
Common Secondary and Tertiary Structural Features of Aptamer–Ligand Interaction Shared by RNA Aptamers with Different Primary Sequences

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
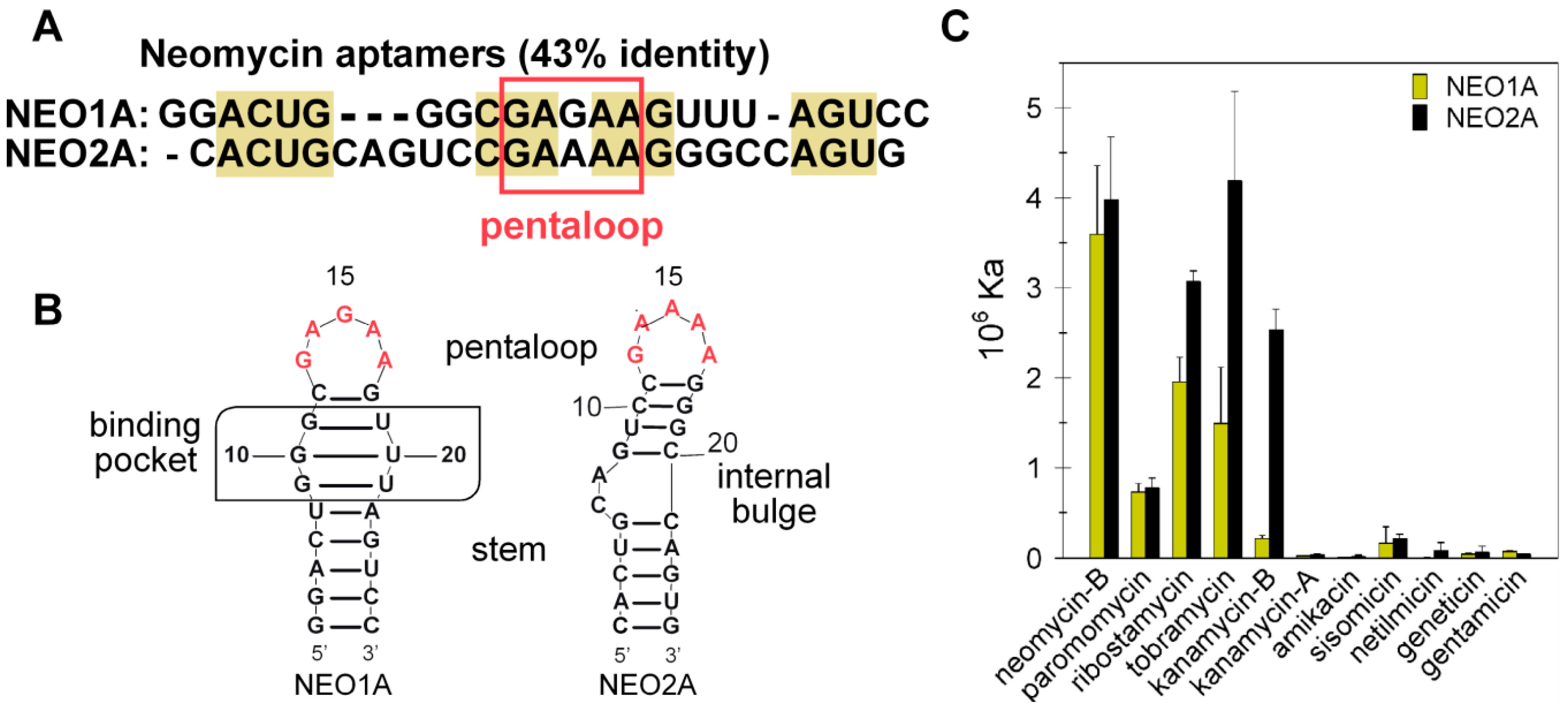
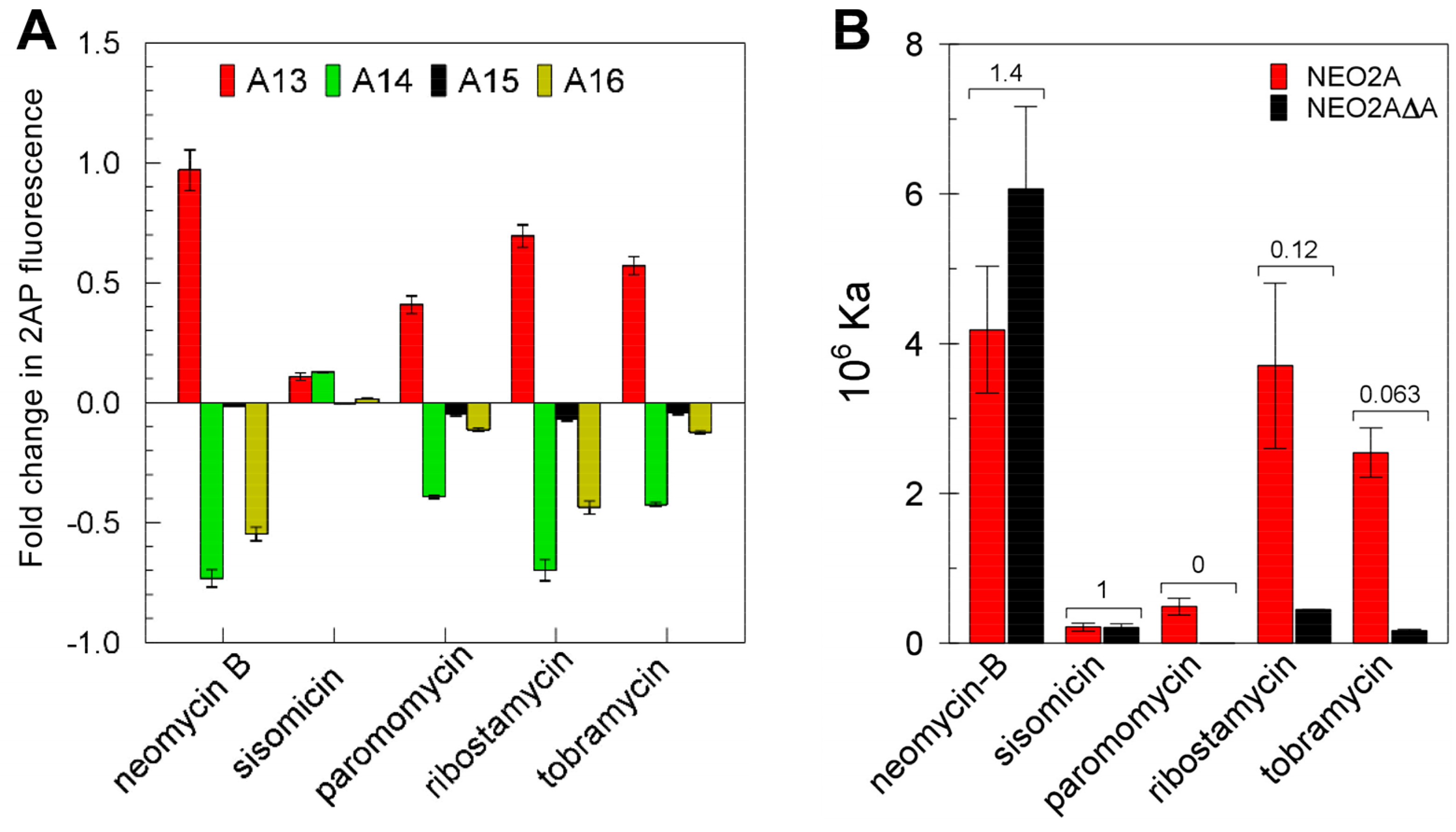
2.1. NEO2A Binds Several Aminoglycosides with a Similar Specificity to NEO1A
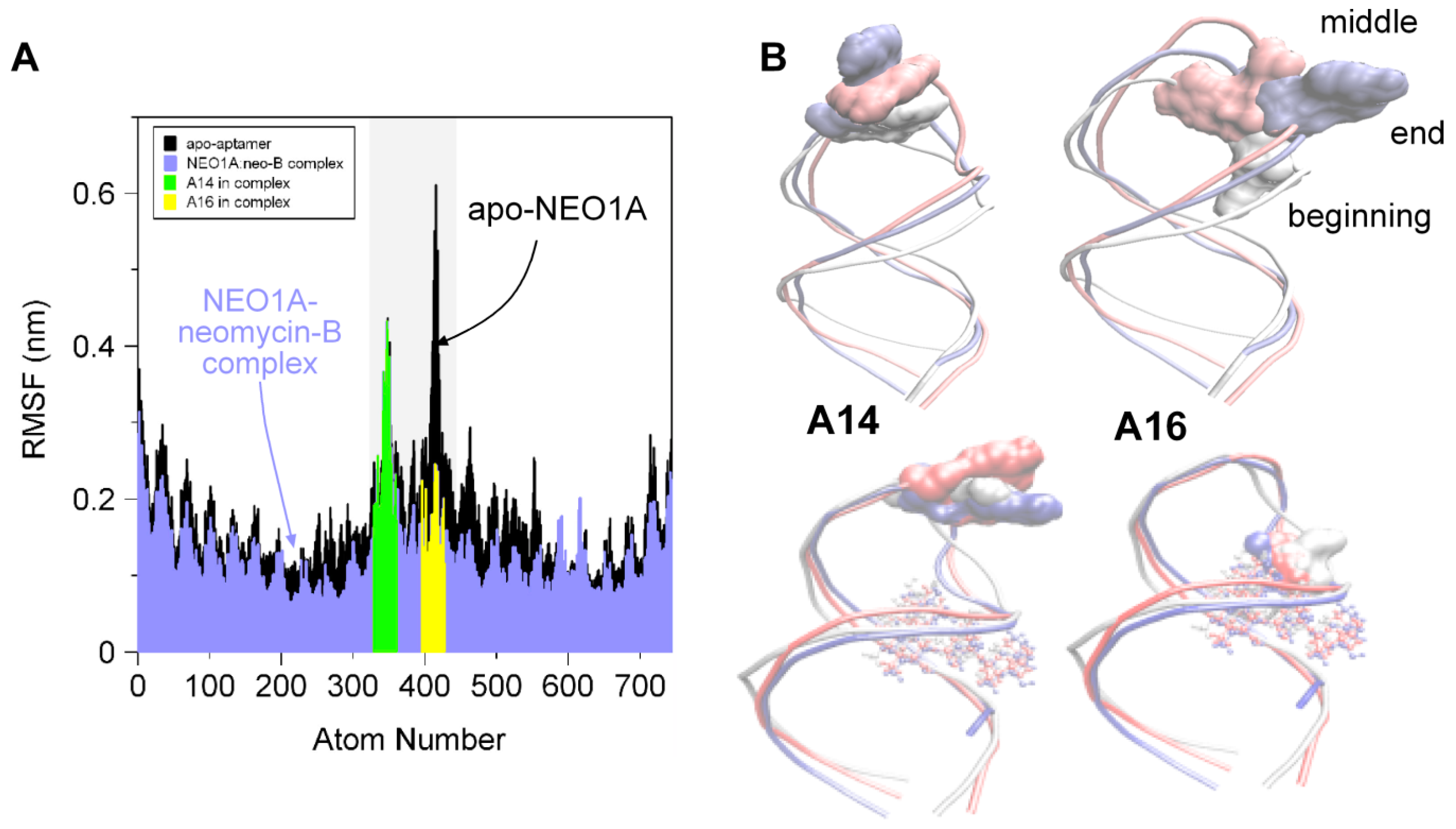
2.2. Molecular Dynamics (MD) Predicts NEO1A Binding to Aminoglycosides Involves Large Loop Movements
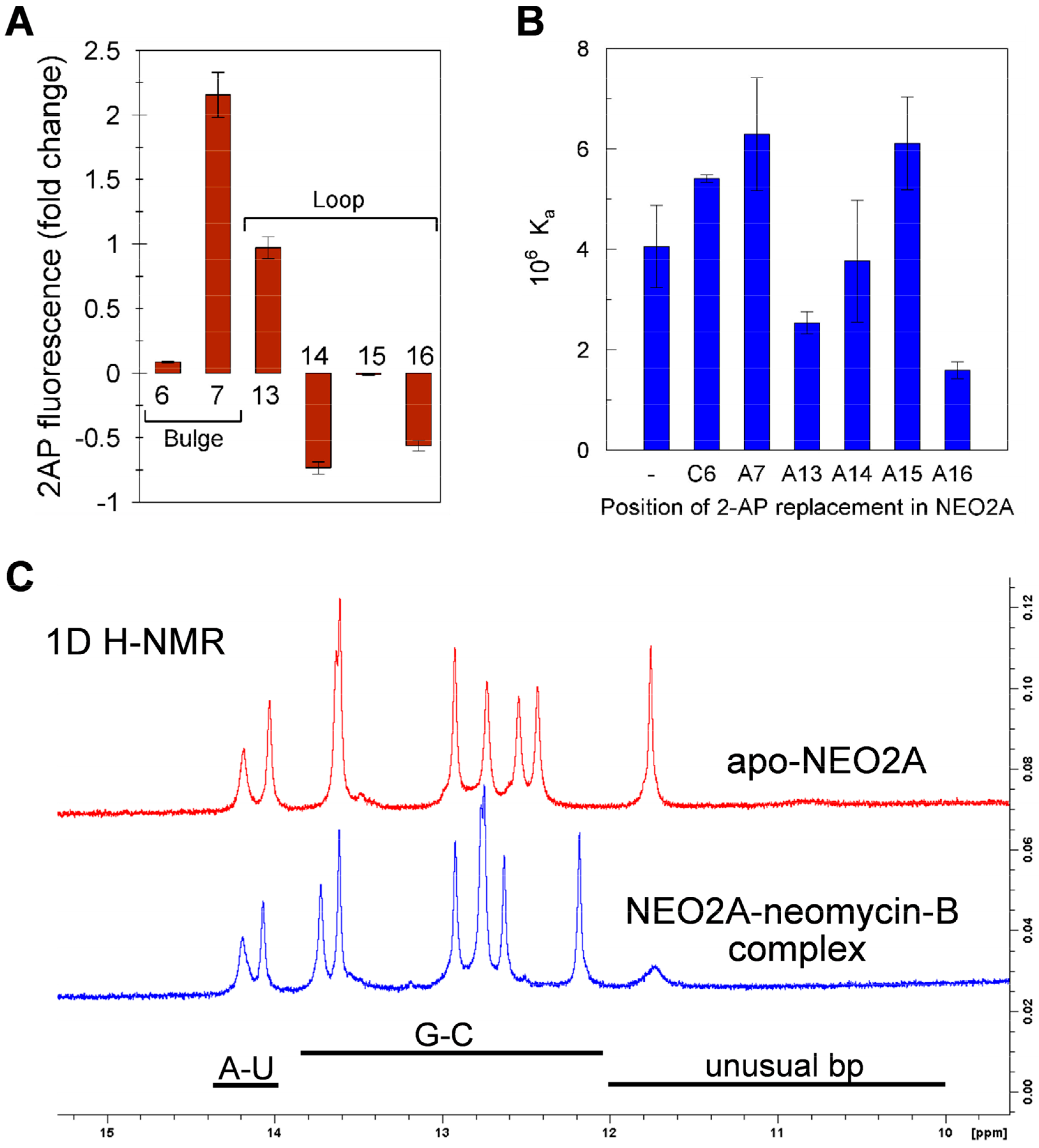
2.3. A global Change in NEO2A with Ligand Binding
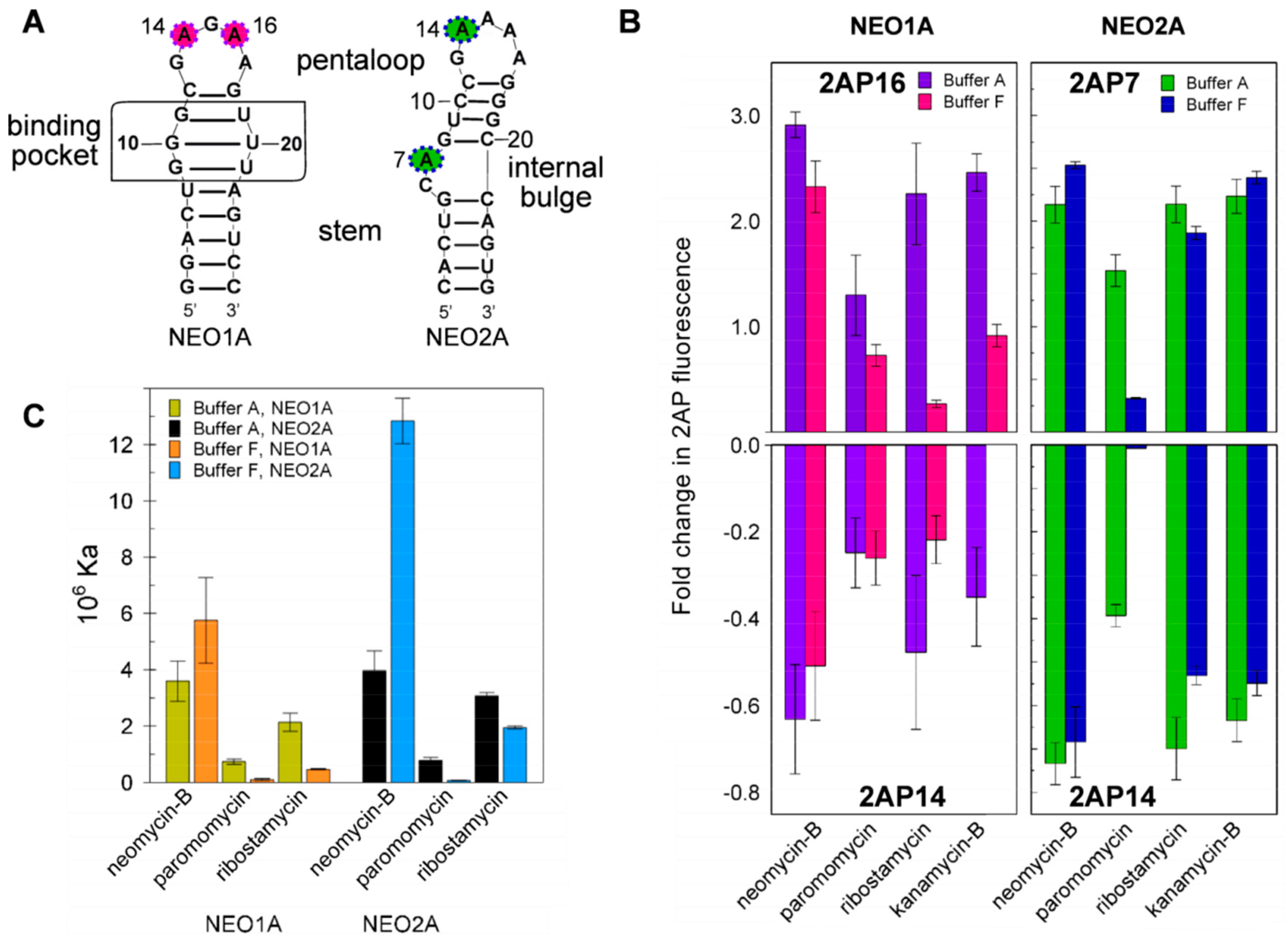
2.4. Variation of Aptamer–Ligand Interactions with Ionic Environment Suggests Similar Ligand Disposition in NEO2A as in NEO1A
2.5. Ligand-Specific Interactions with the Loop Bases of the NEO2A
3. Discussion
4. Materials and Methods
4.1. Materials and Chemicals, Buffers, and RNAs
4.2. Isothermal Titration Calorimetry (ITC)
4.3. Measurement of 2-Aminopurine Fluorescence Intensity
4.4. Molecular Dynamics Simulations
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ditzler, M.A.; Otyepka, M.; Šponer, J.; Walter, N.G. Molecular dynamics and quantum mechanics of RNA: Conformational and chemical change we can believe in. Acc. Chem. Res. 2010, 43, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Dethoff, E.A.; Petzold, K.; Chugh, J.; Casiano-Negroni, A.; Al-Hashimi, H.M. Visualizing transient low-populated structures of RNA. Nature 2012, 491, 724. [Google Scholar] [CrossRef] [PubMed]
