4. Materials and Methods
4.1. General Information
Solvents and chemicals were purchased and used as received without further purification. Products were purified by standard column chromatography on silica gel (230–400 mesh, Merck, Kenilworth, NJ, USA). Unless stated otherwise, yields refer to analytically pure samples. NMR spectra were recorded with a Bruker Avance III 600 MHz instrument (1H-NMR: 600 MHz; 13C-NMR: 151 MHz; Bruker, Billerica, MA, USA). Chemical shifts are reported relative to solvent residual peaks (1H-NMR: δ = 7.26 ppm [CHCl3]; 13C-NMR: δ = 77.0 ppm [CDCl3]). IR spectra were recorded with a FTIR NEXUS spectrometer (as film or KBr pellets) or with a Cary 630 FTIR (Agilent Technologies, Santa Clara, CA, USA) spectrometer, in neat. ESI-MS spectra were performed with a Varian 500-MS LC Ion Trap. High-resolution mass spectrometry (HRMS) measurements were performed using Synapt G2-Si mass spectrometer (Waters, Milford, MA, USA) equipped with an ESI source and quadrupole-Time-of-flight mass analyzer. Elemental analyses were obtained with a Vario EL III (Elementar Analysensysteme GmbH, Langenselbold, Germany) instrument. Optical rotations were determined with an Anton Paar MCP 500 polarimeter (Anton Paar, Graz, Austria) at the temperatures indicated. Melting points were determined in capillaries with a Stuart SMP30 apparatus with automatic temperature monitoring or with a polarizing optical microscope (Opta-Tech, Warszawa, Poland), and are uncorrected.
4.2. Starting Materials
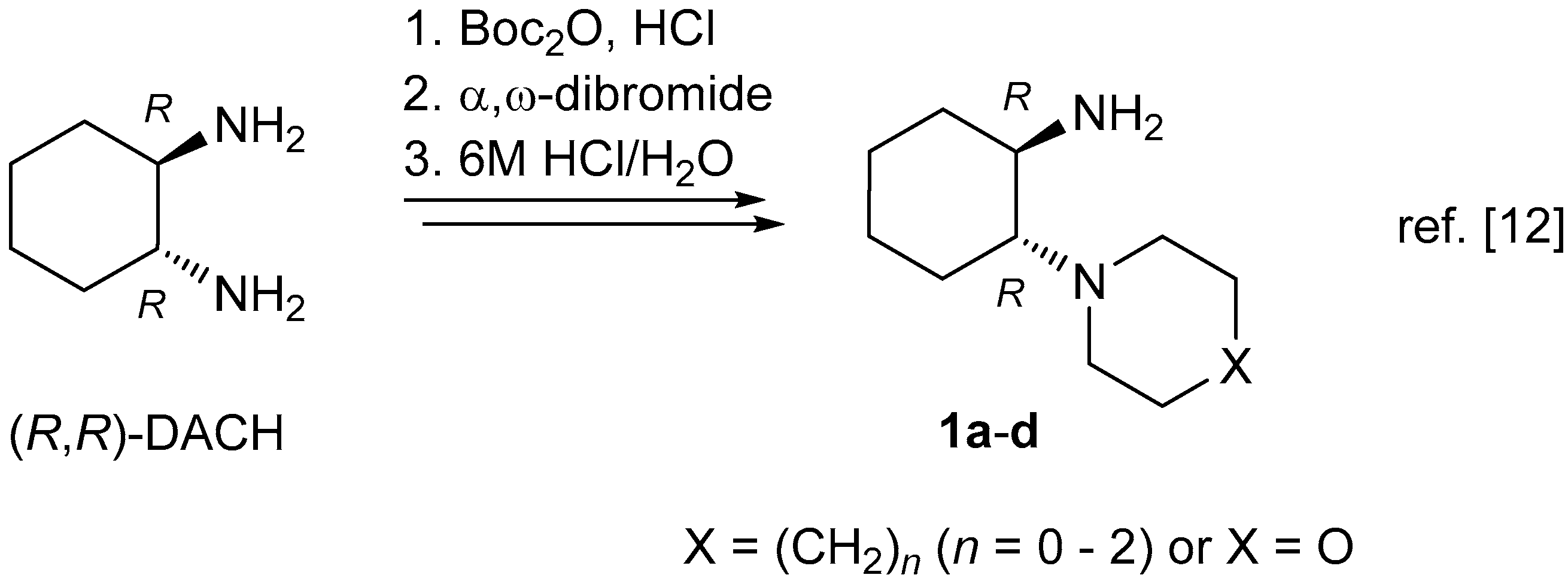
The ‘primary–tertiary’ amines
1 (i.e., compounds (
R,
R)-
1a, (
R,
R)-
1b, (
S,
S)-
1b, (
R,
R)-
1c, and (
R,
R)-
1d) were prepared from the respective, enantiopure diastereomers of
trans-1,2-diaminocyclohexane (
trans-DACH), according to the literature procedure [
12]. α-Hydroxyiminoketones
5 were prepared by nitrozation of ethyl methyl ketone in the case of
7a [
38] and by oximation of dibenzoyl in the case of
7b [
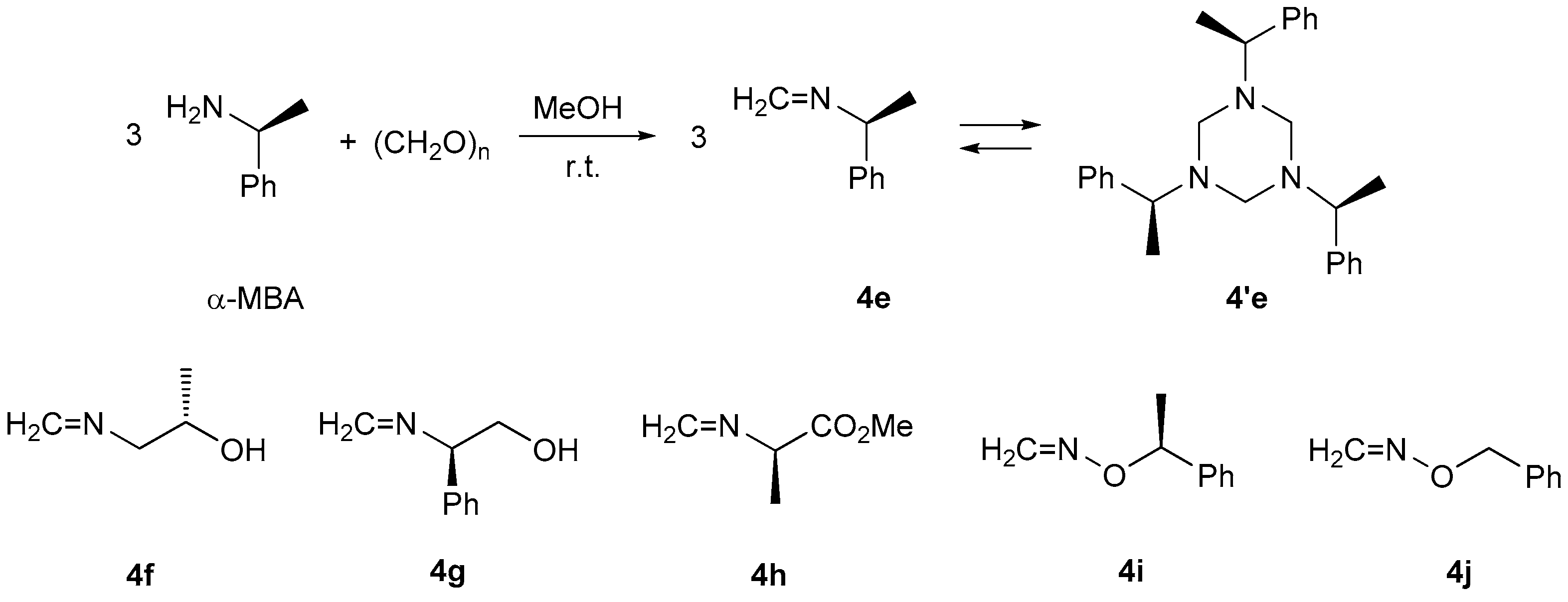
39] based on the published procedures. α-Methylbenzyloxyamine (α-MBOA) [
27] and benzyloxyamine (BOA) [
40] were prepared based on modified literature procedures. Formaldimines
4e [
20],
4f [
21],
4g [
21], and
4h [
22] were synthetized from corresponding amino compounds and formaldehyde based on the published procedures.
