
Synthesis and Pharmacological Evaluation of Hybrids Targeting Opioid and Neurokinin Receptors



 ,
,
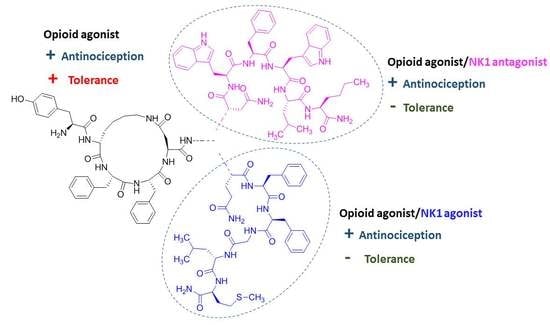
Abstract
:
1. Introduction
2. Results
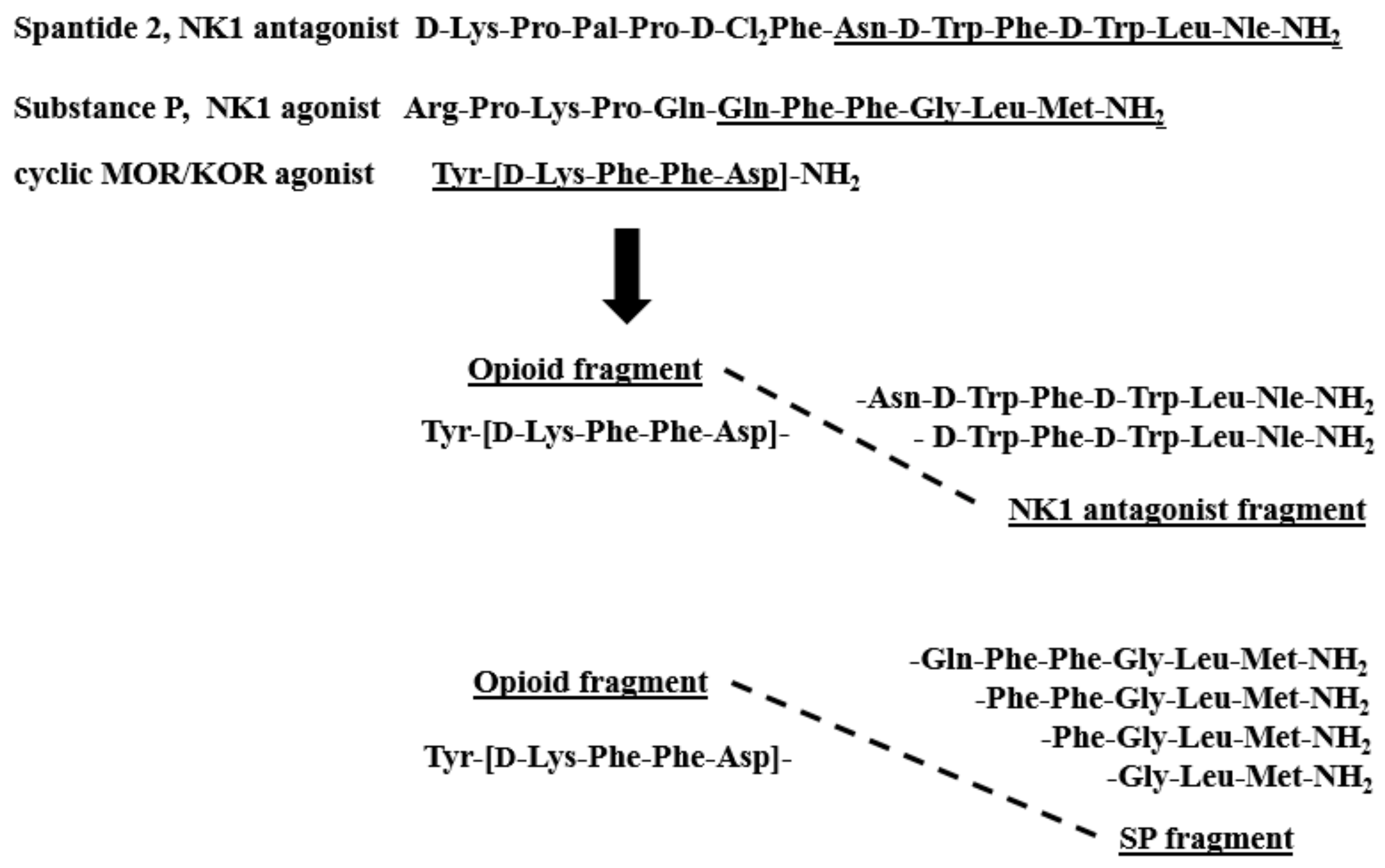
2.1. Chemistry
2.2. Receptor Binding Affinity
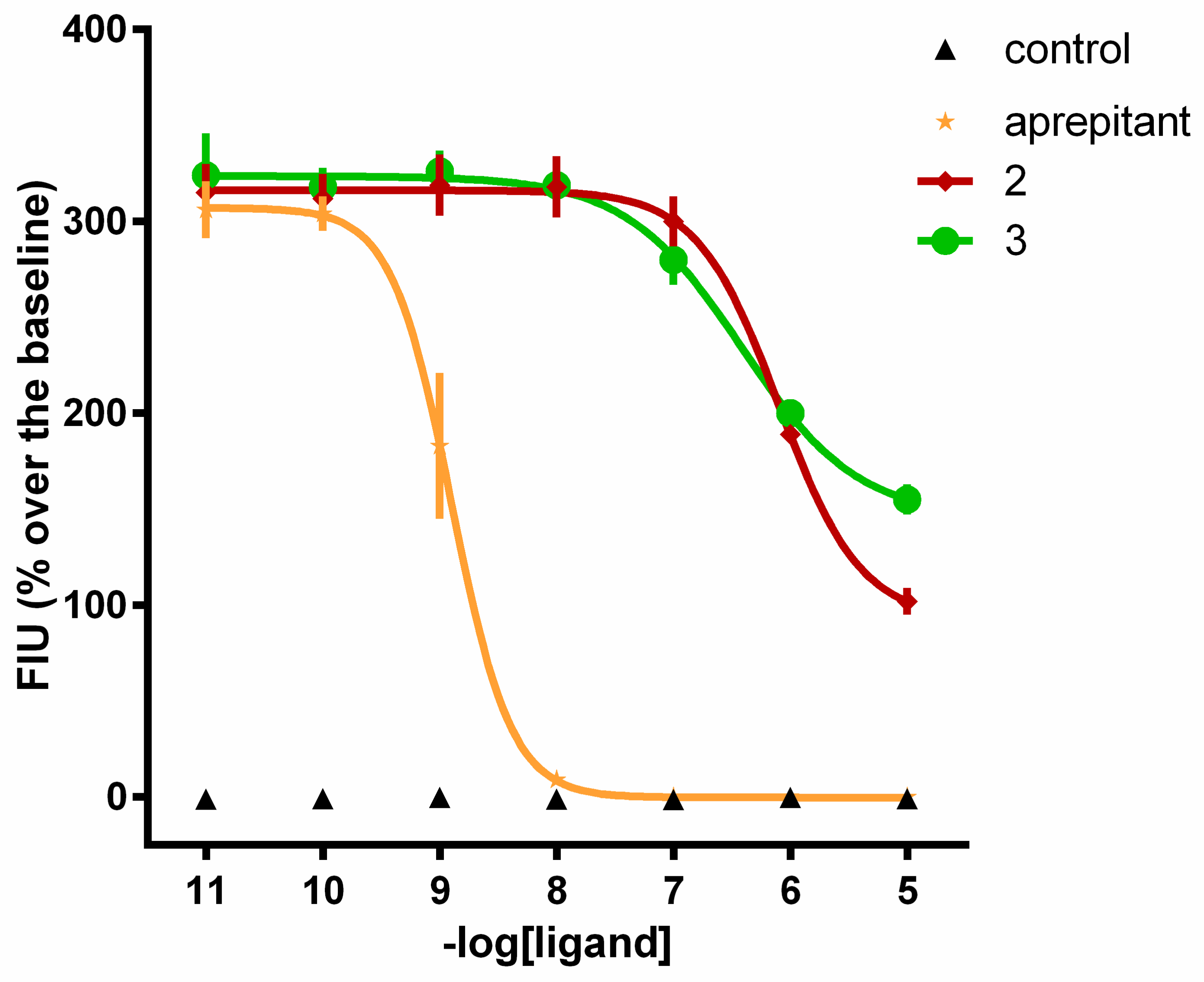
2.3. Calcium Mobilization Functional Assay
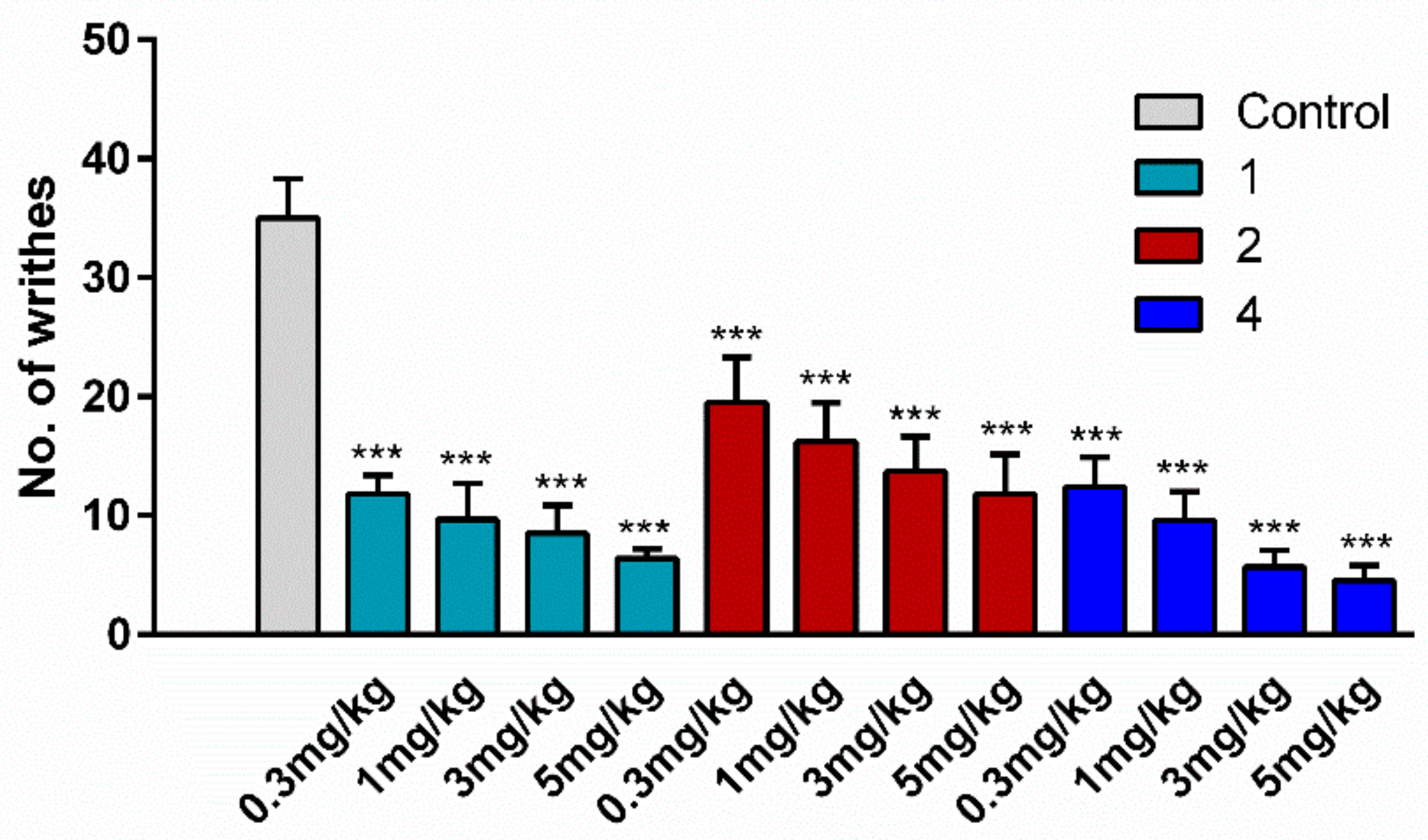
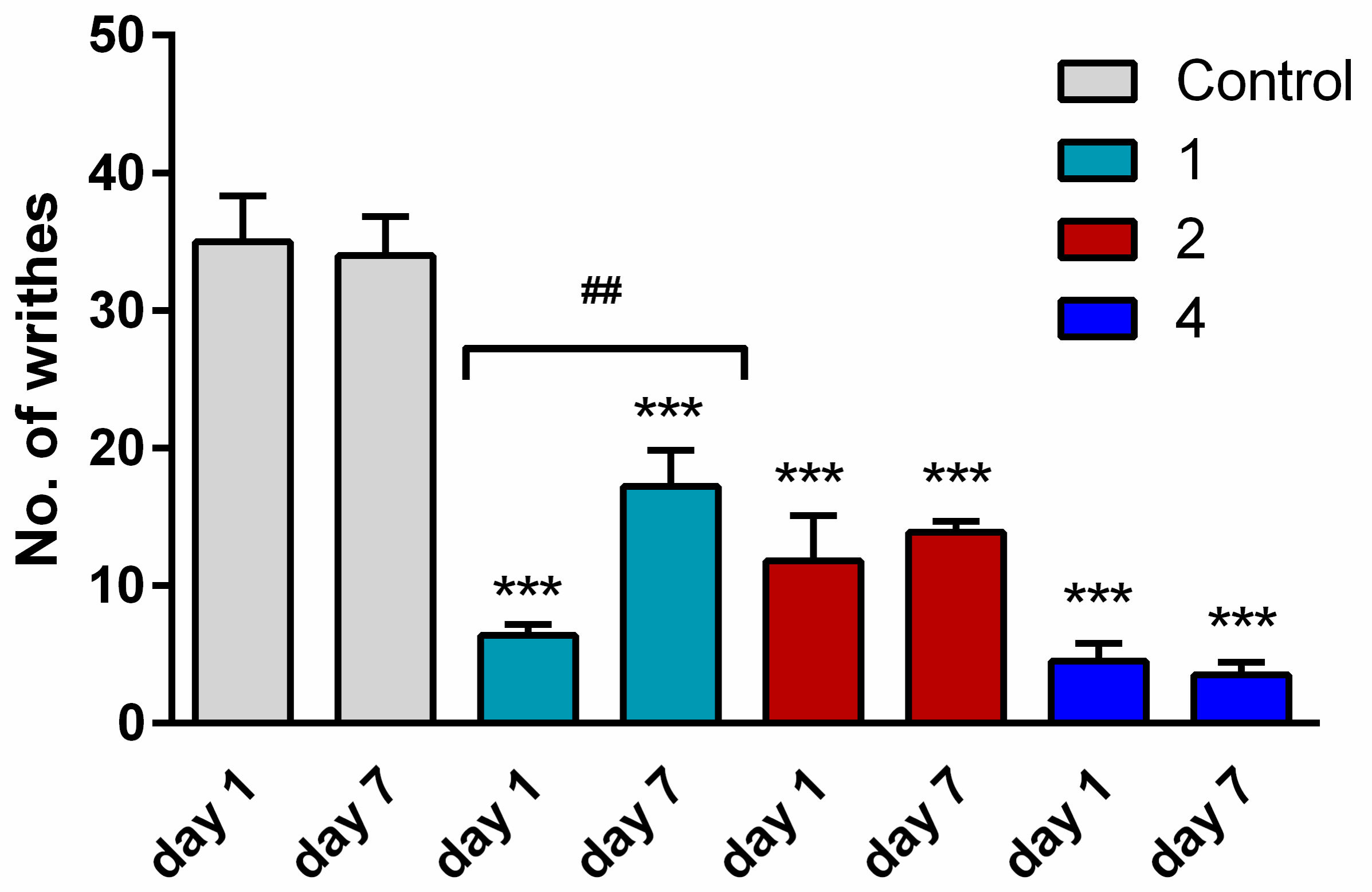
2.4. Antinociceptive Activity
2.5. Tolerance
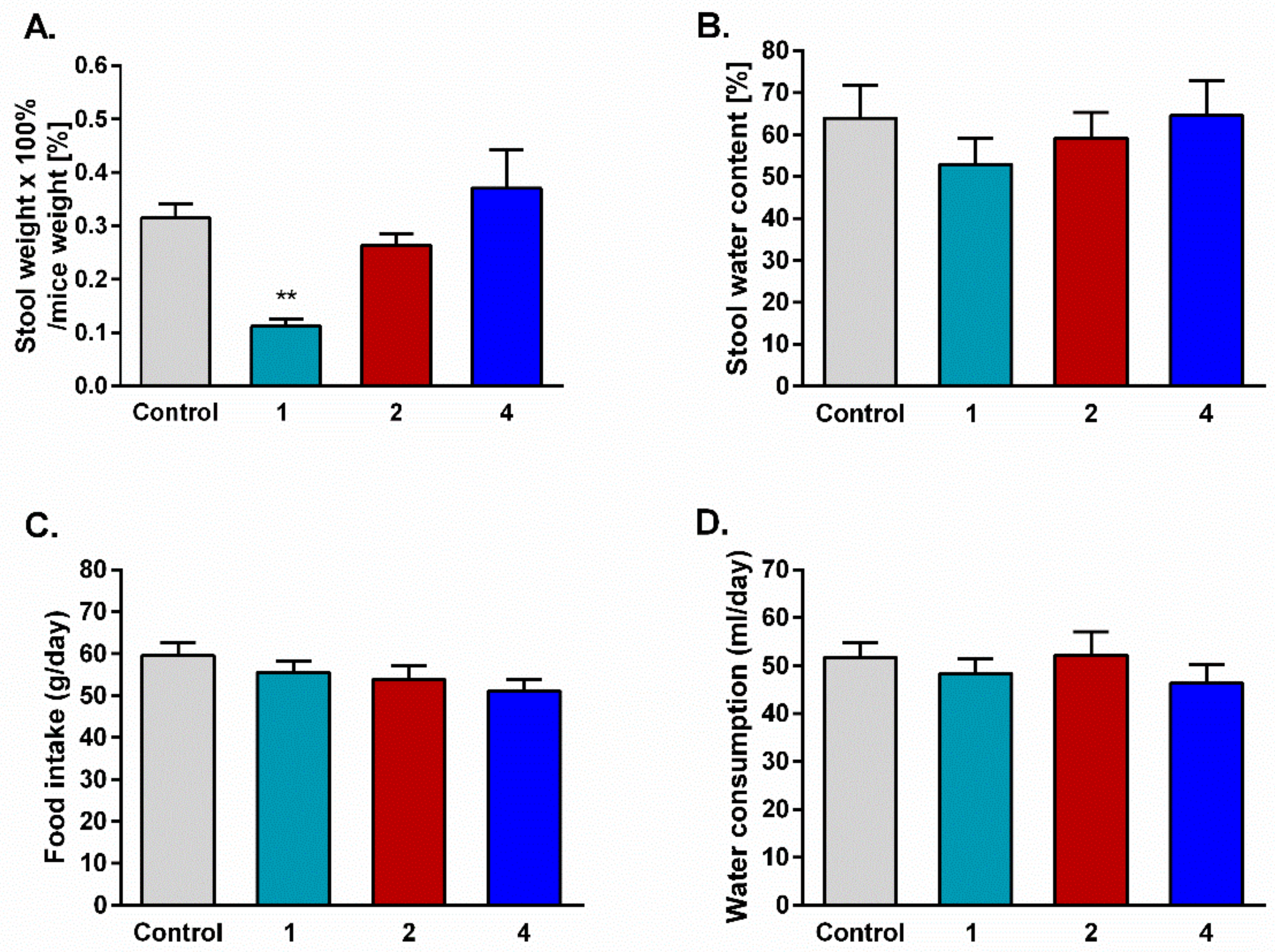
2.6. Stool Mass and Consumption of Food and Water
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Peptide Synthesis
4.3. Radioligand Binding Assays
4.4. Cell Culture
4.5. Calcium Mobilization Functional Assay
4.6. Animals
4.7. Assessment of Antinociception by Writhing Test
4.8. Assessment of Tolerance Development
4.9. Stool Collection
4.10. Food and Water Consumption
4.11. Statistical Analysis and Terminology
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Spetea, M.; Asim, M.F.; Wolber, G.; Schmidhammer, H. The µ opioid receptor and ligands acting at the µ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. [Google Scholar] [CrossRef]
- Smith, H.S.; Peppin, J.F. Toward a systematic approach to opioid rotation. J. Pain Res. 2014, 7, 589–608. [Google Scholar] [PubMed] [Green Version]
- Ueda, H.; Ueda, M. Mechanisms underlying morphine analgesic tolerance and dependence. Front. Biosci. 2009, 14, 5260–5272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.M.; Christie, M.J. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br. J. Pharmacol. 2011, 164, 1322–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnaturi, R.; Aricò, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. [Google Scholar] [CrossRef]
- Giri, A.K.; Hruby, V.J. Investigational peptide and peptidomimetic µ and δ opioid receptor agonists in the relief of pain. Expert Opin. Investig. Drugs 2014, 23, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Mollica, A.; Pinnen, F.; Costante, R.; Locatelli, M.; Stefanucci, A.; Pieretti, S.; Davis, P.; Lai, J.; Rankin, D.; Porreca, F.; et al. Biological active analogues of the opioid peptide biphalin: Mixed α/β(3)-peptides. J. Med. Chem. 2013, 56, 3419–3423. [Google Scholar] [CrossRef] [Green Version]
