
The Meta-Position of Phe4 in Leu-Enkephalin Regulates Potency, Selectivity, Functional Activity, and Signaling Bias at the Delta and Mu Opioid Receptors

 , and
, and 
Abstract
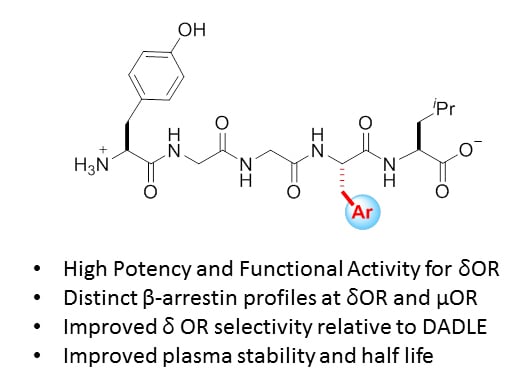
1. Introduction
2. Results and Discussion
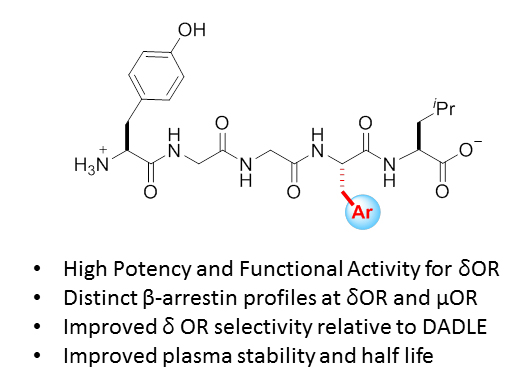
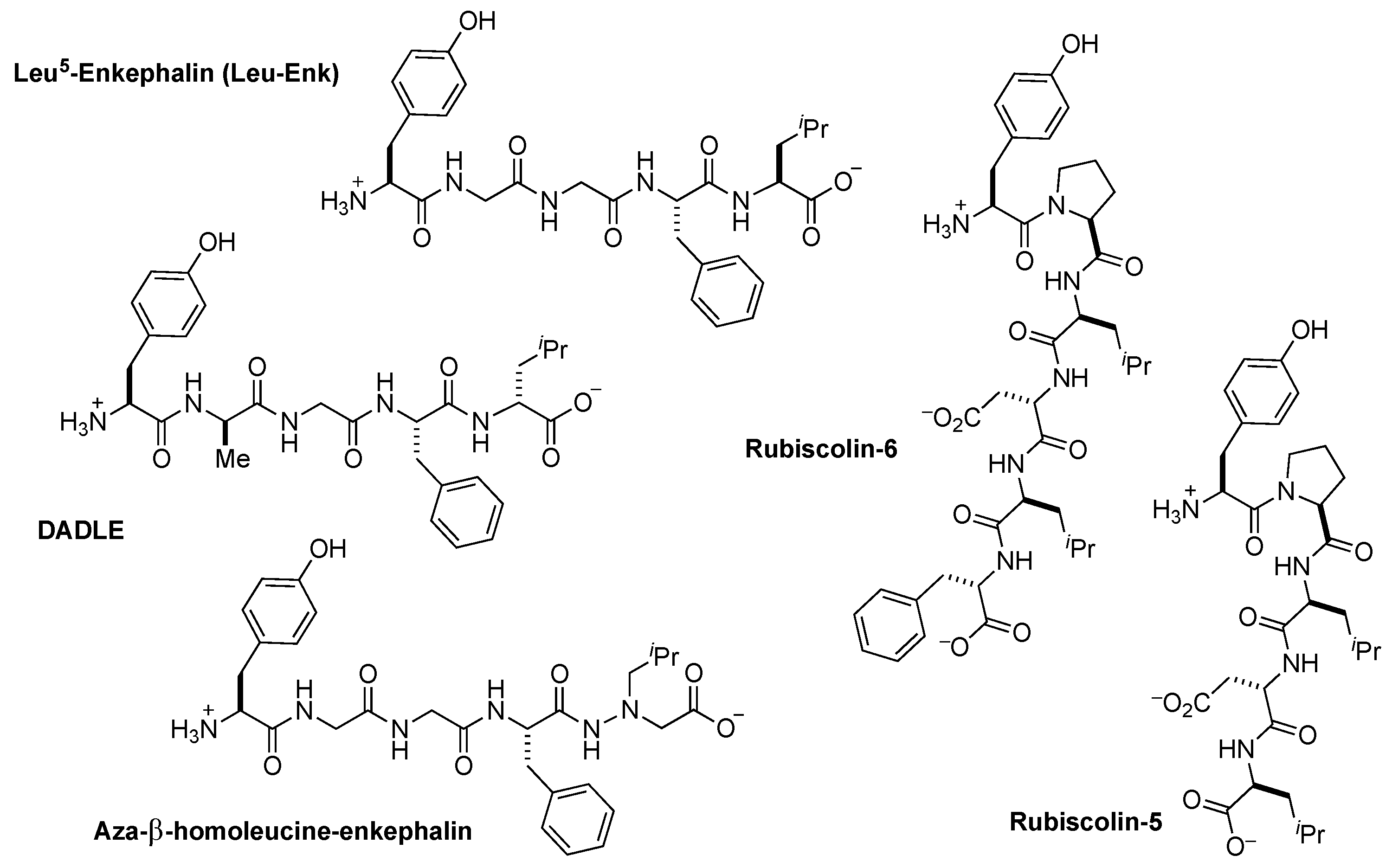
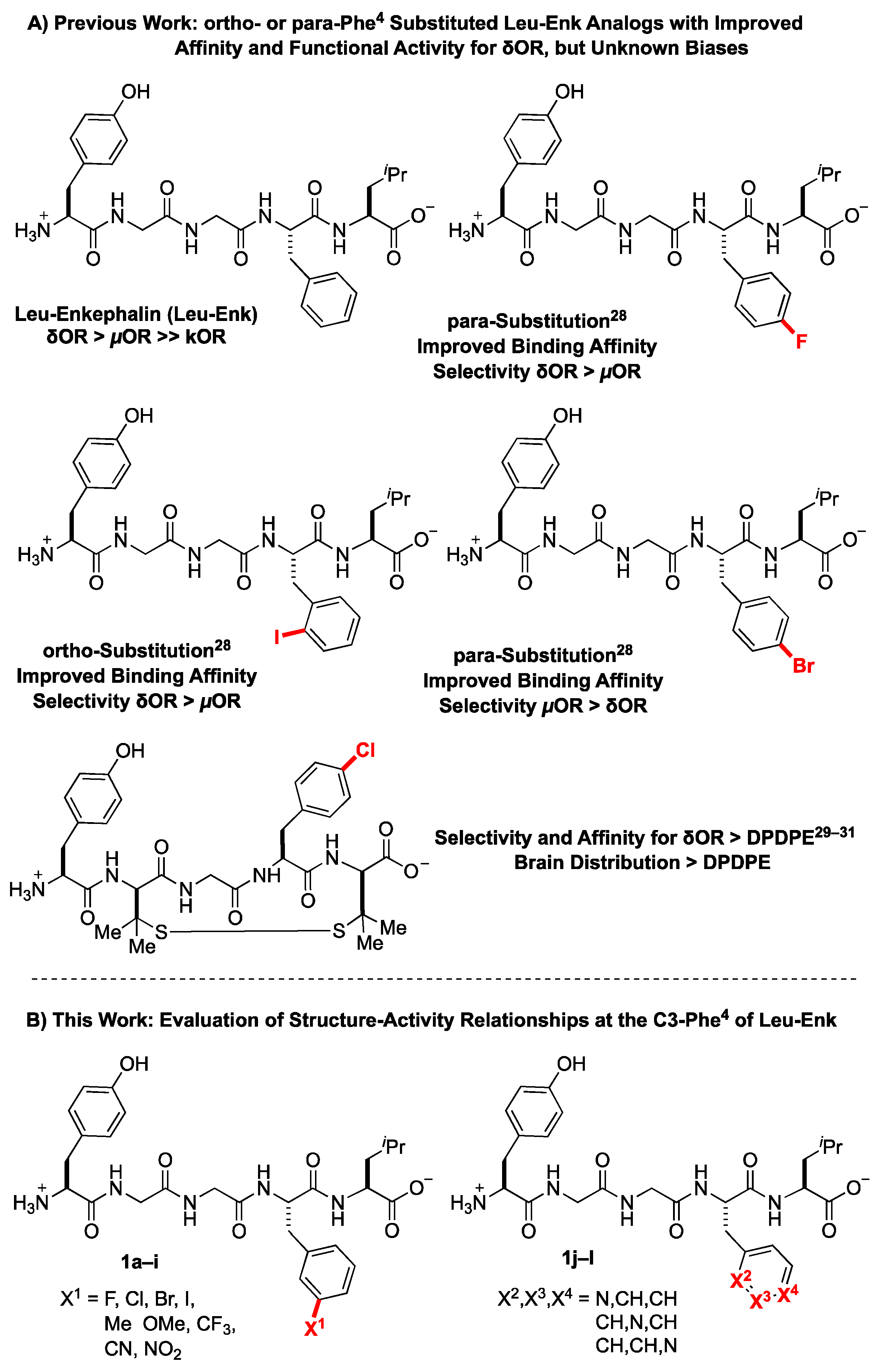
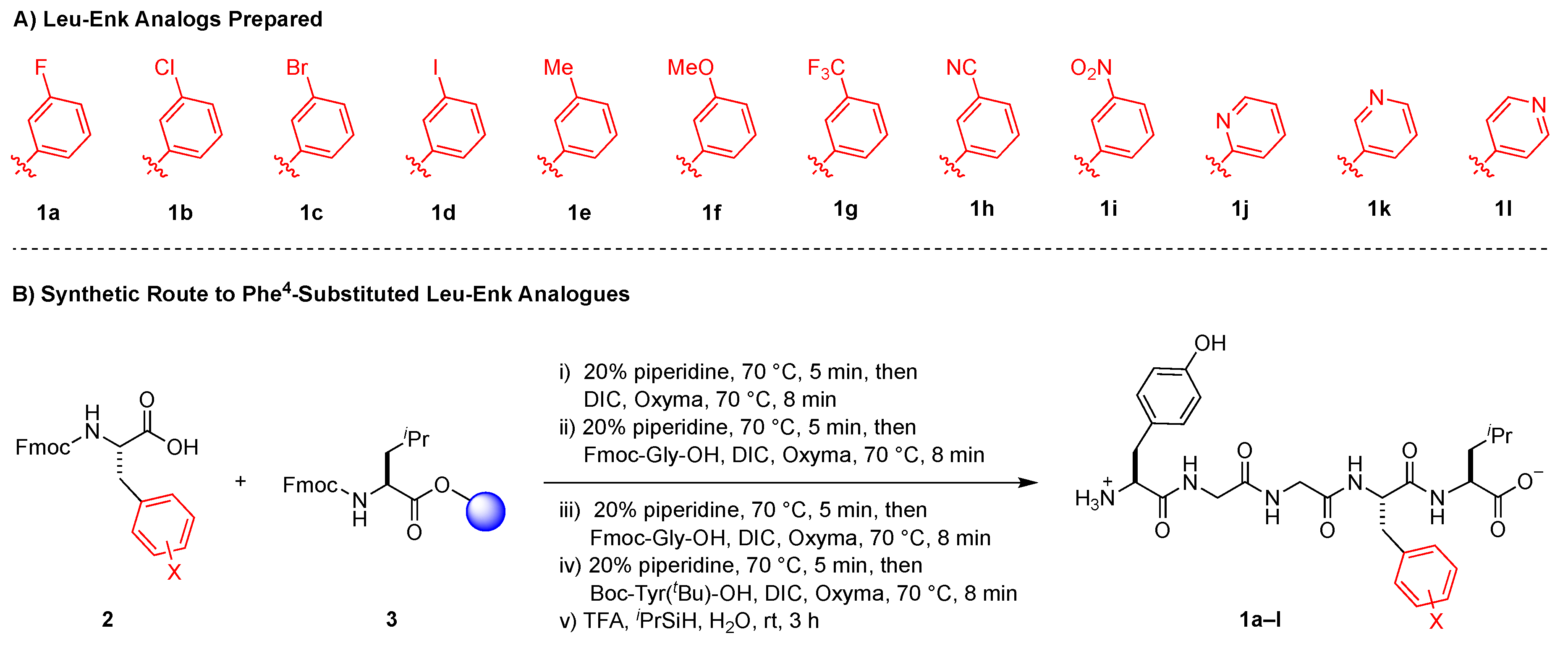
2.1. Design Considerations
2.2. Solid Phase Synthesis of Peptides
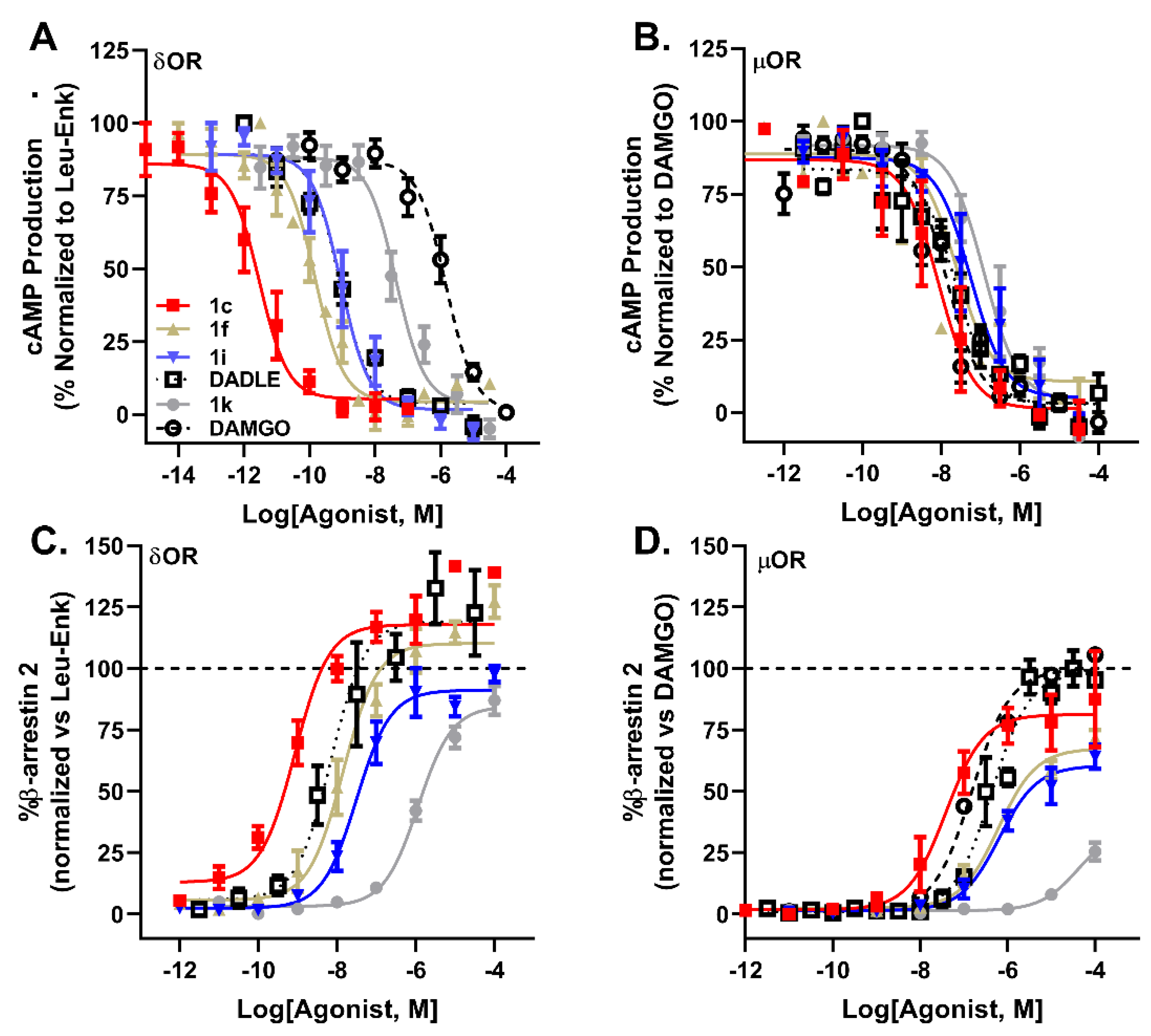
2.3. Pharmacological Characterization
2.4. Stability
3. Methods
3.1. Synthetic Chemistry-General Considerations
3.2. Synthesis of Peptides
3.3. Materials Used in the Cellular Signaling Assays
3.4. Cell Culture and Biased Signaling Assays
3.5. Radioligand Binding Assay
3.6. Calculation of Bias Factor
3.7. Data and Statistical Analysis
3.8. Assessment of Plasma Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–580. [Google Scholar] [CrossRef]
- Comb, M.; Seeburg, P.H.; Adelman, J.; Eiden, L.; Herbert, E. Primary structure of the human Met- and Leu-enkephalin precursor and its mRNA. Nature 1982, 295, 663–666. [Google Scholar] [CrossRef]
- Ikeda, Y.; Nakao, K.; Yoshimasa, T.; Yanaihara, N.; Numa, S.; Imura, H. Existence of Met-enkephalin-Arg6-Gly7-Leu8 with Met-enkephalin, Leu-enkephalin and Met-enkephalin-Arg6-Phe7 in the brain of guinea pig, rat and golden hamster. Biochem. Biophys. Res. Commun. 1982, 107, 656–662. [Google Scholar] [CrossRef]
- Simantov, R.; Kuhar, M.J.; Pasternak, G.W.; Snyder, S.H. The regional distribution of a morphine-like factors enkephalin in monkey brain. Brain Res. 1976, 106, 189–197. [Google Scholar] [CrossRef]
- Akil, H.; Owens, C.; Gutstein, H.; Taylor, L.; Curran, E.; Watson, S. Endogenous opioids: Overview and current issues. Drug Alcohol Depend. 1998, 51, 127–140. [Google Scholar] [CrossRef]
- Raynor, K.; Kong, H.; Chen, Y.; Yasuda, K.; Yu, L.; Bell, G.I.; Reisine, T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol. 1994, 45, 330–334. [Google Scholar]