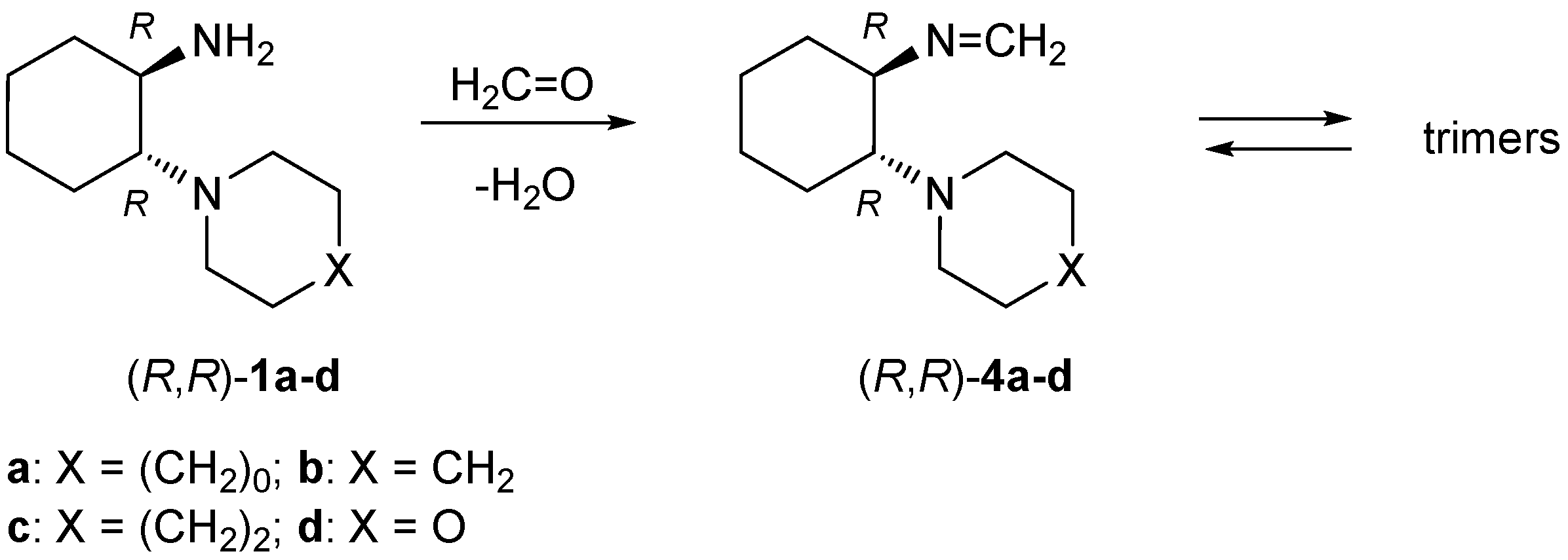
4.3. Synthesis of Formaldimines 4
4.3.1. Reactions of ‘Primary-Tertiary’ Amines 1 with Formaldehyde—General Procedure
A solution of 5 mmol of the respective diamine 1 and 450 mg (15 mmol) of formaldehyde (used as an aqueous solution) in 50 mL of benzene was heated in the Dean-Stark apparatus over 1.5 h. Next, the solvent was evaporated and the crude products were obtained as colorless solids. Analytically pure samples were obtained by recrystallization.
(R,R)-N-[2-(Pyrrolidin-1-yl)cyclohexyl]methanimine ((R,R)-4a): Yield 855 mg (95%). Colorless crystals (diisopropyl ether). M.p. 97–98 °C. = −135.30 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.39 (d, J = 17.2 Hz, 1H), 7.13 (d, J = 17.2 Hz, 1H), 2.93–2.97 (m, 1H), 2.66–2.71 (m, 1H), 2.52–2.62 (m, 4H), 1.86–1.91 (m, 1H), 1.72–1.77 (m, 1H), 1.60–1.69 (m, 4H), 1.52–1.58 (m, 2H), 1.23–1.30 (m, 3H), 1.12–1.20 (m, 1H) ppm. 13C-NMR (CDCl3): δ = 151.1, 74.4, 63.4, 48.4, 34.0, 24.6, 24.2, 23.9, 23.5 ppm. IR: ν 2920 (vs), 2851 (s), 2801 (s), 1457 (m), 1384 (m), 1369 (m), 1183 (s), 1135 (vs), 997 (vs), 898 (m), 875 (m), 766 (m) cm−1. C11H20N2 (180.29): calcd. C 73.28, H 11.18, N 15.54; found C 73.27, H 11.15, N 15.60.
(R,R)-N-[2-(Piperidin-1-yl)cyclohexyl]methanimine ((R,R)-4b): Yield 756 mg (78%). Colorless crystals (diethyl ether). M.p. 126–128 °C. = −130.92 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.28 (d, J = 17.2 Hz, 1H), 7.13 (d, J = 17.2 Hz, 1H), 2.93 (td, J = 9.9, 5.2 Hz, 1H), 2.45–2.62 (m, 3H), 2.29–2.37 (m, 2H), 1.8–11.86 (m, 1H), 1.72–1.79 (m, 1H), 1.58–1.69 (m, 3H), 1.44–1.51 (m, 2H), 1.30–1.40 (m, 4H), 1.10–1.25 (m, 3H) ppm. 13C-NMR (CDCl3): δ = 151.0, 72.4, 67.3, 49.6, 34.6, 26.6, 25.6, 25.1, 24.6, 23.6 ppm. IR: ν 2924 (vs), 2849 (s), 1450 (m), 1381 (m), 1360 (m), 1191 (s), 1127 (vs), 1114 (vs), 1004 (vs), 911 (m), 876 (m), 766 (m), 641 (m) cm–1. C12H22N2 (194.32): calcd. C 74.17, H 11.41, N 14.42; found C 74.17, H 11.38, N 14.46.
(S,S)-N-[2-(Piperidin-1-yl)cyclohexyl]methanimine ((S,S)-4b): Yield 775 mg (80%). Colorless crystals (diethyl ether). M.p. 125–127 °C. = +140.97 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.28 (d, J = 17.2 Hz, 1H), 7.14 (d, J = 17.2 Hz, 1H), 2.94 (td, J = 9.9, 5.3 Hz, 1H), 2.46–2.61 (m, 3H), 2.28–2.36 (m, 2H), 1.82–1.86 (m, 1H), 1.73–1.79 (m, 1H), 1.59–1.70 (m, 3H), 1.44–1.52 (m, 2H), 1.31–1.40 (m, 4H), 1.12–1.26 (m, 3H) ppm. 13C-NMR (CDCl3): δ = 151.1, 72.4, 67.3, 49.6, 34.6, 26.6, 25.6, 25.1, 24.6, 23.6 ppm. IR: ν 2924 (vs), 2849 (s), 1450 (m), 1381 (m), 1360 (m), 1191 (s), 1127 (vs), 1114 (vs), 1004 (vs), 911 (m), 876 (m), 641 (m) cm–1. C12H22N2 (194.32): calcd. C 74.17, H 11.41, N 14.42; found C 73.99, H 11.55, N 14.38.