- Sponer, J.; Bussi, G.; Krepl, M.; Banas, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurečka, P.; et al. RNA structural dynamics as captured by molecular simulations: A comprehensive overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.-Z.; Zhang, D.; Chen, S.-J. Theory and Modeling of RNA Structure and Interactions with Metal Ions and Small Molecules. Annu. Rev. Biophys. 2017, 46, 227–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef]
- Dahlberg, A.E. The functional role of ribosomal RNA in protein synthesis. Cell 1989, 57, 525–529. [Google Scholar] [CrossRef]
- Lee, J.; Kwon, M.; Lee, K.H.; Jeong, S.; Hyun, S.; Shin, K.J.; Yu, J. An Approach to Enhance Specificity against RNA Targets Using Heteroconjugates of Aminoglycosides and Chloramphenicol (or Linezolid). J. Am. Chem. Soc. 2004, 126, 1956–1957. [Google Scholar] [CrossRef]
- Tok, J.B.H.; Cho, J.; Rando, R.R. Aminoglycoside antibiotics are able to specifically bind the 5′- untranslated region of thymidylate synthase messenger RNA. Biochemistry 1999, 38, 199–206. [Google Scholar] [CrossRef]
- Wang, S.; Huber, P.W.; Cui, M.; Czarnik, A.W.; Mei, H.Y. Binding of neomycin to the TAR element of HIV-1 RNA induces dissociation of Tat protein by an allosteric mechanism. Biochemistry 1998, 37, 5549–5557. [Google Scholar] [CrossRef]
