]
- Kooistra, A.J.; Kuhne, S.; de Esch, I.J.P.; Leurs, R.; de Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013, 170, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Łażewska, D.; Kieć-Kononowicz, K. New developments around histamine H3 receptor antagonists/inverse agonists: A patent review (2010–present). Expert Opin. Ther. Pat. 2013, 24, 89–111. [Google Scholar] [CrossRef]
- Nakamura, T.; Kawai, Y.; Kitamoto, N.; Osawa, T.; Kato, Y. Covalent modification of lysine residues by allyl isothiocyanate in physiological conditions: Plausible transformation of isothiocyanate from thiol to amine. Chem. Res. Toxicol. 2009, 22, 536–542. [Google Scholar] [CrossRef]
- Karlsson, I.; Samuelsson, K.; Ponting, D.J.; Törnqvist, M.; Ilag, L.L.; Nilsson, U. Peptide Reactivity of Isothiocyanates-Implications for Skin Allergy. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Shi, L.; Javitch, J.A. The Binding Site of Aminergic G Protein-Coupled Receptors: The Transmembrane Segments and Second Extracellular Loop. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 437–467. [Google Scholar] [CrossRef] [PubMed]
- Uveges, A.J.; Kowal, D.; Zhang, Y.; Spangler, T.B.; Dunlop, J.; Semus, S.; Philip, G.J. The Role of Transmembrane Helix 5 in Agonist Binding to the Human H3 Receptor. J. Pharmacol. Exp. Ther. 2002, 301, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Francom, P.; Janeba, Z.; Shibuya, S.; Robins, M.J. Nucleic acid related compounds. 116. Nonaqueous diazotization of aminopurine nucleosides. Mechanistic considerations and efficient procedures with tert-butyl nitrite or sodium nitrite. J. Org. Chem. 2002, 67, 6788–6796. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liang, Y.L.; Qu, J. Boiling water-catalyzed neutral and selective N-Boc deprotection. Chem. Commun. 2009, 5144–5146. [Google Scholar] [CrossRef]
- Burke, T.R.; Bajwa, B.S.; Jacobson, A.E.; Rice, K.C.; Streaty, R.A.; Klee, W.A. Probes for Narcotic Receptor Mediated Phenomena. 7. Synthesis and Pharmacological Properties of Irreversible Ligands Specific for μ or δ Opiate Receptors. J. Med. Chem. 1984, 27, 1570–1574. [Google Scholar] [CrossRef]
- Mocking, T.A.M.; Verweij, E.W.E.; Vischer, H.F.; Leurs, R. Homogeneous, Real-Time NanoBRET Binding Assays for the Histamine H3 and H4 Receptors on Living Cells. Mol. Pharmacol. 2018, 94, 1371–1381. [Google Scholar] [CrossRef]
- Mercier, R.W.; Pei, Y.; Pandainathan, L.; Janero, D.R.; Zhang, J.; Makriyannis, A. hCB2 ligand-interaction landscape: Cysteine residues critical to biarylpyrazole antagonist binding motif and receptor modulation. Chem. Biol. 2011, 17, 1132–1142. [Google Scholar] [CrossRef]
- Pei, Y.; Mercier, R.W.; Anday, J.K.; Thakur, G.A.; Zvonok, A.M.; Hurst, D.; Reggio, P.H.; Janero, D.R.; Makriyannis, A. Ligand-Binding Architecture of Human CB2 Cannabinoid Receptor: Evidence for Receptor Subtype-Specific Binding Motif and Modeling GPCR Activation. Chem. Biol. 2008, 15, 1207–1219. [Google Scholar] [CrossRef]
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W.; et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–72. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.F.; Choi, H.J.; Devree, B.T.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 2011, 469, 236–242. [Google Scholar] [CrossRef]