(R,R)-N-[2-(Azepan-1-yl)cyclohexyl]methanimine ((R,R)-4c): Yield 969 mg (93%). Colorless crystals (diisopropyl ether). M.p. 112–114 °C. = –81.57 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.32 (d, J = 17.3 Hz, 1H), 7.13 (d, J = 17.3 Hz, 1H), 2.90 (td, J = 10.0, 5.0 Hz, 1H), 2.64–2.68 (m, 2H), 2.59–2.64 (m, 1H), 2.50–2.55 (m, 2H), 1.79–1.82 (m, 1H), 1.71–1.76 (m, 1H), 1.63–1.68 (m, 1H), 1.41–1.62 (m, 10H), 1.15–1.24 (m, 3H) ppm. 13C-NMR (CDCl3): δ = 151.1, 73.7, 68.6, 51.4, 34.5, 30.0, 27.0, 25.6, 25.3, 24.6 ppm. IR: ν 2920 (vs), 2849 (s), 1448 (m), 1355 (m), 1166 (m), 1131 (s), 993 (m), 907 (m), 874 (m) cm−1. C13H24N2 (208.34): calcd. C 74.94, H 11.62, N 13.44; found C 75.10, H 11.73, N 13.61.
(R,R)-N-(2-Morpholinocyclohexyl)methanimine ((R,R)-4d): Yield 1.02 g (98%). Colorless crystals (diisopropyl ether). M.p. 127–129 °C. = −124.52 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.30 (d, J = 17.1 Hz, 1H), 7.14 (d, J = 17.1 Hz, 1H), 3.59–3.66 (m, 2H), 3.51–3.55 (m, 2H), 2.93 (td, J = 9.8, 5.5 Hz, 1H), 2.50–2.60 (m, 3H), 2.40–2.46 (m, 2H), 1.83–1.88 (m, 1H), 1.75–1.79 (m, 1H), 1.65–1.70 (m, 1H), 1.57–1.63 (m, 2H), 1.16–1.26 (m, 3H) ppm. 13C-NMR (CDCl3): δ = 151.4, 72.5, 67.6, 67.0, 48.9, 34.6, 25.5, 24.6, 23.9 ppm. IR: ν 2920 (s), 2851 (s), 1450 (m), 1258 (m), 1112 (vs), 995 (s), 872 (m) cm–1. C11H20N2O (196.29): calcd. C 67.31, H 10.27, N 14.27; found C 67.07, H 10.39, N 14.23.
4.3.2. Synthesis of Alkoxyformaldimines 4i–j—General Procedure
A solution of 3 mmol of the corresponding primary amine ((S)-MBOA or BOA) and 0.25 mL (ca. 3 mmol) of formaldehyde (as aqueous solution (37%)) in 40 mL benzene was heated in the Dean-Stark apparatus until no formation of water was observed. After cooling to room temperature, the solvent was evaporated and the colorless, oily liquids were used for further reactions without purification.
(S)-(α-Methylbenzyloxy)methanimine (4i): Yield 404 mg (90%). Colorless liquid. = −11.90 (c 0.51, CHCl3). 1H-NMR (CDCl3): δ = 7.38–7.33, 7.30–7.27 (2m, 4H, 1H, Ph), 7.08, 6.43 (2d, 2H, J = 8.5 Hz, =CH2), 5.27 (q, J = 6.7 Hz, 1H), 1.56 (d, J = 6.7 Hz, 3H, Me) ppm. 13C-NMR (CDCl3): δ = 143.0 (t, N=CH2), 137.4, 128.4, 127.6, 126.3 (s, 3d, Ph), 81.1 (d, CH), 21.8 (q, Me) ppm. HRMS (CI+): calcd for C9H13NO [M + H]+ 150.0917; found 150.0919.
(Benzyloxy)methanimine (
4j): Yield 360 mg (88%). Colorless liquid (ref. [
41], bp 76 °C/15 mmHg).
1H-NMR (CDCl
3): δ = 7.40–7.35, 7.34–7.30 (2m, 4H, 1H, Ph), 7.09, 6.47 (2d, 2H,
J = 8.2 Hz, =CH
2), 5.14 (s, 2H, -C
H2-) ppm.
13C-NMR (CDCl
3): δ = 137.4 (t, N=
CH
2), 137.3 (s, Ph), 128.3, 128.1, 127.8 (3d, Ph), 75.8 (t, -CH
2-) ppm.
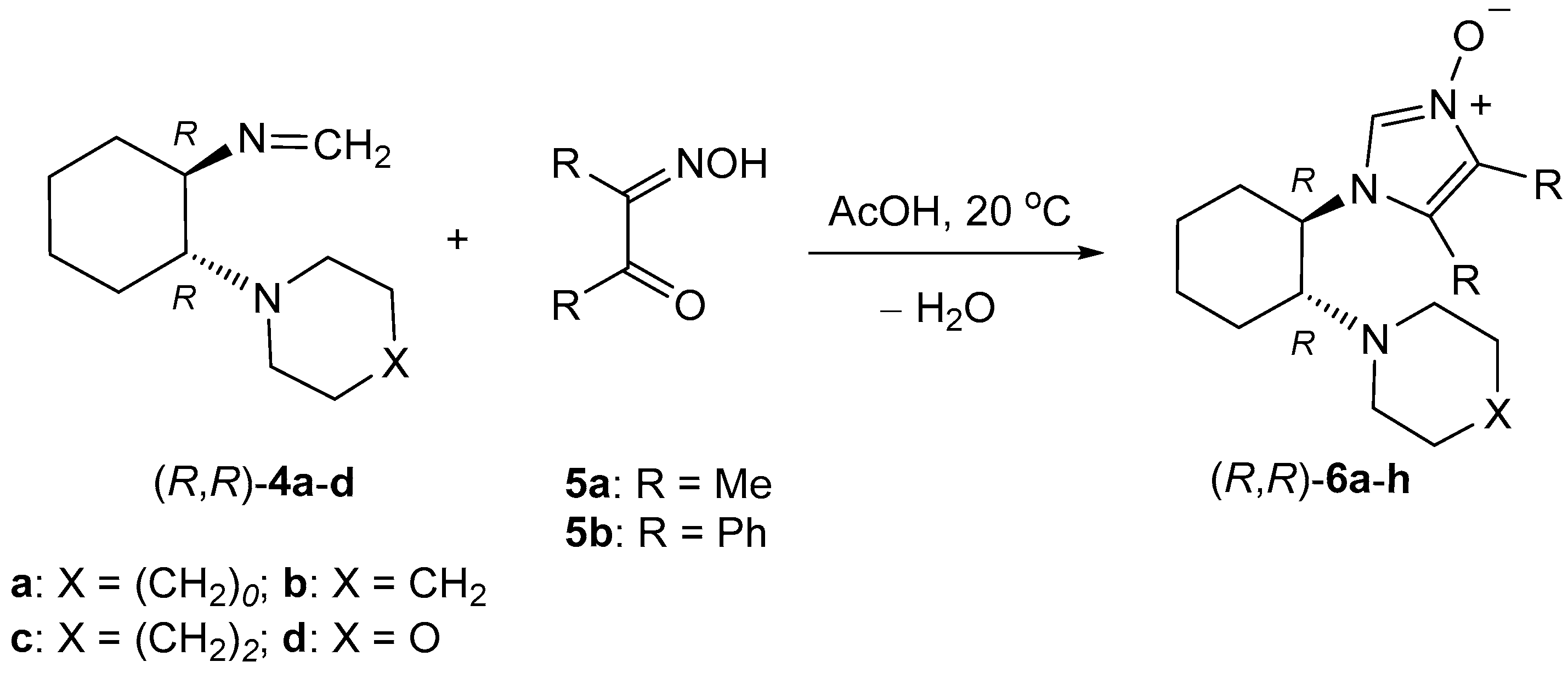
4.4. Synthesis of Imidazole N-Oxides 6—General Procedure
A solution of 1.1 mmol of the respective formaldimine 4 and 1 mmol of α-hydroxyiminoketone 5 in 3 mL of glacial acetic acid was stirred magnetically overnight. The next day, 1 mL of aqueous hydrochloric acid was added and the solution was stirred for 15 min. Then, the resulting solution was evaporated to dryness, the solid residue was dissolved in 20 mL of dichloromethane, and the obtained solution was neutralized with 20 mL of diluted aqueous sodium hydroxide. The organic layer was separated, dried over MgSO4, filtrated, and evaporated. The resulting crude products were washed with two portions (each ca. 10 mL) of diethyl ether, and after separation, the obtained thick oils or amorphous solids were characterized spectroscopically and used for further transformations.