- Belluzzi, J.D.; Grant, N.; Garsky, V.; Sarantakis, D.; Wise, C.D.; Stein, L. Analgesia induced in vivo by central administration of enkephalin in rat. Nature 1976, 260, 625–626. [Google Scholar] [CrossRef]
- Schaz, K.; Stock, G.; Simon, W.; Schlor, K.H.; Unger, T.; Rockhold, R.; Ganten, D. Enkephalin effects on blood pressure, heart rate, and baroreceptor reflex. Hypertens. (Dallas, Tex. 1979) 1980, 2, 395–407. [Google Scholar] [CrossRef]
- Violin, J.D.; Crombie, A.L.; Soergel, D.G.; Lark, M.W. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 2014, 35, 308–316. [Google Scholar] [CrossRef]
- Vicente-Sanchez, A.; Dripps, I.J.; Tipton, A.F.; Akbari, H.; Akbari, A.; Jutkiewicz, E.M.; Pradhan, A.A. Tolerance to high-internalizing delta opioid receptor agonist is critically mediated by arrestin 2. Br. J. Pharmacol. 2018, 175, 3050–3059. [Google Scholar] [CrossRef]
- Robins, M.T.; Chiang, T.; Mores, K.L.; Alongkronrusmee, D.; van Rijn, R.M. Critical Role for Gi/o-Protein Activity in the Dorsal Striatum in the Reduction of Voluntary Alcohol Intake in C57Bl/6 Mice. Front. Psychiatry 2018, 9, 112. [Google Scholar] [CrossRef]
- Chiang, T.; Sansuk, K.; Van Rijn, R.M. β-Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake. Br. J. Pharmacol. 2016, 173, 332–343. [Google Scholar] [CrossRef]
- Remesic, M.; Lee, Y.S.; Hruby, V.J. Cyclic Opioid Peptides. Curr. Med. Chem. 2016, 23, 1288–1303. [Google Scholar] [CrossRef]
- Zhang, J.; Ferguson, S.S.G.; Barak, L.S.; Bodduluri, S.R.; Laporte, S.A.; Law, P.-Y.; Caron, M.G. Role for G protein-coupled receptor kinase in agonist-specific regulation of -opioid receptor responsiveness. Proc. Natl. Acad. Sci. 1998, 95, 7157–7162. [Google Scholar] [CrossRef]
- Borlongan, C.V. Delta opioid peptide (D-ALA 2, D-LEU 5) enkephalin: Linking hibernation and neuroprotection. Front. Biosci. 2004, 9, 3392. [Google Scholar] [CrossRef]
- Wolf, A.; Lusczek, E.R.; Beilman, G.J. Hibernation-Based Approaches in the Treatment of Hemorrhagic Shock. SHOCK 2018, 50, 14–23. [Google Scholar] [CrossRef]
