
Amelioration of Hyperglycemia-Induced Nephropathy by 3,3′-Diindolylmethane in Diabetic Mice


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
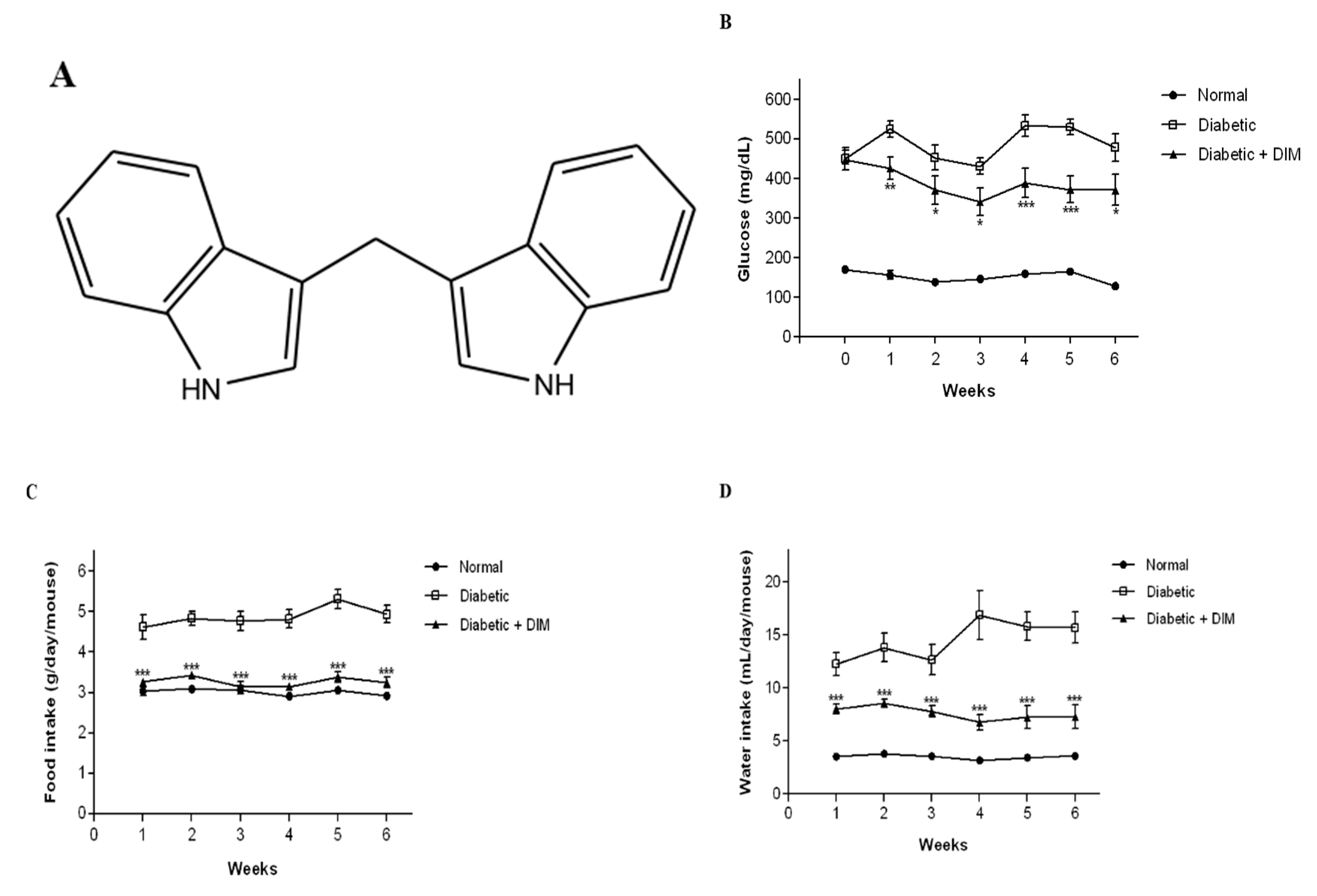
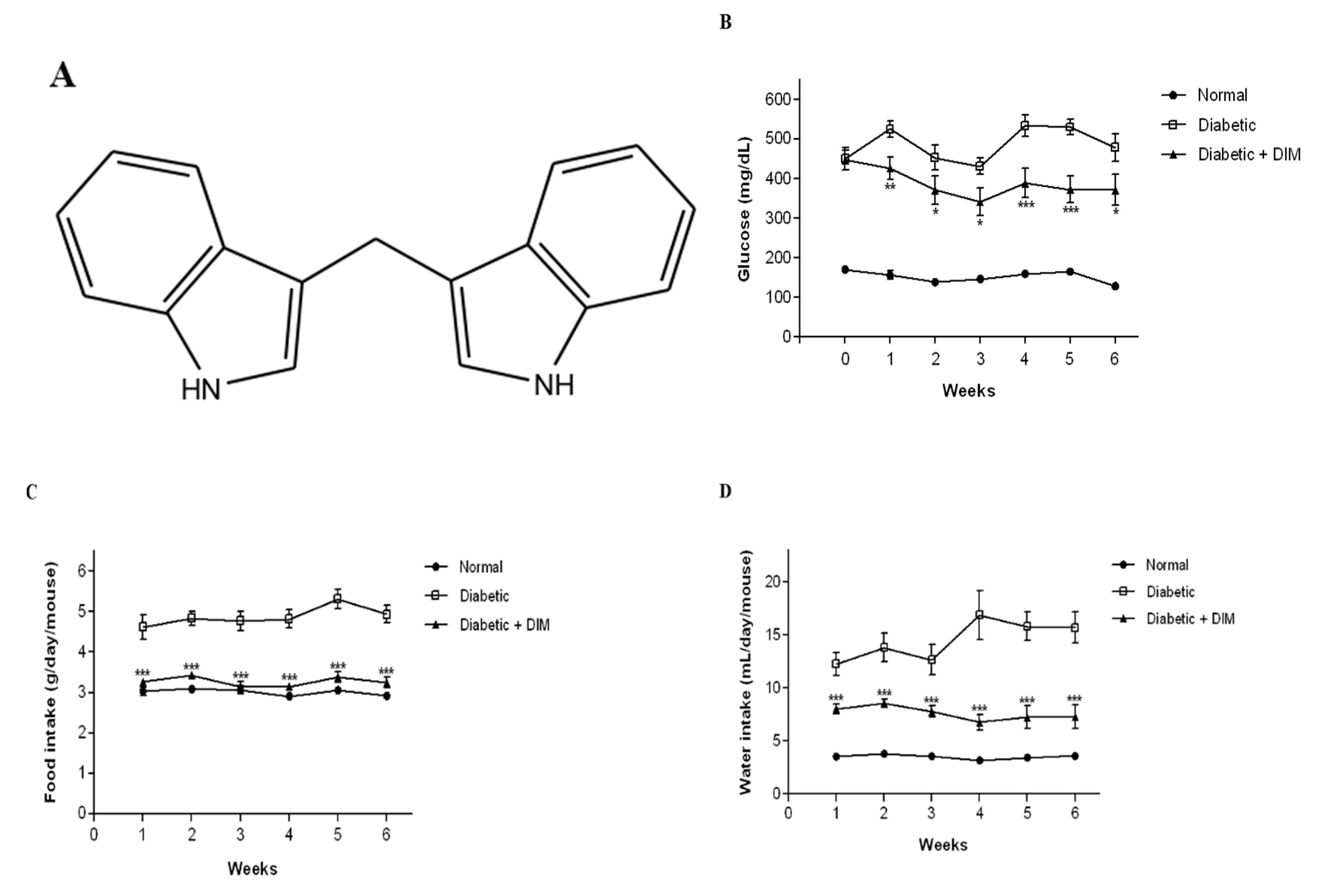
2.1. DIM Improves Blood Glucose Levels and Food and Water Intakes in Diabetic Mice
2.2. DIM Inhibits Hyperglycemia-Induced Kidney Damage of Diabetic Mice
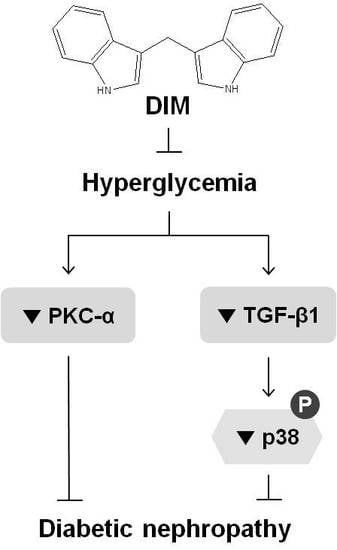
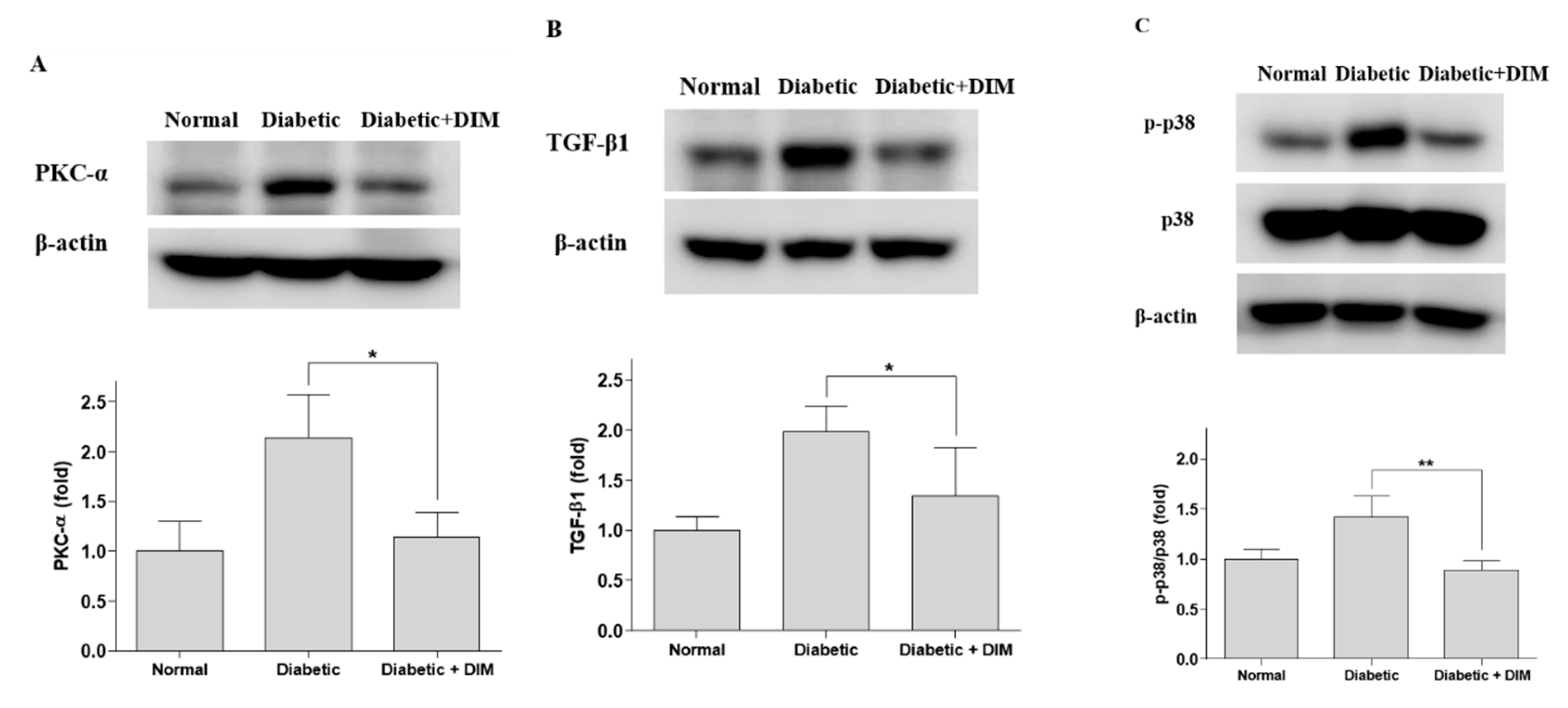
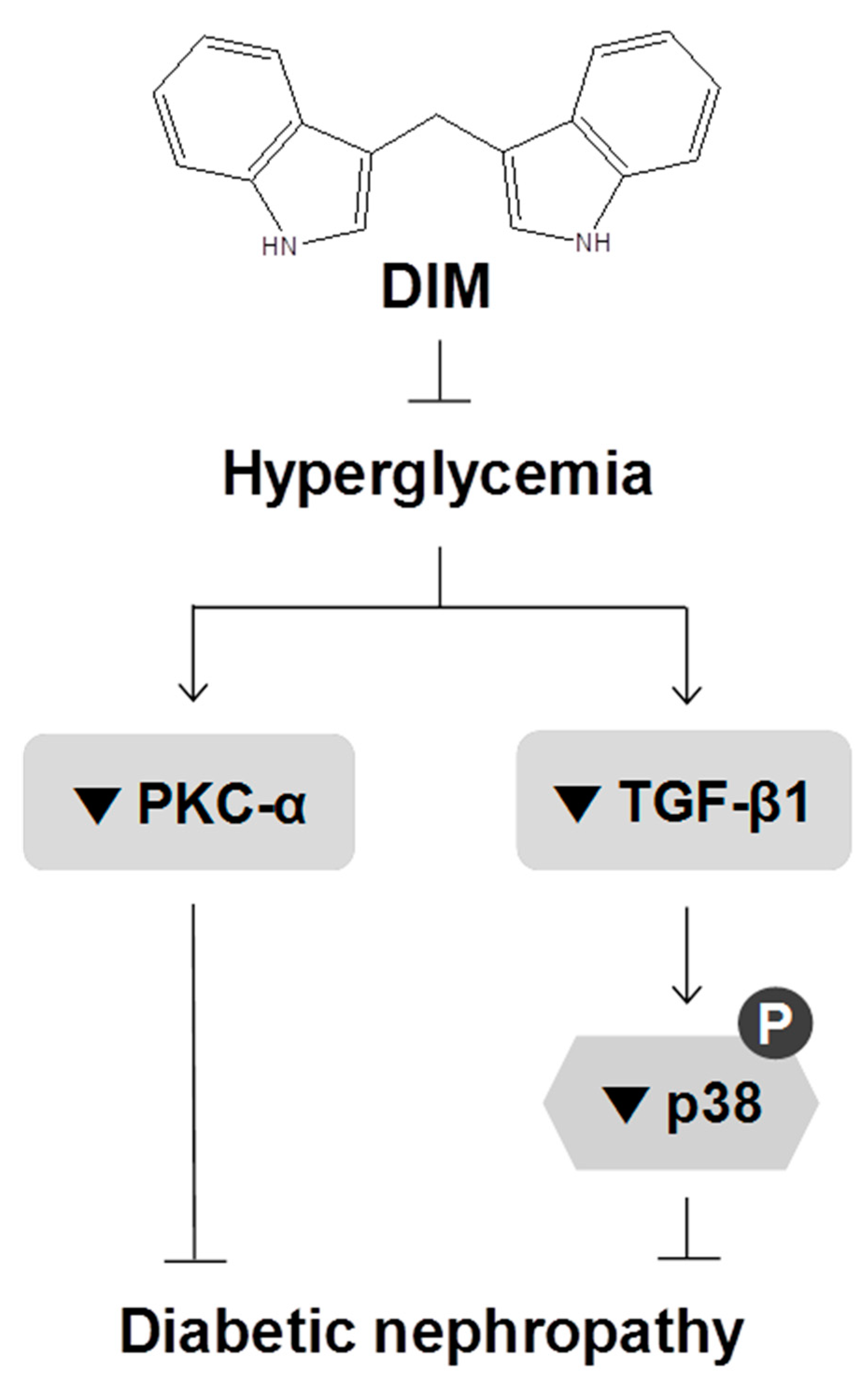
2.3. DIM Inhibits Hyperglycemia-Induced Activation of Pkc-α and Tgf-β1 in the Kidneys
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Treatment
4.3. Creatinine Assay
4.4. Western Blot Analysis
4.5. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou-Marketou, N.; Chrousos, G.P.; Kanaka-Gantenbein, C. Diabetic nephropathy in type 1 diabetes: A review of early natural history, pathogenesis, and diagnosis. Diabetes Metab. Res. Rev. 2017, 33, e2841. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.K.; Singh, U.K. Molecular mechanisms in the pathogenesis of diabetic nephropathy: An update. Vascul. Pharmacol. 2013, 58, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Dalla Vestra, M.; Saller, A.; Bortoloso, E.; Mauer, M.; Fioretto, P. Structural involvement in type 1 and type 2 diabetic nephropathy. Diabetes Metab. 2000, 26, 8–14. [Google Scholar]