- Morphy, R.; Rankovic, Z. Designing multiple ligands-medicinal chemistry strategies and challenges. Curr. Pharm. Des. 2009, 15, 587–600. [Google Scholar] [CrossRef]
- Ballet, S.; Pietsch, M.; Abell, A.D. Multiple ligands in opioid research. Protein Pept. Lett. 2008, 15, 668–682. [Google Scholar] [CrossRef]
- Fujii, H. Twin and triplet drugs in opioid research. Top. Curr. Chem. 2011, 299, 239–275. [Google Scholar]
- Kleczkowska, P.; Lipkowski, A.W.; Tourwé, D.; Ballet, S. Hybrid opioid/non-opioid ligands in pain research. Curr. Pharm. Des. 2013, 19, 7435–7450. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowska, P.; Nowicka, K.; Bujalska-Zadrozny, M.; Hermans, E. Neurokinin-1 receptor-based bivalent drugs in pain management: The journey to nowhere? Pharmacol. Ther. 2019, 196, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Dvoracsko, S.; Stefanucci, A.; Novellino, E.; Mollica, A. The design of multitarget ligands for chronic and neuropathic pain. Future Med. Chem. 2015, 7, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Recio, S.; Gascón, P. Biological and pharmacological aspects of the NK1-receptor. Biomed. Res. Int. 2015, 2015, 495704. [Google Scholar] [CrossRef] [Green Version]
- Corrêa, J.M.X.; Soares, P.C.L.R.; Niella, R.V.; Costa, B.A.; Ferreira, M.S.; Junior, A.C.S.; Sena, A.S.; Sampaio, K.M.O.R.; Silva, E.B.; Silva, F.L.; et al. Evaluation of the antinociceptive effect of maropitant, a neurokinin-1 receptor antagonist, in cats undergoing ovariohysterectomy. Vet. Med. Int. 2019, 2019, 9352528. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhu, Y.; Zheng, W.; Qian, T.; Wang, H.; Hou, X. Antagonism of NK-1R using aprepitant suppresses inflammatory response in rheumatoid arthritis fibroblast-like synoviocytes. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1628–1634. [Google Scholar] [CrossRef] [Green Version]
- Prasoon, P.; Gupta, S.; Kumar, R.; Gautam, M.; Kaler, S.; Ray, S.B. Role of fosaprepitant, a neurokinin Type 1 receptor antagonist, in morphine-induced antinociception in rats. Indian J. Pharmacol. 2016, 48, 394–398. [Google Scholar]