- Uchiyama, T.; Kotani, A.; Kishida, T.; Tatsumi, H.; Okamoto, A.; Fujita, T.; Murakami, M.; Muranishi, S.; Yamamoto, A. Effects of various protease inhibitors on the stability and permeability of [D-Ala2,D-Leu5]enkephalin in the rat intestine: Comparison with leucine enkephalin. J. Pharm. Sci. 1998, 87, 448–452. [Google Scholar] [CrossRef]
- Beddell, C.R.; Clark, R.B.; Lowe, L.A.; Wilkinson, S.; Chang, K.J.; Cuatrecasas, P.; Miller, R. A conformational analysis for leucine-enkephalin using activity and binding data of synthetic analogues. Br. J. Pharmacol. 1977, 61, 351–356. [Google Scholar] [CrossRef]
- Kovoor, A.; Nappey, V.; Kieffer, B.L.; Chavkin, C. μ and δ Opioid Receptors Are Differentially Desensitized by the Coexpression of β-Adrenergic Receptor Kinase 2 and β-Arrestin 2 in Xenopus Oocytes. J. Biol. Chem. 1997, 272, 27605–27611. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Whistler, J.L.; Chuang, H.; Chu, P.; Jan, L.Y.; von Zastrow, M. Functional Dissociation of μ Opioid Receptor Signaling and Endocytosis. Neuron 1999, 23, 737–746. [Google Scholar] [CrossRef]
- Cassell, R.J.; Mores, K.L.; Zerfas, B.L.; Mahmoud, A.H.; Lill, M.A.; Trader, D.J.; van Rijn, R.M. Rubiscolins are naturally occurring G protein-biased delta opioid receptor peptides. Eur. Neuropsychopharmacol. 2019, 29, 450–456. [Google Scholar] [CrossRef]
- Bella Ndong, D.; Blais, V.; Holleran, B.J.; Proteau-Gagné, A.; Cantin-Savoie, I.; Robert, W.; Nadon, J.-F.; Beauchemin, S.; Leduc, R.; Piñeyro, G.; et al. Exploration of the fifth position of leu-enkephalin and its role in binding and activating delta (DOP) and mu (MOP) opioid receptors. Pept. Sci. 2018, 111, e24070. [Google Scholar] [CrossRef]
- Vezzi, V.; Onaran, H.O.; Molinari, P.; Guerrini, R.; Balboni, G.; Calo, G.; Costa, T.; Calò, G.; Costa, T. Ligands raise the constraint that limits constitutive activation in G protein-coupled opioid receptors. J. Biol. Chem. 2013, 288, 23964–23978. [Google Scholar] [CrossRef]