- Mocking, T.A.M.; Buzink, M.C.M.L.; Leurs, R.; Vischer, H.F. Bioluminescence Resonance Energy Transfer Based G Protein-Activation Assay to Probe Duration of Antagonism at the Histamine H3 Receptor. Int. J. Mol. Sci. 2019, 20, 3724. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C.; Prusoff, W.H. Relationship Between the Inhibition Constant (Ki) and the Concentration of Inhibitor which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Hauwert, N.J.; Mocking, T.A.M.; Da Costa Pereira, D.; Kooistra, A.J.; Wijnen, L.M.; Vreeker, G.C.M.; Verweij, E.W.E.; de Boer, A.H.; Smit, M.J.; de Graaf, C.; et al. Synthesis and Characterization of a Bidirectional Photoswitchable Antagonist Toolbox for Real-Time GPCR Photopharmacology. J. Am. Chem. Soc. 2018, 140, 4232–4243. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound 44 are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd. | Central Ring | R1 | R2 | R3 | pKi | % Unlabeled Receptors |
| 2 | Pyrimidine | A | Me | phenyl | 7.1 ± 0.1 | 84 ± 16 |
| 1 | Pyrimidine | A | CH=CH2 | phenyl | < 5 | 41 ± 6 (SD) |
| 10 | Pyrimidine | B | NH2 | iPr | 7.3 ± 0.1 | 100 ± 0.0 |
| 34 | Pyrimidine | B | Me | iPr | < 5 | 87 ± 13 |
| 35 | Pyrimidine | B | Et | iPr | < 5 | 93 ± 7 |
| 21 | Pyrimidine | B | CH=CH2 | iPr | < 5 | 85 ± 15 |
| 11 | Pyrimidine | C | NH2 | iPr | 6.7 ± 0.1 | 96 ± 4 |
| 36 | Pyrimidine | C | Me | iPr | 5.9 ± 0.1 | 96 ± 3 |
| 37 | Pyrimidine | C | Et | iPr | 5.5 ± 0.1 | 89 ± 11 |
| 22 | Pyrimidine | C | CH=CH2 | iPr | 5.5 ± 0.0 | 41 ± 9 |
| 12 | Pyrimidine | C | NH2 | Me | 6.5 ± 0.1 | 91 ± 5 |
| 23 | Pyrimidine | C | CH=CH2 | Me | 5.4 ± 0.1 | 40 ± 8 |
| 20 | Pyrimidine | C | C≡CH | Me | < 5 | 80 ± 2 |
| 42 | Phenyl | C | NH2 | - | 5.9 ± 0.3 | 72 ± 8 |
| 43 | Phenyl | C | NH-CO-CH2-Cl | - | 6.2 ± 0.1 | 86 ± 5 |
| 44 | Phenyl | C | N=C=S | - | 6.5 ± 0.3 | 15 ± 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wágner, G.; Mocking, T.A.M.; Kooistra, A.J.; Slynko, I.; Ábrányi-Balogh, P.; Keserű, G.M.; Wijtmans, M.; Vischer, H.F.; de Esch, I.J.P.; Leurs, R. Covalent Inhibition of the Histamine H3 Receptor. Molecules 2019, 24, 4541. https://doi.org/10.3390/molecules24244541
Wágner G, Mocking TAM, Kooistra AJ, Slynko I, Ábrányi-Balogh P, Keserű GM, Wijtmans M, Vischer HF, de Esch IJP, Leurs R. Covalent Inhibition of the Histamine H3 Receptor. Molecules. 2019; 24(24):4541. https://doi.org/10.3390/molecules24244541
Chicago/Turabian StyleWágner, Gábor, Tamara A. M. Mocking, Albert J. Kooistra, Inna Slynko, Péter Ábrányi-Balogh, György M. Keserű, Maikel Wijtmans, Henry F. Vischer, Iwan J. P. de Esch, and Rob Leurs. 2019. "Covalent Inhibition of the Histamine H3 Receptor" Molecules 24, no. 24: 4541. https://doi.org/10.3390/molecules24244541
APA StyleWágner, G., Mocking, T. A. M., Kooistra, A. J., Slynko, I., Ábrányi-Balogh, P., Keserű, G. M., Wijtmans, M., Vischer, H. F., de Esch, I. J. P., & Leurs, R. (2019). Covalent Inhibition of the Histamine H3 Receptor. Molecules, 24(24), 4541. https://doi.org/10.3390/molecules24244541