- Schena, F.P.; Gesualdo, L. Pathogenetic mechanisms of diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16, S30–S33. [Google Scholar] [CrossRef]
- Yao, L.; Wang, J.; Mao, Y.; Zhu, H.; Deng, A.; Zhu, Z. Different expressions of protein kinase C-alpha, beta I and beta II in glomeruli of diabetic nephropathy patients. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 651–653. [Google Scholar] [CrossRef]
- Wolf, G.; Ziyadeh, F.N. Molecular mechanisms of diabetic renal hypertrophy. Kidney Int. 1999, 56, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Lan, H.Y.; Chung, A.C. TGF-beta/Smad signaling in kidney disease. Semin. Nephrol. 2012, 32, 236–243. [Google Scholar] [CrossRef]
- Anderton, M.J.; Manson, M.M.; Verschoyle, R.D.; Gescher, A.; Lamb, J.H.; Farmer, P.B.; Steward, W.P.; Williams, M.L. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin. Cancer Res. 2004, 10, 5233–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grose, K.R.; Bjeldanes, L.F. Oligomerization of indole-3-carbinol in aqueous acid. Chem. Res. Toxicol. 1992, 5, 188–193. [Google Scholar] [CrossRef]
- Choi, K.M.; Yoo, H.S. 3,3′-Diindolylmethane Enhances Glucose Uptake Through Activation of Insulin Signaling in 3T3-L1 Adipocytes. Obesity 2018, 26, 1153–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poornima, J.; Mirunalini, S. Regulation of carbohydrate metabolism by indole-3-carbinol and its metabolite 3,3′-diindolylmethane in high-fat diet-induced C57BL/6J mice. Mol. Cell. Biochem. 2014, 385, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Deeds, M.C.; Anderson, J.M.; Armstrong, A.S.; Gastineau, D.A.; Hiddinga, H.J.; Jahangir, A.; Eberhardt, N.L.; Kudva, Y.C. Single dose streptozotocin-induced diabetes: Considerations for study design in islet transplantation models. Lab. Anim. 2011, 45, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar] [PubMed]
- Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Awar, A.; Kupai, K.; Veszelka, M.; Szucs, G.; Attieh, Z.; Murlasits, Z.; Torok, S.; Posa, A.; Varga, C. Experimental Diabetes Mellitus in Different Animal Models. J. Diabetes Res. 2016, 2016, 9051426. [Google Scholar] [CrossRef] [Green Version]
- Brito-Casillas, Y.; Melian, C.; Wagner, A.M. Study of the pathogenesis and treatment of diabetes mellitus through animal models. Endocrinol. Nutr. 2016, 63, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Ohara, N.; Kobayashi, M.; Ikeda, Y.; Hoshi, T.; Morita, S.; Kanefuji, T.; Yagi, K.; Suda, T.; Takada, T.; Hasegawa, G.; et al. Non-insulin-dependent Diabetes Mellitus Induced by Immune Checkpoint Inhibitor Therapy in an Insulinoma-associated Antigen-2 Autoantibody-positive Patient with Advanced Gastric Cancer. Intern. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Buko, V.; Zavodnik, I.; Kanuka, O.; Belonovskaya, E.; Naruta, E.; Lukivskaya, O.; Kirko, S.; Budryn, G.; Zyzelewicz, D.; Oracz, J.; et al. Antidiabetic effects and erythrocyte stabilization by red cabbage extract in streptozotocin-treated rats. Food Funct. 2018, 9, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9, eaah4477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahadoran, Z.; Tohidi, M.; Nazeri, P.; Mehran, M.; Azizi, F.; Mirmiran, P. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: A randomized double-blind clinical trial. Int. J. Food Sci. Nutr. 2012, 63, 767–771. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, T.; Zhao, L. 3,3′Diindolylmethane mitigates lipopolysaccharideinduced acute kidney injury in mice by inhibiting NOXmediated oxidative stress and the apoptosis of renal tubular epithelial cells. Mol. Med. Rep. 2019, 19, 5115–5122. [Google Scholar]
- Lee, H.B.; Yu, M.R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, S241–S245. [Google Scholar] [CrossRef] [Green Version]
- Menne, J.; Park, J.K.; Boehne, M.; Elger, M.; Lindschau, C.; Kirsch, T.; Meier, M.; Gueler, F.; Fiebeler, A.; Bahlmann, F.H.; et al. Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-alpha-deficient diabetic mice. Diabetes 2004, 53, 2101–2109. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Jin, Y.; Guo, J.; Ziyadeh, F.N. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 1996, 45, 522–530. [Google Scholar] [CrossRef]
- Xia, Z.E.; Xi, J.L.; Shi, L. 3,3′-Diindolylmethane ameliorates renal fibrosis through the inhibition of renal fibroblast activation in vivo and in vitro. Ren. Fail. 2018, 40, 447–454. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, K.-M.; Yoo, H.-S. Amelioration of Hyperglycemia-Induced Nephropathy by 3,3′-Diindolylmethane in Diabetic Mice. Molecules 2019, 24, 4474. https://doi.org/10.3390/molecules24244474
Choi K-M, Yoo H-S. Amelioration of Hyperglycemia-Induced Nephropathy by 3,3′-Diindolylmethane in Diabetic Mice. Molecules. 2019; 24(24):4474. https://doi.org/10.3390/molecules24244474
Chicago/Turabian StyleChoi, Kyeong-Mi, and Hwan-Soo Yoo. 2019. "Amelioration of Hyperglycemia-Induced Nephropathy by 3,3′-Diindolylmethane in Diabetic Mice" Molecules 24, no. 24: 4474. https://doi.org/10.3390/molecules24244474
APA StyleChoi, K.-M., & Yoo, H.-S. (2019). Amelioration of Hyperglycemia-Induced Nephropathy by 3,3′-Diindolylmethane in Diabetic Mice. Molecules, 24(24), 4474. https://doi.org/10.3390/molecules24244474