- Aguila, B.; Coulbault, L.; Boulouard, M.; Leveille, F.; Davis, A.; Toth, G.; Borsodi, A.; Balboni, G.; Salvadori, S.; Jauzac, P.; et al. In vitro and in vivo pharmacological profile of UFP-512, a novel selective delta-opioid receptor agonist; correlations between desensitization and tolerance. Br. J. Pharmacol. 2007, 152, 1312–1324. [Google Scholar] [CrossRef]
- Raehal, K.M.; Walker, J.K.L.; Bohn, L.M. Morphine Side Effects in β-Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef]
- Rosa, M.; Caltabiano, G.; Barreto-Valer, K.; Gonzalez-Nunez, V.; Gómez-Tamayo, J.C.; Ardá, A.; Jiménez-Barbero, J.; Pardo, L.; Rodríguez, R.E.; Arsequell, G.; et al. Modulation of the Interaction between a Peptide Ligand and a G Protein-Coupled Receptor by Halogen Atoms. ACS Med. Chem. Lett. 2015, 6, 872–876. [Google Scholar] [CrossRef]
- Toth, G.; Kramer, T.H.; Knapp, R.; Lui, G.; Davis, P.; Burks, T.F.; Yamamura, H.I.; Hruby, V.J. [D-Pen2,D-Pen5]enkephalin analogues with increased affinity and selectivity for delta opioid receptors. J. Med. Chem. 1990, 33, 249–253. [Google Scholar] [CrossRef]
- Weber, S.J.; Greene, D.L.; Sharma, S.D.; Yamamura, H.I.; Kramer, T.H.; Burks, T.F.; Hruby, V.J.; Hersh, L.B.; Davis, T.P. Distribution and analgesia of [3H][D-Pen2, D-Pen5]enkephalin and two halogenated analogs after intravenous administration. J. Pharmacol. Exp. Ther. 1991, 259, 1109–1117. [Google Scholar]
- Weber, S.J.; Greene, D.L.; Hruby, V.J.; Yamamura, H.I.; Porreca, F.; Davis, T.P. Whole body and brain distribution of [3H]cyclic [D-Pen2,D-Pen5] enkephalin after intraperitoneal, intravenous, oral and subcutaneous administration. J. Pharmacol. Exp. Ther. 1992, 263, 1308–1316. [Google Scholar] [PubMed]
- Grant Liska, M.; Crowley, M.G.; Lippert, T.; Corey, S.; Borlongan, C.V. Delta Opioid Receptor and Peptide: A Dynamic Therapy for Stroke and Other Neurological Disorders. In Handbook of Experimental Pharmacology, 1st ed.; Emily, J., Ed.; Springer: Cham, Switzerland, 2017; pp. 277–299. [Google Scholar]
- He, X.; Sandhu, H.K.; Yang, Y.; Hua, F.; Belser, N.; Kim, D.H.; Xia, Y. Neuroprotection against hypoxia/ischemia: δ-opioid receptor-mediated cellular/molecular events. Cell. Mol. Life Sci. 2013, 70, 2291–2303. [Google Scholar] [CrossRef]