- Yamamoto, T.; Nair, P.; Largent-Milnes, T.M.; Jacobsen, N.E.; Davis, P.; Ma, S.W.; Yamamura, H.I.; Vanderah, T.W.; Porreca, F.; Lai, J.; et al. Discovery of a potent and efficacious peptide derivative for δ/µ opioid agonist/neurokinin 1 antagonist activity with a 2′,6′-dimethyl-L-tyrosine: In vitro, in vivo, and NMR-based structural studies. J. Med. Chem. 2011, 54, 2029–2038. [Google Scholar] [CrossRef] [Green Version]
- Bonney, I.M.; Foran, S.E.; Marchand, J.E.; Lipkowski, A.W.; Carr, D.B. Spinal antinociceptive effects of AA501, a novel chimeric peptide with opioid receptor agonist and tachykinin receptor antagonist moieties. Eur. J. Pharmacol. 2004, 488, 91–99. [Google Scholar] [CrossRef]
- Guillemyn, K.; Kleczkowska, P.; Novoa, A.; Vandormael, B.; Van den Eynde, I.; Kosson, P.; Asim, M.F.; Schiller, P.W.; Spetea, M.; Lipkowski, A.W.; et al. In vivo antinociception of potent mu opioid agonist tetrapeptide analogues and comparison with a compact opioid agonist-neurokinin 1 receptor antagonist chimera. Mol. Brain 2012, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Giri, A.K.; Apostol, C.R.; Wang, Y.; Forte, B.L.; Largent-Milnes, T.M.; Davis, P.; Rankin, D.; Molnar, G.; Olson, K.M.; Porreca, F.; et al. Discovery of novel multifunctional ligands with µ/δ opioid agonist/neurokinin-1 (NK1) antagonist activities for the treatment of pain. J. Med. Chem. 2015, 58, 8573–8583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, P.; Yamamoto, T.; Cowell, S.; Kulkarni, V.; Moye, S.; Navratilova, E.; Davis, P.; Ma, S.W.; Vanderah, T.W.; Lai, J.; et al. Discovery of tripeptide-derived multifunctional ligands possessing delta/mu opioid receptor agonist and neurokinin 1 receptor antagonist activities. Bioorg. Med. Chem. Lett. 2015, 25, 3716–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemyn, K.; Kleczkowska, P.; Lesniak, A.; Dyniewicz, J.; Van der Poorten, O.; Van den Eynde, I.; Keresztes, A.; Varga, E.; Lai, J.; Porreca, F.; et al. Synthesis and biological evaluation of compact, conformationally constrained bifunctional opioid agonist-neurokinin-1 antagonist peptidomimetics. Eur. J. Med. Chem. 2015, 92, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kream, R.M.; Kato, T.; Shimonaka, H.; Marchand, J.E.; Wurm, W.H. Substance P markedly