(R,R)-4,5-Dimethyl-1-[2-(pyrrolidin-1-yl)cyclohexyl]-1H-imidazole 3-oxide ((R,R)-6a): Yield 171.0 mg (65%). Colorless, viscous oil. 1H-NMR (CDCl3): δ = 7.88 (s, 1H), 3.73 (td, J = 11.2, 3.5 Hz, 1H), 2.81–2.87 (m, 1H), 2.45–2.49 (m, 2H), 2.37–2.41 (m, 2H), 2.14 (s, 3H), 2.10 (s, 3H), 2.00–2.03 (m, 1H), 1.93–1.97 (m, 1H), 1.80–1.84 (m, 1H), 1.76–1.79 (m, 1H), 1.44–1.60 (m, 4H), 1.14–1.35 (m, 4H) ppm. 13C-NMR (CDCl3): δ = 125.2, 123.0, 120.6, 62.1, 58.7, 48.0, 35.0, 25.2, 24.8, 24.6, 23.6, 8.8, 7.1 ppm. IR: ν 2927 (s), 1655 (m), 1448 (m), 1377 (m), 1332 (vs), 1191 (m), 1142 (m), 883 (s), 702 (s), 579 (s) cm−1. HRMS (ESI+): calcd for C15H26N3O [M + H]+ 264.2076; found 264.2082.
(R,R)-4,5-Diphenyl-1-[2-(pyrrolidin-1-yl)cyclohexyl]-1H-imidazole 3-oxide ((R,R)-6b): Yield 360.0 mg (93%). Yellowish crystals. M.p. 189–191 °C. 1H-NMR (CDCl3): δ = 8.12 (s, 1H), 7.49 (d, J = 7.9 Hz), 7.35–7.39 (m, 3H), 7.18–7.23 (m, 5H), 3.74 (td, J = 11.7, 4.2 Hz, 1H), 2.86 (td, J = 11.1, 3.5 Hz, 1H), 2.32–2.36 (m, 2H), 2.25–2.29 (m, 2H), 2.06–2.11 (m, 1H), 1.89–1.94 (m, 1H), 1.47–1.78 (m, 8H), 1.21–1.29 (m, 1H), 1.08–1.18 (m, 3H) ppm. 13C-NMR (CDCl3): δ = 130.7, 129.6, 129.3, 129.1, 128.9, 128.0, 127.9, 127.8, 127.3, 127.0, 124.2, 62.3, 58.7, 47.6, 35.4, 25.2, 24.6, 24.0, 23.56, 23.51 ppm. IR: ν 2931 (m), 2808 (s), 1448 (m), 1341 (m), 1321 (m), 1224 (m), 881 (m), 754 (s), 710 (s), 689 (vs), 658 cm−1. C25H29N3O·0.5 H2O (387.52 + 9): calcd. C 75.72, H 7.62, N 10.60; found C 75.50, H 7.59, N 10.61.
(R,R)-4,5-Dimethyl-1-[2-(piperidin-1-yl)cyclohexyl]-1H-imidazole 3-oxide ((R,R)-6c): Yield 249.5 mg (90%). Yellowish crystals. M.p. 68–71 °C. 1H-NMR (CDCl3): δ = 7.70 (s, 1H), 3.75 (td, J = 11.5, 3.8 Hz, 1H), 2.47–2.54 (m, 3H), 2.23–2.27 (m, 2H), 2.19 (s, 3H), 2.11 (s, 3H), 1.99–2.02 (m, 2H), 1.78–1.87 (m, 2H), 1.45–1.52 (m, 1H), 1.37–1.42 (m, 2H), 1.24–1.33 (m, 7H) ppm. 13C-NMR (CDCl3): δ = 124.9, 123.4, 120.8, 67.7, 56.6, 50.0, 34.9, 26.5, 25.4, 25.0, 24.7 (2C signals overlap), 8.9, 7.2 ppm. IR: ν 2926 (vs), 2853 (m), 1655 (br.m), 1450 (s), 1377 (m), 1331 (vs), 1103 (m), 881 (m), 702 (s), 579 (s) cm–1. HRMS (ESI+): calcd for C16H28N3O [M + H]+ 278.2232; found 278.2234.
(S,S)-4,5-Dimethyl-1-[2-(piperidin-1-yl)cyclohexyl]-1H-imidazole 3-oxide ((S,S)-6c): Yield: 263.5 g (95%). Yellowish crystals. M.p. 51–54 °C. 1H-NMR (CDCl3): δ = 7.87 (s, 1H), 3.70 (td, J = 11.5, 3.8 Hz, 1H), 2.51–2.56 (m, 1H), 2.41–2.46 (m, 2H), 2.27–2.21 (m, 2H), 2.12 (s, 3H), 2.06 (s, 3H), 1.92–1.96 (m, 2H), 1.71–1.80 (m, 2H), 1.46–1.53 (m, 1H), 1.29–1.35 (m, 2H), 1.18–1.25 (m, 7H) ppm. 13C-NMR (CDCl3): δ = 124.7, 122.8, 120.6, 67.6, 56.3, 49.9, 34.8, 26.3, 25.2, 24.9, 24.51, 24.46, 8.7, 7.1 ppm. IR: ν 2928 (vs), 2855 (m), 1664 (br, m), 1450 (s), 1379 (s), 1332 (vs), 1101 (m), 881 (m), 704 (s), 581 (s) cm−1. HRMS (ESI+): calcd for C16H28N3O [M + H]+ 278.2232; found 278.2238.