- Shenmar, K.; Sharma, K.K.; Wangoo, N.; Maurya, I.K.; Kumar, V.; Khan, S.I.; Jacob, M.R.; Tikoo, K.; Jain, R. Synthesis, stability and mechanistic studies of potent anticryptococcal hexapeptides. Eur. J. Med. Chem. 2017, 132, 192–203. [Google Scholar] [CrossRef]
- Mahindra, A.; Sharma, K.K.; Jain, R. Rapid microwave-assisted solution-phase peptide synthesis. Tetrahedron Lett. 2012, 53, 6931–6935. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Weinberger, S.B.; Martinez, J.L.J. Characterization of Hydrolysis of [ Leu ] enkephalin in Rat Plasma. J. Pharmacol. Exp. Ther. 1988, 247, 129–135. [Google Scholar]
- Roques, B.; Noble, F. Dual inhibitors of enkephalin-degrading enzymes (neutral endopeptidase 24.11 and aminopeptidase N) as potential new medications in the management of pain and opioid addiction. NIDA Res. Monogr. 1995, 147, 104–145. [Google Scholar]
- Suzanne, E.T.; Kenneth, L.A. Leucine-Enkephalin Metabolism in Brain Microvessel Endothelial Cells. Peptides 1994, 15, 109–116. [Google Scholar]
- Sarin, V.K.; Kent, S.B.; Tam, J.P.; Merrifield, R.B. Quantitative monitoring of solid-phase peptide synthesis by the ninhydrin reaction. Anal. Biochem. 1981, 117, 147–157. [Google Scholar] [CrossRef]
- Beaudeau, J.L.; Blais, V.; Holleran, B.J.; Bergeron, A.; Pineyro, G.; Guérin, B.; Gendron, L.; Dory, Y.L. N-Guanidyl and C-Tetrazole Leu-Enkephalin Derivatives: Efficient Mu and Delta Opioid Receptor Agonists with Improved Pharmacological Properties. ACS Chem. Neurosci. 2019, 10, 1615–1626. [Google Scholar] [CrossRef]
Sample Availability: Upon reasonable request and based on material in hand, compounds 1a–1l can be distributed in a timely fashion by Dr. Altman. However, as supplies are finite, we cannot guarantee every request can be accommodated. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pKi ± SEM (δOR) | Ki (nM) | pKi ± SEM (µOR) | Ki (nM) | Binding Selectivity (δOR vs µOR) | |
|---|---|---|---|---|---|
| 1a (F) | 9.48 ± 0.1 | 0.33 | 9.12 ± 0.4 | 0.76 | 2.3 |
| 1b (Cl) | 9.87 ± 0.1 | 0.13 | 10.14 ± 0.2 | 0.072 | 0.55 |