- Wang, Y.; Hamasaki, K.; Rando, R.R. Specificity of aminoglycoside binding to RNA constructs derived from the 16S rRNA decoding region and the HIV-RRE activator region. Biochemistry 1997, 36, 768–779. [Google Scholar] [CrossRef]
- Zapp, M.L.; Stern, S.; Green, M.R. Small molecules that selectively block RNA binding of HIV-1 rev protein inhibit rev function and viral production. Cell 1993, 74, 969–978. [Google Scholar] [CrossRef]
- Von Ahsen, U.; Davies, J.; Schroeder, R. Antibiotic inhibition of group I ribozyme function. Nature 1991, 353, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, N.E.; Johansson, K.; Virtanen, A.; Kirsebom, L.A. Aminoglycoside binding displaces a divalent metal ion in a tRNA-neomycin B complex. Nat. Struct. Biol. 2001, 8, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Chang, A.H.; Von Ahsen, U.; Schroeder, R.; Davies, J. Inhibition of the self-cleavage reaction of the human hepatitis delta virus ribozyme by antibiotics. J. Mol. Biol. 1996, 259, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.S.; Wu, H.L.; Wang, H.W.; Chen, D.S.; Chen, P.J. Inhibition of hepatitis delta virus genomic ribozyme self-cleavage by aminoglycosides. J. Biomed. Sci. 1997, 4, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Tor, Y.; Hermann, T.; Westhof, E. Deciphering RNA recognition: Aminoglycoside binding to the hammerhead ribozyme. Chem. Biol. 1998, 5, R277–R283. [Google Scholar] [CrossRef] [Green Version]