(R,R)-4,5-Diphenyl-1-[2-(piperidin-1-yl)cyclohexyl]-1H-imidazole 3-oxide ((R,R)-6d): Yield 261.0 mg (65%). Yellowish crystals. M.p. 192–194 °C. 1H-NMR (CDCl3): δ = 8.00 (s, 1H), 7.57 (d, J = 7.5 Hz, 2H), 7.39–7.45 (m, 3H), 7.23–7.29 (m, 5H), 3.82 (td, J = 11.5, 3.8 Hz, 1H), 2.61 (td, J = 11.3, 3.3 Hz, 1H), 2.25–2.29 (m, 2H), 2.17–2.22 (m, 2H), 1.93–1.97 (m, 1H), 1.78–1.83 (m, 2H), 1.63–1.70 (m, 1H), 1.00–1.44 (m, 10H) ppm. 13C-NMR (CDCl3): δ = 130.6, 129.6, 129.2, 128.9, 128.2, 127.9, 127.7, 127.36, 127.28, 124.1, 68.2, 56.7, 49.7, 35.7, 26.5, 25.3, 25.0, 24.7, 24.0 ppm. IR: ν 2929 (s), 2855 (m), 1444 (m), 1342 (m), 1205 (w), 1101 (w), 1034 (w), 784 (s), 710 (s), 691 (s) cm−1. C26H31N3O (401.54): calcd. C 77.77, H 7.78, N 10.46; found C 77.56, H 7.88, N 10.59.
(S,S)-4,5-Diphenyl-1-[2-(piperidin-1-yl)cyclohexyl]-1H-imidazole 3-oxide ((S,S)-6d): Yield: 333.0 mg (83%) after purification on a short column. Yellowish crystals. M.p. 198–199 °C. 1H-NMR (CDCl3): δ = 8.03 (s, 1H), 7.56 (d, J = 7.5 Hz, 2H), 7.40–7.45 (m, 3H), 7.23–7.29 (m, 5H), 3.81 (td, J = 11.6, 3.8 Hz, 1H), 2.61 (td, J = 11.3, 3.4 Hz, 1H), 2.24–2.28 (m, 2H), 2.17–2.22 (m, 2H), 1.92–1.97 (m, 1H), 1.78–1.83 (m, 2H), 1.68 (qd, J = 12.5, 3.3 Hz, 1H), 1.06–1.44 (m, 10H) ppm. 13C-NMR (CDCl3): δ = 130.5, 129.6, 129.3, 128.95, 128.89, 127.95, 127.91, 127.79, 127.4, 127.1, 68.0, 56.9, 49.6, 35.4, 26.5, 25.3, 24.9, 24.6, 24.0 ppm. IR: ν 2928 (s), 2853 (m), 1444 (m), 1342 (m), 1205 (m), 1103 (m), 1034 (w), 786 (s), 712 (s), 693 (vs) cm−1. HRMS (ESI+): calcd for C26H32N3O [M + H]+ 402.2545; found 402.2546.
(R,R)-1-[2-(Azepan-1-yl)cyclohexyl]-4,5-dimethyl-1H-imidazole 3-oxide ((R,R)-6e): Yield 227.1 g (78 %). Colorless, viscous oil. 1H-NMR (CDCl3): δ = 7.74 (s, 1H), 3.66 (td, J = 11.2, 3.9 Hz, 1H), 2.63–2.67 (m, 1H), 2.53–2.58 (m, 2H), 2.37–2.42 (m, 2H), 2.11 (s, 3H), 2.06 (s, 3H), 1.91–1.96 (m, 2H), 1.71–1.79 (m, 2H), 1.15–1.53 (m, 12H) ppm. 13C-NMR (CDCl3): δ = 124.7, 123.4, 120.1, 67.2, 57.3, 50.4, 35.0, 29.5, 26.5, 25.2, 25.0, 24.7, 8.6, 6.9 ppm. IR: ν 2924 (vs), 2853 (m), 1654 (s), 1448 (s), 1332 (s), 725 (vs), 704 (vs), 581 (m) cm–1. HRMS (ESI+): calcd for C17H30N3O [M + H]+ 292.2389; found 292.2398.
(R,R)-1-[2-(Azepan-1-yl)cyclohexyl]-4,5-diphenyl-1H-imidazole 3-oxide ((R,R)-6f): Yield: 369.6 mg (89%). Yellowish crystals. M.p. 64–68 °C. 1H-NMR (CDCl3): δ = 8.06 (s, 1H), 7.50 (d, J = 7.7 Hz, 2H), 7.37–7.41 (m, 3H), 7.19–7.24 (m, 5H), 3.75 (td, J = 11.4, 3.7 Hz, 1H), 2.71 (td, J = 11.3, 3.5 Hz, 1H), 2.35–2.44 (m, 4H), 2.13–2.17 (m, 1H), 1.88–1.93 (m, 1H), 1.72–1.78 (m, 3H), 1.09–1.64 (m, 11H) ppm. 13C-NMR (CDCl3): δ = 130.5, 129.7, 129.4, 129.0, 128.00, 127.96, 127.5, 127.0, 126.7, 125.9, 125.5, 68.3, 57.9, 50.5, 35.7, 29.7, 26.7, 25.2, 24.9 ppm (one signal of sp3 C atom not observed due to overlap). IR: ν 2926 (s), 2855 (w), 1672 (m), 1444 (m), 1224 (w), 712 (s), 693 (br, s) 635 (m), 508 (br, m), 451 (br, m) cm−1. HRMS (ESI+): calcd for C17H34N3O [M + H]+ 416.2702; found 416.2708.
(R,R)-1-(2-Morpholinocyclohexyl)-4,5-dimethyl-1H-imidazole 3-oxide ((R,R)-6g): Yield: 240.1 mg (86%). Yellowish crystals. M.p. 85–87 °C. 1H-NMR (CDCl3): δ = 9.06 (s, 1H), 3.76–3.82 (m, 1H), 3.58–3.66 (m, 1H), 3.43–3.48 (m, 2H), 3.36–3.40 (m, 2H), 3.10 (td, J = 11.1, 3.1 Hz, 1H), 2.49–2.54 (m, 2H), 2.39–2.43 (m, 2H), 2.15 (s, 3H), 2.09 (s, 3H), 1.95–2.04 (m, 2H), 1.86–1.93 (m, 1H), 1.76–1.82 (m, 2H), 1.42–1.50 (m, 1H), 1.20–1.28 (m, 2H) ppm. 13C-NMR (CDCl3): δ = 126.2, 124.4, 121.0, 67.3, 66.0, 57.2, 48.8, 34.2, 25.3, 24.9, 24.5, 8.9, 7.1 ppm. IR ν 2929 (s), 2855 (m), 1654 (w), 1627 (br, w), 1450 (m), 1332 (s), 1112 (vs), 926 (m), 861 (m), 725 (vs) cm−1. HRMS (ESI+): calcd for C15H26N3O2 [M + H]+ 280.2025; found 280.2026.
(R,R)-1-(2-Morpholinocyclohexyl)-4,5-diphenyl-1H-imidazole 3-oxide ((R,R)-6h): Yield: 266.3 mg (66%). Yellowish crystals. M.p. 152–155 °C. 1H-NMR (CDCl3): δ = 8.08 (s, 1H), 7.54 (d, J = 7.2 Hz, 2H), 7.38–7.43 (m, 3H), 7.24–7.29 (m, 3H), 7.19 (d, J = 6.9 Hz, 2H), 3.84 (td, J = 11.6, 4.0 Hz, 1H), 3.44–3.51 (m, 4H), 2.62 (td, J = 11.3, 3.1 Hz, 1H), 2.22–2.28 (m, 5H), 1.94–1.98 (m, 1H), 1.81–1.86 (m, 2H), 1.69–1.76 (m, 1H), 1.25–1.33 (m, 1H), 1.16–1.24 (m, 1H), 1.08–1.15 (m, 1H) ppm. 13C-NMR (CDCl3): δ = 130.4, 129.7, 129.3, 129.0, 127.99, 127.92, 127.88, 127.4, 127.1, 123.9, 68.1, 67.3, 56.3, 48.6, 35.2, 25.2, 24.8, 24.2 ppm (one signal of an sp2 C atom not observed due to overlap). IR: ν 2926 (s), 2857 (m), 1448 (m), 1341 (m), 1109 (s), 861 (s), 766 (s), 695 (vs), 665 (s), 633 (s) cm−1. C25H29N3O2·0.5 H2O (412.52): calcd. C 72.79, H 7.33, N 10.18; found C 72.68, H 7.25, N 9.90.