potentiates the antinociceptive effects of morphine sulfate administered at the spinal level. Proc. Natl. Acad. Sci. USA 1993, 90, 3564–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foran, S.E.; Carr, D.B.; Lipkowski, A.W.; Maszczynska, I.; Marchand, J.E.; Misicka, A.; Beinborn, M.; Kopin, A.S.; Kream, R.M. A substance P-opioid chimeric peptide as a unique nontolerance-forming analgesic. Proc. Natl. Acad. Sci. USA 2000, 97, 7621–7626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kream, R.M.; Liu, N.L.; Zhuang, M.; Esposito, P.L.; Esposito, T.R.; Stefano, G.B.; Witmeyer, J.J., 3rd. Synthesis and pharmacological analysis of a morphine/substance P chimeric molecule with full analgesic potency in morphine-tolerant rats. Med. Sci. Monit. 2007, 13, 25–31. [Google Scholar]
- Varamini, P.; Hussein, W.M.; Mansfeld, F.M.; Toth, I. Synthesis, biological activity and structure-activity relationship of endomorphin-1/substance P derivatives. Bioorg. Med. Chem. 2012, 20, 6335–6343. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Kleczkowska, P.; Rękawek, M.; Kulik, K.; Lesniak, A.; Erdei, A.; Borics, A.; Martin, C.; Pawlik, K.; Lipkowski, A.W.; et al. Biological evaluation and molecular docking studies of AA3052, a compound containing a µ-selective opioid peptide agonist DALDA and d-Phe-Phe-d-Phe-Leu-Leu-NH2, a substance P analogue. Eur. J. Pharm. Sci. 2016, 93, 11–20. [Google Scholar] [CrossRef]
- Piekielna, J.; Perlikowska, R.; Gach, K.; Janecka, A. Cyclization in opioid peptides. Curr. Drug Targets 2013, 14, 798–816. [Google Scholar] [CrossRef]
- Perlikowska, R.; do-Rego, J.C.; Cravezic, A.; Fichna, J.; Wyrebska, A.; Toth, G.; Janecka, A. Synthesis and biological evaluation of cyclic endomorphin-2 analogs. Peptides 2010, 31, 339–345. [Google Scholar] [CrossRef]
- Perlikowska, R.; Malfacini, D.; Cerlesi, M.C.; Calo’, G.; Piekielna, J.; Floriot, L.; Henry, T.; do-Rego, J.C.; Tömböly, C.; Kluczyk, A. Pharmacological characterization of endomorphin-2-based cyclic pentapeptides with methylated phenylalanine residues. Peptides 2014, 55, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Ye, L.; Liu, H.; Shi, Y.; Zhou, N. An overview of Ca2+ mobilization assays in GPCR drug discovery. Expert Opin. Drug Discov. 