- Stage, T.K.; Hertel, K.J.; Uhlenbeck, O.C. Inhibition of the hammerhead ribozyme by neomycin. RNA 1995, 1, 95–101. [Google Scholar]
- Mikkelsen, N.E.; Brännvall, M.; Virtanen, A.; Kirsebom, L.A. Inhibition of RNase P RNA cleavage by aminoglycosides. Proc. Natl. Acad. Sci. USA 1999, 96, 6155–6160. [Google Scholar] [CrossRef] [Green Version]
- Wallis, M.G.; von Ahsen, U.; Schroeder, R.; Famulok, M. A novel RNA motif for neomycin recognition. Chem. Biol. 1995, 2, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Killian, J.; Hamasaki, K.; Rando, R.R. RNA molecules that specifically and stoichiometrically bind aminoglycoside antibiotics with high affinities. Biochemistry 1996, 35, 12338–12346. [Google Scholar] [CrossRef]
- Werstuck, G. Controlling Gene Expression in Living Cells Through Small Molecule-RNA Interactions. Science 1998, 282, 296–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lato, S.M.; Boles, A.R.; Ellington, A.D. In vitro selection of RNA lectins: Using combinatorial chemistry to interpret ribozyme evolution. Chem. Biol. 1995, 2, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Chun, S.M.; Jeong, S.; Yu, J. In vitro selection of RNA against kanamycin B. Mol. Cells 2001, 11, 303–311. [Google Scholar] [PubMed]