| 1c (Br) | 10.35 ± 0.2 | 0.045 | 9.90 ± 0.3 | 0.13 | 2.9 |
| 1d (I) | 10.64 ± 0.2 | 0.023 | 9.86 ± 0.5 | 0.14 | 6.1 |
| 1e (Me) | 9.86 ± 0.1 | 0.14 | 9.86 ± 0.1 | 0.14 | 1.0 |
| 1f (OMe) | 9.31 ± 0.1 | 0.49 | 9.07 ± 0.1 | 0.85 | 1.7 |
| 1g (CF3) | 9.93 ± 0.1 | 0.12 | 10.23 ± 0.3 | 0.059 | 0.49 |
| 1h (CN) | 9.17 ± 0.3 | 0.68 | 9.52 ± 0.1 | 0.30 | 0.44 |
| 1i (NO2) | 9.03 ± 0.2 | 0.93 | 9.01 ± 0.1 | 0.98 | 1.05 |
| 1j (2-pyr) | 8.21 ± 0.1 | 6.17 | 8.04 ± 0.2 | 9.12 | 1.5 |
| 1k (3-pyr) | 7.48 ± 0.1 | 33.1 | 6.80 ± 0.1 | 158 | 4.8 |
| 1l (4-pyr) | 7.69 ± 0.1 | 20.4 | 7.41 ± 0.1 | 38.9 | 1.9 |
| DADLE | 9.01 ± 0.1 | 0.98 | 8.80 ± 0.1 | 1.58 | 1.6 |
| Leu5-enkephalin | 8.90 ± 0.1 | 1.26 | 8.77 ± 0.1 | 1.70 | 1.3 |
| DAMGO | - | - | 9.01 ± 0.1 | 0.98 | - |
| cAMP | β-Arrestin 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | pIC50 ± SEM (δOR) | IC50 (nM) | pIC50 ± SEM (µOR) | IC50 (nM) | pEC50 ± SEM (δOR) | EC50 (nM) | δOR Efficacy (% ± SEM) | pEC50 ± SEM (µOR) | EC50 (nM) | µOR Efficacy (% + SEM) |
| 1a (F) | 9.47 ± 0.2 | 0.39 | 8.10 ± 0.2 | 7.94 | 8.01 ± 0.2 | 9.77 | 107 ± 9 | 6.24 ± 0.1 | 575 | 75 ± 13 |
| 1b (Cl) | 10.66 ± 0.3 | 0.022 | 7.52 ± 0.1 | 30.2 | 8.77 ± 0.2 | 1.70 | 130 ± 10 | 7.17 ± 0.2 | 67.6 | 96 ± 8 |
| 1c (Br) | 10.52 ± 0.3 | 0.030 | 8.00 ± 0.4 | 10 | 9.25 ± 0.2 | 0.56 | 116 ± 7 | 7.45 ± 0.2 | 35.5 | 79 ± 6 |
| 1d (I) | 10.61 ± 0.2 | 0.025 | 8.37 ± 0.3 | 4.27 | 8.95 ± 0.2 | 1.12 | 111 ± 7 | 7.33 ± 0.2 | 46.8 | 70 ± 3 |
| 1e (Me) | 10.41 ± 0.3 | 0.039 | 8.43 ± 0.3 | 3.72 | 8.46 ± 0.2 | 3.46 | 105 ± 5 | 6.73 ± 0.2 | 186 | 71 ± 3 |
| 1f (OMe) | 9.84 ± 0.3 | 0.14 | 8.09 ± 0.3 | 8.12 | 7.93 ± 0.2 | 11.7 | 106 ± 5 | 6.23 ± 0.1 | 589 | 67 ± 3 |
| 1g (CF3) | 9.97 ± 0.4 | 0.11 | 8.61 ± 0.4 | 2.45 | 8.39 ± 0.2 | 4.07 | 108 ± 9 | 7.25 ± 0.2 | 56.2 | 71 ± 3 |
| 1h (CN) | 9.46 ± 0.3 | 0.35 | 7.92 ± 0.3 | 12.0 | 7.77 ± 0.2 | 17.0 | 91 ± 5 | 6.8 ± 0.2 | 158 | 68 ± 4 |
| 1i (NO2) | 9.33 ± 0.3 | 0.47 | 7.31 ± 0.4 | 49.0 | 7.31 ± 0.2 | 49.0 | 96 ± 6 | 6.27 ± 0.1 | 537 | 60 ± 6 |
| 1j (2-pyr) | 8.34 ± 0.2 | 4.57 | 7.39 ± 0.4 | 40.7 | 7.00 ± 0.2 | 100 | 92 ± 13 | 5.87 ± 0.1 | 1349 | 70 ± 4 |