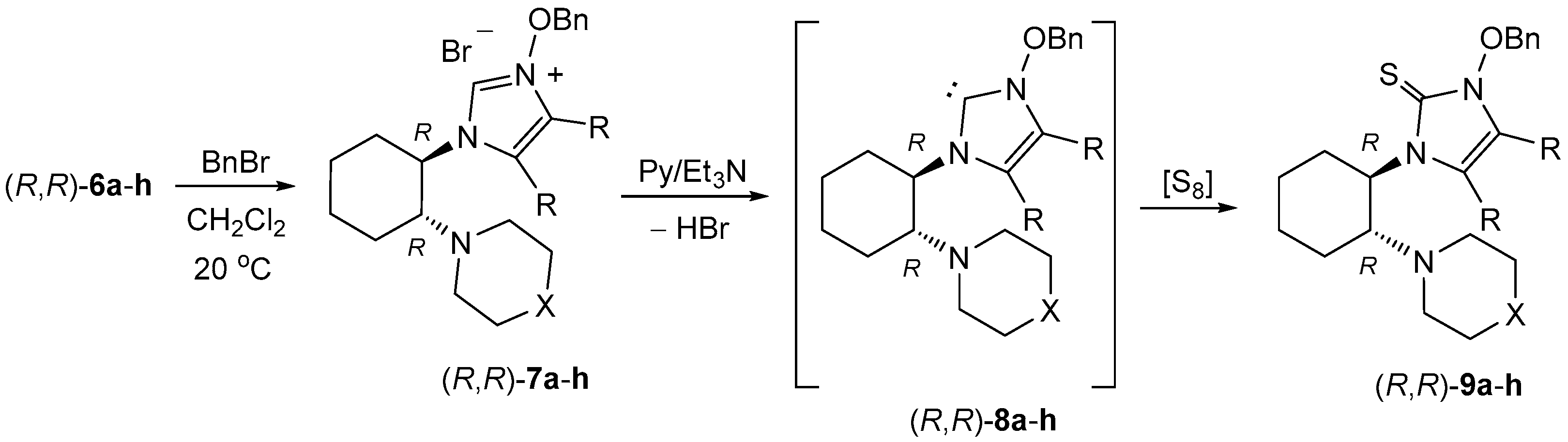
4.5. Synthesis of 3-Benzyloxyimidazolium Bromides 7—General Procedure
A solution of 0.5 mmol of crude imidazole
N-oxide
6 and 86 mg (0.5 mmol) benzyl bromide in 1 mL of CHCl
3 was stirred magnetically overnight at room temperature. The next day, the solvent was evaporated and the obtained crude imidazolium salts were triturated with diethyl ether. The ethereal phase was separated and the non-soluble imidazolium salts
7 were used for the generation of carbenes
8 and their reaction with elemental sulfur without further purification. Whereas di-Me substituted imidazolium salts formed viscous, thick oils, the corresponding di-Ph derivatives were obtained as amorphous solids. The structures of selected imidazolium salts were confirmed by running the
1H-NMR spectra. A representative example of the
1H-NMR spectra registered for
7e is described below and the scanned spectrum is presented in the
Supplementary Materials.
(R,R)-1-[2-(Azepan-1-yl)cyclohexyl]-4,5-dimethyl-3-benzyloxy-1H-imidazolium bromide((R,R)-7e): 1H-NMR (CDCl3): δ = 11.09 (s, 1H), 7.51 (d, J = 6.9 Hz, 2H), 7.27–7.37 (m, 3H), 5.70 (d, J= 10.0 Hz, 1H), 5.57 (d, J= 10.0 Hz, 1H), 3.91 (br., 1H), 3.62 (br., 1H), 2.60 (br., 4H), 2.13–2.17 (m, 1H), 2.13 (s, 3H), 2.02–2.06 (m, 1H), 1.96–2.01 (m, 1H), 1.91 (s, 3H), 1.80–1.85 (m, 1H), 1.74–1.79 (m, 1H), 1.55–1.63 (m, 1H), 1.47 (br., 2H), 1.29 (br., 6H), 1.18 (br., 2H) ppm.
4.6. Synthesis of Non-Symmetric Imidazole-2-Thiones 9—General Procedure
The crude imidazolium salt 7 obtained from 0.5 mmol of the corresponding imidazole N-oxide 6 and 86 mg (0.5 mmol) of benzyl bromide according to the general procedure (see above) was dissolved in 1 mL of dry pyridine. Next, 38 mg (1.2 mmol) of elemental sulfur and 122 mg (1.2 mmol) of triethylamine were added to the magnetically stirred, homogenous solution. Stirring at room temperature was continued overnight. Next day, pyridine was removed under reduced pressure and the residual semi-solid material was purified by preparative thin layer chromatography with silica gel. A mixture of dichloromethane and methanol (99:1) was used as an eluent. Imidazole-2-thiones 9 formed a single fraction with Rf ca. 0.3. Solid products were additionally purified by crystallization.
(R,R)-3-Benzyloxy-4,5-dimethyl-1-[2-(pyrrolidin-1-yl)cyclohexyl]-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9a): Yield: 238.8 mg (62%). Colorless, viscous oil, purified by chromatography. = −91.15 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.32–7.44 (m, 5H), 5.75–5.80 (m, 1H), 5.42 (d, J = 10.0 Hz, 1H), 5.28 (d, J = 10.0 Hz, 1H), 4.57–4.63 (m, 1H), 3.06–3.11 (m, 1H), 2.83–3.02 (m, 3H), 2.36 (s, 3H), 2.26–2.31 (m, 1H), 1.72–2.13 (m, 8H), 1.81 (s, 3H), 1.38–1.51 (m, 3H) ppm. 13C-NMR (CDCl3): δ = 155.1, 133.3, 130.2, 129.5, 128.5, 121.1, 118.4, 78.1, 60.7, 58.4, 52.2, 29.6, 29.4, 24.1, 23.7, 22.6, 9.9, 7.7 ppm. IR ν 2929 (s), 2862 (m), 1377 (br, s), 1401 (br, s), 1321 (vs), 1140 (br, m), 1025 (m),954 (m), 913 (m), 836 (w), 751 (s), 697 (vs), 606 (w), 483 (s) cm−1. HRMS (ESI+): calcd for C22H32N3OS [M + H]+ 386.2266; found 386.2274.
(R,R)-3-Benzyloxy-4,5-diphenyl-1-[2-(pyrrolidin-1-yl)cyclohexyl]-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9b): Yield: 356.8 g (70%). Colorless crystals. M.p. 285–286 °C (from MeOH/CH2Cl2) (decomp.). = −36.65 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = (signals for both rotamers) 6.96–7.45 (m, 15H), 4.95–5.47 (m, 3H), 3.44 and 3.74 (br., 1H), 2.14–2.70 (m, 4H), 0.80–1.96 (m, 12H) ppm. 13C-NMR (CDCl3): δ = (signals for both rotamers) 157.3, 156.1, 133.2, 132.6, 131.1, 130.6, 129.4, 129.3, 129.0, 128.7, 128.2, 128.0, 127.8, 126.4, 125.2, 124.5, 77.4, 61.7, 58.2, 55.0, 46.4, 33.3, 29.4, 26.0, 25.8, 25.0, 24.8, 23.8, 23.6, 22.1 ppm. IR: ν 2929 (m), 2797 (m), 1425 (m), 1358 (m), 1332 (m), 1306 (br, m), 1187 (w), 1073 (w), 965 (m), 911 (m), 786 (m), 753 (s), 695 (vs), 598 (m), 506 (m) cm−1. HRMS (ESI+): calcd. for C32H36N3OS [M + H]+ 510.2579; found 510.2595. C32H35N3OS (509.70): calcd. C 75.40, H 6.92, N 8.24, S 6.30; found C 75.30, H 6.91, N 8.24, S 6.14.