- Weigand, J.E.; Sanchez, M.; Gunnesch, E.B.; Zeiher, S.; Schroeder, R.; Suess, B. Screening for engineered neomycin riboswitches that control translation initiation. RNA 2008, 14, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Suri, A.K.; Fiala, R.; Patel, D.J. Saccharide-RNA recognition in an aminoglycoside antibiotic-RNA aptamer complex. Chem. Biol. 1997, 4, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Majumdar, A.; Hu, W.; Jaishree, T.J.; Xu, W.; Patel, D.J. Saccharide-RNA recognition in a complex formed between neomycin B and an RNA aptamer. Structure 1999, 7, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Duchardt-Ferner, E.; Weigand, J.E.; Ohlenschläger, O.; Schmidtke, S.R.; Suess, B.; Wöhnert, J. Highly modular structure and ligand binding by conformational capture in a minimalistic riboswitch. Angew. Chem. Int. Ed. 2010, 49, 6216–6219. [Google Scholar] [CrossRef]
- Wang, Y.; Rando, R.R. Specific binding of aminoglycoside antibiotics to RNA. Chem. Biol. 1995, 2, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Ilgu, M.; Fulton, D.B.; Yennamalli, R.M.; Lamm, M.H.; Sen, T.Z.; Nilsen-Hamilton, M. An adaptable pentaloop defines a robust neomycin-B RNA aptamer with conditional ligand-bound structures. RNA 2014, 20, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Stampfl, S.; Lempradl, A.; Koehler, G.; Schroeder, R. Monovalent ion dependence of neomycin B binding to an RNA aptamer characterized by spectroscopic