| 1k (3-pyr) | 7.32 ± 0.2 | 47.9 | 6.63 ± 0.4 | 234 | 5.95 ± 0.1 | 1122 | 85 ± 6 | 4.38 ± 0.1 | 41687 | 36 ± 7 |
| 1l (4-pyr) | 7.52 ± 0.2 | 30.2 | 6.52 ± 0.3 | 302 | 6.40 ± 0.2 | 398 | 98 ± 6 | 4.43 ± 0.1 | 37153 | 64 ± 11 |
| DADLE | 9.09 ± 0.2 | 0.81 | 7.45 ± 0.1 | 35.5 | 7.92 ± 0.4 | 12.0 | 126 ± 10 | 6.33 ± 0.1 | 468 | 99 ± 3 |
| Leu5-enkephalin | 8.99 ± 0.1 | 1.02 | 7.34 ± 0.2 | 45.7 | 8.05 ± 0.1 | 8.91 | 100 | 6.01 ± 0.1 | 977 | 61 ± 3 |
| DAMGO | 5.91 ± 0.2 | 1230 | 7.82 ± 0.1 | 15.1 | <5 | - | 6.80 ± 0.1 | 158 | 100 | |
| Compound | G Protein Selectivity (δOR vs µOR) | Bias Factor | ||
|---|---|---|---|---|
| δOR | µOR (DG = Ref) | µOR (LE = Ref) | ||
| 1a (F) | 20 | 1.8 | 22.2 | 0.36 |
| 1b (Cl) | 1372 | 3.2 | 0.3 | 0.004 |
| 1c (Br) | 333 | 1.3 | 1.2 | 0.02 |
| 1d (I) | 171 | 3.5 | 3.2 | 0.05 |
| 1e (Me) | 95 | 4.3 | 11.5 | 0.19 |
| 1f (OMe) | 58 | 7.5 | 8.4 | 0.14 |
| 1g (CF3) | 22 | 2.0 | 4.3 | 0.07 |
| 1h (CN) | 34 | 6.8 | 5.4 | 0.09 |
| 1i (NO2) | 104 | 6.9 | 11.1 | 0.18 |
| 1j (2-pyr) | 8.9 | 4.0 | 17.2 | 0.27 |
| 1k (3-pyr) | 4.9 | 6.7 | 331.1 | 5.89 |
| 1l (4-pyr) | 10 | 2.2 | 63.7 | 1.08 |
| DADLE | 44 | 0.5 | 2.3 | 0.035 |
| Leu5-enkephalin | 45 | 1 | 4.9 | 1 |
| DAMGO | 0.012 | - | 1 | 0.003 |
| Aza-β-homoleucine ¶ | 9.9 [23] | 5.2 ¶ | - | 1.2 |
| Rubiscolin-5 | - | 2.0 [22] | - | - |
| Compound | Half-Life (min) | 95% CI | Degradation Products (First Appearance) |
|---|---|---|---|
| Leu5-enkephalin | 9.4 | 6.3−4.5 | Gly-Gly-Phe-Leu (5 min) Phe-Leu (10 min) |
| 1a (F) | 82.3 | 68.0–102.6 | Gly-Gly-(meta-F)Phe-Leu (5 min) (meta-F)Phe-Leu (60 min) |
| 1b (Cl) | 37.8 | 26.5–56.6 | Gly-Gly-(meta-Cl)Phe-Leu (5 min) (meta-Cl)Phe-Leu (30 min) |
| 1c (Br) | 21.5 | 12.7–38.7 | Gly-Gly-(meta-Br)Phe-Leu (5 min) (meta-Br)Phe-Leu (15 min) |
| 1d (I) | 13.2 | 11.4–15.2 | Gly-Gly-(meta-I)Phe-Leu (5 min) (meta-I)Phe-Leu (15 min) |
| 1e (Me) | 39.5 | 30.0–53.5 | Gly-Gly-(meta-Me)Phe-Leu (5 min) (meta-Me)Phe-Leu (20 min) |
| 1f (OMe) | 46.1 | 30.4–75.6 | Gly-Gly-(meta-OMe)Phe-Leu (5 min) (meta-OMe)Phe-Leu (20 min) |