(R,R)-3-Benzyloxy-4,5-dimethyl-1-[2-(piperidin-1-yl)cyclohexyl]-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9c): Yield: 239.5 mg (60%). Colorless, viscous oil, purified by chromatography. = −16.53 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.47 (d, J = 7.7 Hz, 2H), 7.32–7.37 (m, 3H), 5.46 (d, J = 10.5 Hz, 1H), 5.32 (d, J = 10.5 Hz, 1H), 5.27 (td, J = 11.6, 3.7 Hz, 1H), 2.89 (td, J = 11.4, 2.8 Hz, 1H), 2.74–2.79 (m, 2H), 2.23–2.27 (m, 2H), 2.08 (s, 3H), 1.94–2.02 (m, 2H), 1.83–1.88 (m, 1H), 1.73–1.78 (m, 1H), 1.70 (s, 3H), 1.59 (qd, J = 12.5, 3.7 Hz, 1H), 1.29–1.52 (m, 8H), 1.17–1.25 (m, 1H) ppm. 13C-NMR (CDCl3): δ = 156.5, 134.2, 130.5, 129.1, 128.4, 119.4, 117.9, 77.6, 65.9, 58.5, 49.4, 32.0, 26.8, 25.9, 25.7, 24.9, 24.6, 10.5, 7.2 ppm. IR: ν 2927 (vs), 2853 (m), 1403 (s), 1377 (s), 1330 (s), 1211 (m), 1105 (m), 963 (m), 911 (m), 749 (vs), 699 (vs), 479 (br.s) cm−1. HRMS (ESI+) calcd. for C23H34N3OS [M + H]+ 400.2423; found: 400.2424.
(S,S)-3-Benzyloxy-4,5-dimethyl-1-[2-(piperidin-1-yl)cyclohexyl]-1,3-dihydro-2H-imidazole-2-thione ((S,S)-9c): Yield: 187.6 mg (47%). Colorless, viscous oil, purified by chromatography. = +26.13 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.46 (d, J = 7.5 Hz, 2H), 7.32–7.37 (m, 3H), 5.46 (d, J = 10.5 Hz, 1H), 5.31 (d, J = 10.5 Hz, 1H), 5.26 (td, J = 11.9, 4.2 Hz, 1H), 2.89 (td, J = 11.4, 2.8 Hz, 1H), 2.74–2.78 (m, 2H), 2.23–2.27 (m, 2H), 2.07 (s, 3H), 1.94–2.03 (m, 2H), 1.83–1.87 (m, 1H), 1.73–1.77 (m, 1H), 1.68 (s, 3H), 1.58 (qd, J = 12.5, 3.7 Hz, 1H), 1.27–1.51 (m, 8H), 1.17–1.25 (m, 1H) ppm. 13C-NMR (CDCl3): δ = 156.3, 134.2, 130.5, 129.1, 128.4, 119.4, 117.9, 77.6, 65.9, 58.5, 49.4, 32.0, 26.8, 25.8, 25.7, 24.9, 24.5, 10.5, 7.2 ppm. IR: ν 2927 (vs), 2853 (m), 1403 (s), 1377 (s), 1328 (s), 1211 (m), 1105 (m), 963 (m), 911 (w), 749 (vs), 697 (vs), 479 (br, s) cm−1. HRMS (ESI+): calcd for C23H34N3OS [M + H]+ 400.2423; found 400.2426.
(R,R)-3-Benzyloxy-4,5-diphenyl-1-[2-(piperidin-1-yl)cyclohexyl]-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9d): Yield 429.4 mg (82%). Colorless crystals. M.p. 283–285 °C (MeOH/CH2Cl2) (decomp.). = −26.77 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = (signals for both rotamers in ca. 1:2 ratio) 7.07–7.41 (m, 15H), 5.40 and 5.32 (d, J = 9.4 Hz, 1H), 4.61–4.67 (m) and 5.36 (td, J = 11.8, 3.4 Hz, total: 1H), 4.98 and 5.12 (br. d, J = 9.4 Hz, 1H), 0.77–3.76 (m, 18H) ppm. 13C-NMR (CDCl3): δ = (signals for both rotamers) 157.6, 156.7, 133.2, 133.1, 132.4, 130.6, 130.42, 130.38, 129.5, 129.3, 129.22, 129.15, 128.92, 128.87, 128.84, 128.6, 128.4, 128.3, 128.1, 128.02, 127.97, 127.8, 127.7, 126.9, 126.6, 126.40, 126.31, 124.9, 124.6, 124.3, 77.51, 77.49, 64.4, 59.9, 59.7, 49.1 (br.), 48.8 (br.), 33.2, 29.6, 27.1, 26.8, 26.0, 25.7, 25.2, 24.94, 24.91, 24.6, 23.5 ppm. IR: ν 2924 (s), 2849 (w), 1442 (w), 1397 (s), 1321 (s), 1207 (m), 959 (m), 760 (s), 691 (vs), 596 (m), 568 (m), 475 (w) cm−1. C33H37N3OS (523.73): calcd. C 75.68, H 7.13, N 8.02, S 6.12; found C 75.63, H 7.17, N 8.00. S 6.00.
(S,S)-3-Benzyloxy-4,5-diphenyl-1-[2-(piperidin-1-yl)cyclohexyl]-1,3-dihydro-2H-imidazole-2-thione ((S,S)-9d): Yield 324.7 mg (62%). Viscous oil (after chromatography). = +22.42 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = (signals for both rotamers in ca. 1:2 ratio) 7.07–7.41 (m, 15H), 5.41 and 5.32 (d, J = 9.3 Hz, 1H), 4.61–4.67 and 5.33–5.38 (m, 1H), 4.98 and 5.12 (br. d, J = 9.3 Hz, 1H), 0.78–3.74 (m, 18H) ppm. 13C-NMR (CDCl3): δ = (signals for both rotamers) 157.6, 156.7, 133.14, 133.06, 132.4, 130.6, 130.43, 130.40, 129.5, 129.4, 129.2, 128.93, 128.89, 128.88, 128.7, 128.5, 128.13, 128.05, 127.8, 127.7, 126.9, 126.7, 126.4, 125.9, 124.9, 124.6, 124.4, 77.53, 77.51, 64.4, 60.0, 59.7, 49.2 (br.), 33.2, 29.6, 27.1, 26.8, 26.0, 25.7, 25.2, 24.9, 24.6, 23.5 ppm. IR: ν 2927 (s), 2853 (m), 1444 (w), 1396 (w), 1321 (m), 1207 (m), 959 (m), 753 (s), 693 (vs), 596 (m), 568 (m), 475 (w) cm−1. HRMS (ESI+): calcd for C33H38N3OS [M + H]+ 524.2736; found 524.2743. C33H37N3OS (523.73): calcd. C 75.68, H 7.13, N 8.02, S 6.12; found C 75.64, H 7.10, N 8.06, S 5.92.