2017, 12, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Caers, J.; Peymen, K.; Suetens, N.; Temmerman, L.; Janssen, T.; Schoofs, L.; Beets, I. Characterization of G protein-coupled receptors by a fluorescence-based calcium mobilization assay. J. Vis. Exp. 2014, 89, e51516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarda, V.; Calo’, G. Chimeric G proteins in fluorimetric calcium assays: Experience with opioid receptors. Methods Mol. Biol. 2013, 937, 293–306. [Google Scholar]
- Camarda, V.; Fischetti, C.; Anzellotti, N.; Molinari, P.; Ambrosio, C.; Kostenis, E.; Regoli, D.; Trapella, C.; Guerrini, R.; Severo, S.; et al. Pharmacological profile of NOP receptors coupled with calcium signaling via the chimeric protein G alpha qi5. Naunyn-Schmiedebergs Arch. Pharmacol. 2009, 379, 599–607. [Google Scholar] [CrossRef]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Pinto, M.; Sousa, M.; Lima, D.; Tavares, I. Participation of mu-opioid, GABA(B), and NK1 receptors of major pain control medullary areas in pathways targeting the rat spinal cord: Implications for descending modulation of nociceptive transmission. J. Comp. Neurol. 2008, 510, 175–187. [Google Scholar] [CrossRef]
- Greedy, B.M.; Bradbury, F.; Thomas, M.P.; Grivas, K.; Cami-Kobeci, G.; Archambeau, A.; Bosse, K.; Clark, M.J.; Aceto, M.; Lewis, J.W.; et al. Orvinols with mixed kappa/mu opioid receptor agonist activity. J. Med. Chem. 2013, 56, 3207–3216. [Google Scholar] [CrossRef] [Green Version]
- Lao, L.; Song, B.; Chen, W.; Marvizón, J.C. Noxious mechanical stimulation evokes the segmental release of opioid peptides that induce mopioid receptor internalization in the presence of peptidase inhibitors. Brain Res. 2008, 1197, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Marvizón, J.C.G. Acute inflammation induces segmental, bilateral, supraspinally mediated opioid release in the rat spinal cord, as measured by mu-opioid receptor internalization. Neuroscience 2009, 161, 157–172. [Google Scholar] [CrossRef] [Green Version]