| 1g (CF3) | 44.5 | 35.0–59.1 | Gly-Gly-(meta-CF3)Phe-Leu (5 min) (meta- CF3)Phe-Leu (30 min) |
| 1h (CN) | 33.0 | 24.0–47.0 | Gly-Gly-(meta-CN)Phe-Leu (5 min) (meta-CN)Phe-Leu (20 min) |
| 1i (NO2) | 28.8 | 16.8–55.8 | Gly-Gly-(meta-NO2)Phe-Leu (5 min) (meta- NO2)Phe-Leu (20 min) |
| 1j (2-pyr) | 26.8 | 15.4–53.7 | (2-pyridyl)Ala-Leu (5 min) Gly-Gly-(2-pyridyl)Ala-Leu (30 min) |
| 1k (3-pyr) | 54.0 | 29.0–127.0 | (3-pyridyl)Ala-Leu (10 min) Gly-Gly-(3-pyridyl)Ala-Leu (20 min) |
| 1l (4-pyr) | 78.1 | 47.5–165.4 | (4-pyridyl)Ala-Leu (10 min) Gly-Gly-(4-pyridyl)Ala-Leu (30 min) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassell, R.J.; Sharma, K.K.; Su, H.; Cummins, B.R.; Cui, H.; Mores, K.L.; Blaine, A.T.; Altman, R.A.; van Rijn, R.M. The Meta-Position of Phe4 in Leu-Enkephalin Regulates Potency, Selectivity, Functional Activity, and Signaling Bias at the Delta and Mu Opioid Receptors. Molecules 2019, 24, 4542. https://doi.org/10.3390/molecules24244542
Cassell RJ, Sharma KK, Su H, Cummins BR, Cui H, Mores KL, Blaine AT, Altman RA, van Rijn RM. The Meta-Position of Phe4 in Leu-Enkephalin Regulates Potency, Selectivity, Functional Activity, and Signaling Bias at the Delta and Mu Opioid Receptors. Molecules. 2019; 24(24):4542. https://doi.org/10.3390/molecules24244542
Chicago/Turabian StyleCassell, Robert J., Krishna K. Sharma, Hongyu Su, Benjamin R. Cummins, Haoyue Cui, Kendall L. Mores, Arryn T. Blaine, Ryan A. Altman, and Richard M. van Rijn. 2019. "The Meta-Position of Phe4 in Leu-Enkephalin Regulates Potency, Selectivity, Functional Activity, and Signaling Bias at the Delta and Mu Opioid Receptors" Molecules 24, no. 24: 4542. https://doi.org/10.3390/molecules24244542
APA StyleCassell, R. J., Sharma, K. K., Su, H., Cummins, B. R., Cui, H., Mores, K. L., Blaine, A. T., Altman, R. A., & van Rijn, R. M. (2019). The Meta-Position of Phe4 in Leu-Enkephalin Regulates Potency, Selectivity, Functional Activity, and Signaling Bias at the Delta and Mu Opioid Receptors. Molecules, 24(24), 4542. https://doi.org/10.3390/molecules24244542