methods. Chem. Biol. Chem. 2007, 8, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Allnér, O.; Nilsson, L.; Villa, A. Loop-loop interaction in an adenine-sensing riboswitch: A molecular dynamics study. RNA 2013, 19, 916–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aytenfisu, A.H.; Liberman, J.A.; Wedekind, J.E.; Mathews, D.H. Molecular mechanism for preQ1-II riboswitch function revealed by molecular dynamics. RNA 2015, 21, 1898–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachan, A.; Ilgu, M.; Kempema, A.; Kraus, G.A.; Nilsen-Hamilton, M. Specificity and Ligand Affinities of the Cocaine Aptamer: Impact of Structural Features and Physiological NaCl. Anal. Chem. 2016, 88, 7715–7723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, O.F.; Van Der Spoel, D.; De Groot, B.L. Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR Data. Biophys. J. 2010, 99, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Zhao, Y.; Chen, C.; Xiao, Y. Role of ligand binding in structural organization of add a-riboswitch aptamer: A molecular dynamics simulation. J. Biomol. Struct. Dyn. 2011, 29, 403–416. [Google Scholar] [CrossRef]
- Petrone, P.M.; Dewhurst, J.; Tommasi, R.; Whitehead, L.; Pomerantz, A.K. Atomic-scale characterization of conformational changes in the preQ 1 riboswitch aptamer upon ligand binding. J. Mol. Graph. Model. 2011, 30, 179–185. [Google Scholar] [CrossRef]