- De Felipe, C.; Herrero, J.F.; O’Brien, J.A.; Palmer, J.A.; Doyle, C.A.; Smith, A.J.; Laird, J.M.; Belmonte, C.; Cervero, F.; Hunt, S.P. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature 1998, 392, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Perl, E.R. Ideas about pain, a historical view. Nat. Rev. Neurosci. 2007, 8, 71–80. [Google Scholar] [CrossRef] [PubMed]
- George, D.T.; Gilman, J.; Hersh, J.; Thorsell, A.; Herion, D.; Geyer, C.; Peng, X.; Kielbasa, W.; Rawlings, R.; Brandt, J.E.; et al. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science 2008, 319, 1536–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.C.; Korlipara, V.L. Neurokinin-1 receptor antagonists: A comprehensive patent survey. Expert Opin. Ther. Pat. 2010, 20, 1019–1045. [Google Scholar] [CrossRef]
- Gallantine, E.L.; Meert, T.F. Attenuation of the gerbil writhing response by mu-, kappa-and deltaopioids, and NK-1, -2 and -3 receptor antagonists. Pharmacol. Biochem. Behav. 2004, 79, 125–135. [Google Scholar] [CrossRef]
- Hill, R. NK1 (substance P) receptor antagonists—Why are they not analgesic in humans? Trends Pharmacol. Sci. 2000, 21, 244–246. [Google Scholar] [CrossRef]
- Allouche, S.; Noble, F.; Marie, N. Opioid receptor desensitization: Mechanisms and its link to tolerance. Front. Pharmacol. 2014, 5, 280. [Google Scholar] [CrossRef] [Green Version]
- Bowman, S.L.; Soohoo, A.L.; Shiwarski, D.J.; Schulz, S.; Pradhan, A.A.; Puthenveedu, M.A. Cellautonomous regulation of Mu-opioid receptor recycling by substance P. Cell Rep. 2015, 10, 1925–1936. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Zeng, S.; Wang, X.; Babazada, H.; Li, Z.; Liu, R.; Yu, W. Neurokinin 1 and opioid receptors: Relationships and interactions in nervous system. Transl. Perioper. Pain Med. 2016, 1, 11–21. [Google Scholar]
- Perlikowska, R.; Fichna, J.; Wyrebska, A.; Poels, J.; Vanden Broeck, J.; Toth, G.; Storr, M.; do Rego, J.C.; Janecka, A. Design, synthesis and pharmacological characterization of endomorphin analogues with non-cyclic amino acid residues in position 2. Basic Clin. Pharmacol. Toxicol. 2010, 106, 106–113. [Google Scholar] [CrossRef]
- Wtorek, K.; Artali, R.; Piekielna-Ciesielska, J.; Koszuk, J.; Kluczyk, A.; Gentilucci, L.; Janecka, A. Endomorphin-2 analogs containing modified tyrosines: Biological and theoretical investigation of the influence on conformation and pharmacological profile. Eur. J. Med. Chem. 2019, 179, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. NC3Rs Reporting Guidelines Working Group. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef] [PubMed]