- Di Palma, F.; Colizzi, F.; Bussi, G. Ligand-induced stabilization of the aptamer terminal helix in the add adenine riboswitch. RNA 2013, 19, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors upon request. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilgu, M.; Yan, S.; Khounlo, R.M.; Lamm, M.H.; Nilsen-Hamilton, M. Common Secondary and Tertiary Structural Features of Aptamer–Ligand Interaction Shared by RNA Aptamers with Different Primary Sequences. Molecules 2019, 24, 4535. https://doi.org/10.3390/molecules24244535
Ilgu M, Yan S, Khounlo RM, Lamm MH, Nilsen-Hamilton M. Common Secondary and Tertiary Structural Features of Aptamer–Ligand Interaction Shared by RNA Aptamers with Different Primary Sequences. Molecules. 2019; 24(24):4535. https://doi.org/10.3390/molecules24244535
Chicago/Turabian StyleIlgu, Muslum, Shuting Yan, Ryan M. Khounlo, Monica H. Lamm, and Marit Nilsen-Hamilton. 2019. "Common Secondary and Tertiary Structural Features of Aptamer–Ligand Interaction Shared by RNA Aptamers with Different Primary Sequences" Molecules 24, no. 24: 4535. https://doi.org/10.3390/molecules24244535
APA StyleIlgu, M., Yan, S., Khounlo, R. M., Lamm, M. H., & Nilsen-Hamilton, M. (2019). Common Secondary and Tertiary Structural Features of Aptamer–Ligand Interaction Shared by RNA Aptamers with Different Primary Sequences. Molecules, 24(24), 4535. https://doi.org/10.3390/molecules24244535