- Fichna, J.; Sibaev, A.; Sałaga, M.; Sobczak, M.; Storr, M. The cannabinoid-1 receptor inverse agonist taranabant reduces abdominal pain and increases intestinal transit in mice. Neurogastroenterol. Motil. 2013, 25, e550–e559. [Google Scholar] [CrossRef] [PubMed]
- Neubig, R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Kenakin, T.; Williams, M. Defining and characterizing drug/compound function. Biochem. Pharmacol. 2014, 87, 40–63. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–7 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence | Ki [nM] | |||
|---|---|---|---|---|---|
| MOR a | DOR a | KOR a | NK1 b | ||
| 1 | Tyr-[d-Lys-Phe-Phe-Asp]-NH2 | 0.35 ± 0.02 | 170.8 ± 3.50 | 1.12 ± 0.20 | Inactive |
| 2 | Tyr-[d-Lys-Phe-Phe-Asp]-Asn-D-Trp-Phe-D-Trp-Leu-Nle-NH2 | 5.99 ± 0.70 | 201.6 ± 2.50 | 7.46 ± 0.60 | 10.48 ± 0.60 |
| 3 | Tyr-[d-Lys-Phe-Phe-Asp]-D-Trp-Phe-D-Trp-Leu-Nle-NH2 | 28.24 ± 1.45 | 212.6 ± 6.31 | 2.85 ± 0.44 | 12.82 ± 0.92 |
| 4 | Tyr-[d-Lys-Phe-Phe-Asp]-Gln-Phe-Phe-Gly-Leu-Met-NH2 | 7.98 ± 0.97 | 224.8 ± 14.0 | 10.76 ± 0.85 | 15.6 ± 1.23 |
| 5 | Tyr-[d-Lys-Phe-Phe-Asp]-Phe-Phe-Gly-Leu-Met-NH2 | 4.90 ± 0.34 | 96.6 ± 3.8 | 28.34 ± 1.71 | 51.6 ± 4.2 |
| 6 | Tyr-[d-Lys-Phe-Phe-Asp]-Phe-Gly-Leu-Met-NH2 | 2.98 ± 0.14 | 28.7 ± 1.01 | 2.7 ± 0.12 | 2332 ± 189 |
| 7 | Tyr-[d-Lys-Phe-Phe-Asp]-Gly-Leu-Met-NH2 | 1.14 ± 0.21 | 113.2 ± 4.6 | 0.89 ± 0.04 | 8128 ± 724 |
| 8 | SP | ND | ND | ND | 3.24 ± 0.43 |
| Peptide | MOR | DOR | KOR | |||
|---|---|---|---|---|---|---|
| pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | |
| EM-2 | 8.22 (7.87–8.56) | 1.00 | Inactive | Inactive | ||
| DPDPE | Inactive | 7.29 (7.16–7.43) | 1.00 | Inactive | ||
| Dynorphin A | 6.67 (6.17–7.17) | 0.83 ± 0.10 | 7.73 (7.46–8.00) | 0.99 ± 0.04 | 8.86 (8.59–9.12) | 1.0 |
| 1 | 8.98 (8.50–9.45) | 0.98 ± 0.01 | Crc incomplete | 8.66 (8.56–8.76) | 0.96 ± 0.02 | |
| 2 | 7.50 (7.28–7.71) | 0.66 ± 0.04 | Crc incomplete | 8.01 (7.56–8.46) | 0.99 ± 0.05 | |
| 3 | 6.76 (6.46–7.06) | 0.72 ± 0.04 | Crc incomplete | 8.45 (7.56–9.34) | 1.05 ± 0.05 | |
| 4 | 7.46 (7.26–8.00) | 0.90 ± 0.04 | Crc incomplete | 7.85 (7.60–8.11) | 0.92 ± 0.05 | |
| 5 | 7.63 (7.12–7.80) | 0.86 ± 0.03 | 6.41 (5.82–7.01) | 0.50 ± 0.01 | 7.17 (7.02–7.31) | 0.64 ± 0.04 |
| 6 | 8.04 (7.89–8.20) | 0.81 ± 0.05 | 6.82 (5.97–7.66) | 0.65 ± 0.05 | 8.33 (7.96–8.69) | 0.92 ± 0.02 |
| 7 | 8.69 (8.23–9.16) | 0.85 ± 0.05 | 6.44 (5.80–7.08) | 0.38 ± 0.04 | 8.80 (8.46–9.14) | 1.06 ± 0.06 |
| No. | NK1 | |
|---|---|---|
| pEC50 (CL95%) | α ± SEM | |
| SP | 9.08 (8.83–9.34) | 1.00 |
| 2 | Inactive | |
| 3 | Inactive | |
| 4 | 8.45 (8.00–8.91) | 0.96 ± 0.05 |
| 5 | 7.80 (7.45–8.15) | 1.11 ± 0.05 |
| 6 | 6.11 (5.73–6.49) | 1.03 ± 0.05 |
| 7 | 5.68 (5.35–6.01) | 0.99 ± 0.08 |
| No. | pKB (CL95%) |
|---|---|
| aprepitant | 10.11 (9.48–10.74) |
| 2 | 7.33 (7.03–7.63) |
| 3 | 7.63 (7.25–8.00) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wtorek, K.; Adamska-Bartłomiejczyk, A.; Piekielna-Ciesielska, J.; Ferrari, F.; Ruzza, C.; Kluczyk, A.; Piasecka-Zelga, J.; Calo’, G.; Janecka, A. Synthesis and Pharmacological Evaluation of Hybrids Targeting Opioid and Neurokinin Receptors. Molecules 2019, 24, 4460. https://doi.org/10.3390/molecules24244460
Wtorek K, Adamska-Bartłomiejczyk A, Piekielna-Ciesielska J, Ferrari F, Ruzza C, Kluczyk A, Piasecka-Zelga J, Calo’ G, Janecka A. Synthesis and Pharmacological Evaluation of Hybrids Targeting Opioid and Neurokinin Receptors. Molecules. 2019; 24(24):4460. https://doi.org/10.3390/molecules24244460
Chicago/Turabian StyleWtorek, Karol, Anna Adamska-Bartłomiejczyk, Justyna Piekielna-Ciesielska, Federica Ferrari, Chiara Ruzza, Alicja Kluczyk, Joanna Piasecka-Zelga, Girolamo Calo’, and Anna Janecka. 2019. "Synthesis and Pharmacological Evaluation of Hybrids Targeting Opioid and Neurokinin Receptors" Molecules 24, no. 24: 4460. https://doi.org/10.3390/molecules24244460
APA StyleWtorek, K., Adamska-Bartłomiejczyk, A., Piekielna-Ciesielska, J., Ferrari, F., Ruzza, C., Kluczyk, A., Piasecka-Zelga, J., Calo’, G., & Janecka, A. (2019). Synthesis and Pharmacological Evaluation of Hybrids Targeting Opioid and Neurokinin Receptors. Molecules, 24(24), 4460. https://doi.org/10.3390/molecules24244460