(R,R)-3-Benzyloxy-1-[2-(azepan-1-yl)cyclohexyl]-4,5-dimethyl-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9e): Yield: 173.7 mg (42 %).Viscous oil. = −28.85 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.46–7.48 (m, 2H), 7.33–7.36 (m, 3H), 5.38 (d, J = 10.4 Hz, 1H), 5.32 (d, J = 10.4 Hz, 1H), 5.21 (td, J = 11.6, 3.4 Hz, 1H), 2.97 (td, J = 11.6, 2.7 Hz, 1H), 2.79–2.84 (m, 2H), 2.40–2.44 (m, 2H), 2.11 (s, 3H), 1.93–2.01 (m, 2H), 1.82–1.86 (m, 1H), 1.73–1.77 (m, 1H), 1.74 (s, 3H), 1.57 (qd, J = 12.4, 3.3 Hz, 1H), 1.31–1.52 (m, 10H) 1.16–1.25 (m, 1H) ppm. 13C-NMR (CDCl3): δ = 156.6, 134.2, 130.3, 129.1, 128.4, 119.2, 118.1, 77.9, 67.1, 59.4, 51.2, 32.1, 30.0, 26.6, 25.8, 25.7, 25.5, 10.6, 7.2 ppm. IR: ν 2922 (vs), 2853 (m), 1403 (s), 1377 (s), 1328 (s), 1170 (w), 1133 (w), 959 (m), 907 (m), 749 (vs), 697 (vs) cm−1. C24H35N3OS (413.62): calcd. C 69.69, H 8.53, N 10.16., S 7.75; found C 69.47, H 8.62, N 10.21, S 7.59.
(R,R)-3-Benzyloxy-1-[2-(azepan-1-yl)cyclohexyl]-4,5-diphenyl-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9f): Yield: 376.4 mg (70%). Colorless crystals. M.p. 264–267 °C (MeOH/CH2Cl2) (decomp.). = −27.17 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = (signals for both rotamers in ca. 1:2 ratio) 7.05–7.38 (m, 15H), 5.32 and 4.72 (td, J = 11.5, 3.6 Hz, 1H), 5.31 and 5.21 (d, J = 9.3 Hz, 1H), 5.02 (br.) and 5.15 (d, J = 9.3 Hz, 1H), 3.69 (td, J = 11.7, 3.0 Hz, 0.3H), 3.28–3.36 (m, 0.3H), 2.75–2.80 (m, 1H), 2.43–2.47 (m, 0.7H), 2.29–2.32 (m, 0.7H), 1.34–2.21 (m, 15H), 1.16 and 0.98 (qd, J = 12.8, 3.5 Hz, 1H), 1.03–1.11 and 0.73–0.82 (m, 1H) ppm. 13C-NMR (CDCl3): δ = 158.0, 157.0, 133.2, 133.1, 132.5, 130.6, 130.37, 130.3, 129.6, 129.4, 129.2, 129.1, 128.9, 128.6, 128.15, 128.13, 128.10, 128.07, 127.9, 127.8, 127.7, 126.6, 126.4, 126.0, 125.0, 124.7, 77.62, 77.58, 66.1, 61.4, 60.5, 51.4 (br.), 50.5, 33.3, 30.05, 29.97, 29.8, 26.9, 26.6, 26.4, 26.0, 25.8, 25.23, 25.17 ppm. IR: ν 2924 (s), 2853 (m), 1446 (m), 1356 (m), 1332 (m), 1073 (w), 963 (w), 752 (s), 693 (vs) cm−1. C34H39N3OS (537.76): calcd. C 75.94, H 7.31, N 7.81, S 5.96; found C 75.91, H 7.45, N 8.01, S 5.86.
(R,R)-3-Benzyloxy-1-(2-morpholinocyclohexyl)-4,5-dimethyl-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9g): Yield 135 mg (67%). Colorless crystals. M.p. 122–124 °C (after chromatographic separation). = −21.22 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.43–7.46 (m, 2H), 7.33–7.37 (m, 3H), 5.44 (d, J = 10.4 Hz, 1H), 5.30 (d, J = 10.4 Hz, 1H), 5.27 (td, J = 11.9, 3.7 Hz, 1H), 3.52–3.57 (m, 2H), 3.45–3.50 (m, 2H), 2.92 (dt, J = 11.4, 3.2 Hz, 1H), 2.85–2.90 (m, 2H), 2.30–2.34 (m, 2H), 2.08 (s, 3H), 1.97–2.02 (m, 2H), 1.85–1.90 (m, 1H), 1.75–1.80 (m, 1H), 1.72 (s, 3H), 1.62 (qd, J = 12.4, 3.8 Hz, 1H), 1.42–1.51 (m, 1H), 1.32–1.40 (m, 1H), 1.17–1.27 (m, 1H) ppm. 13C-NMR (CDCl3): δ = 156.5, 134.1, 130.5, 129.3, 128.6, 119.9, 117.7, 77.8, 67.7, 65.4, 58.1, 48.6, 31.9, 25.8, 25.5, 24.5, 10.7, 7.3 ppm. IR: ν 2935 (br, m), 2851 (m) 2816 (w), 1451 (m), 1407 (s), 1334 (s), 1269 (s), 1146 (m), 1108 (vs), 915 (m), 855 (m), 754 (vs), 702 (m) cm−1. C22H31N3O2S (401.56): calcd. C 65.80, H 7.78, N 10.46, S 7.98; found C 65.71, H 7.90, N 10.59, S 7.84.
(R,R)-3-Benzyloxy-1-(2-morpholinocyclohexyl)-4,5-diphenyl-1,3-dihydro-2H-imidazole-2-thione ((R,R)-9h): Yield 213 mg (81%). Colorless crystals. M.p. 279–281 °C (MeOH/CH2Cl2) (decomp.). = −43.58 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = (signals for both rotamers in ca. 1:1.2 ratio) 7.07–7.43 (m, 15H), 5.40 and 5.33 (d, J = 9.4 Hz, 1H), 5.37 and 4.72 (td, J = 11.8, 3.3 Hz, 1H), 5.09 and 4.92 (d, J = 9.4 Hz, 1H), 3.75 and 3.43 (td, J = 11.7, 3.7 Hz, 1H), 3.44–3.69 (m, 4H), 2.78 and 2.43 (br., 2H), 2.09–2.28 (m, 3H), 1.87–1.93 and 2.01–2.06 (m, 1H), 1.66–1.78 (m, 3H), 1.40–1.49 (m, 1H), 1.20–1.27 and 0.92–1.00 (m, 1H), 1.09–1.17 and 0.81–0.89 (m, 1H) ppm. 13C-NMR (CDCl3): δ = (signals for both rotamers) 157.9, 156.9, 133.13, 133.07, 132.5, 130.6, 130.5, 130.4, 129.6, 129.5, 129.4, 129.3, 129.1, 129.04, 129.02, 128.8, 128.3, 128.0, 127.9, 126.5, 126.34, 126.29, 125.3, 124.5, 77.72, 77.66, 68.0, 67.8, 63.8, 59.8, 59.47, 59.43, 48.3, 33.2, 29.6, 25.93, 25.75, 25.2, 25.0, 24.9, 23.7 ppm. IR: ν 2924 (br m), 2853 (m), 1401 (m), 1360 (w), 1323 (m), 1112 (s), 965 (m), 861 (m), 762 (s), 699 (vs) cm−1. C32H35N3O2S (525.70): calcd. C 73.11, H 6.72, N 7.99; S 6.10; found C 73.12, H 6.76, N 7.93, S 6.03.
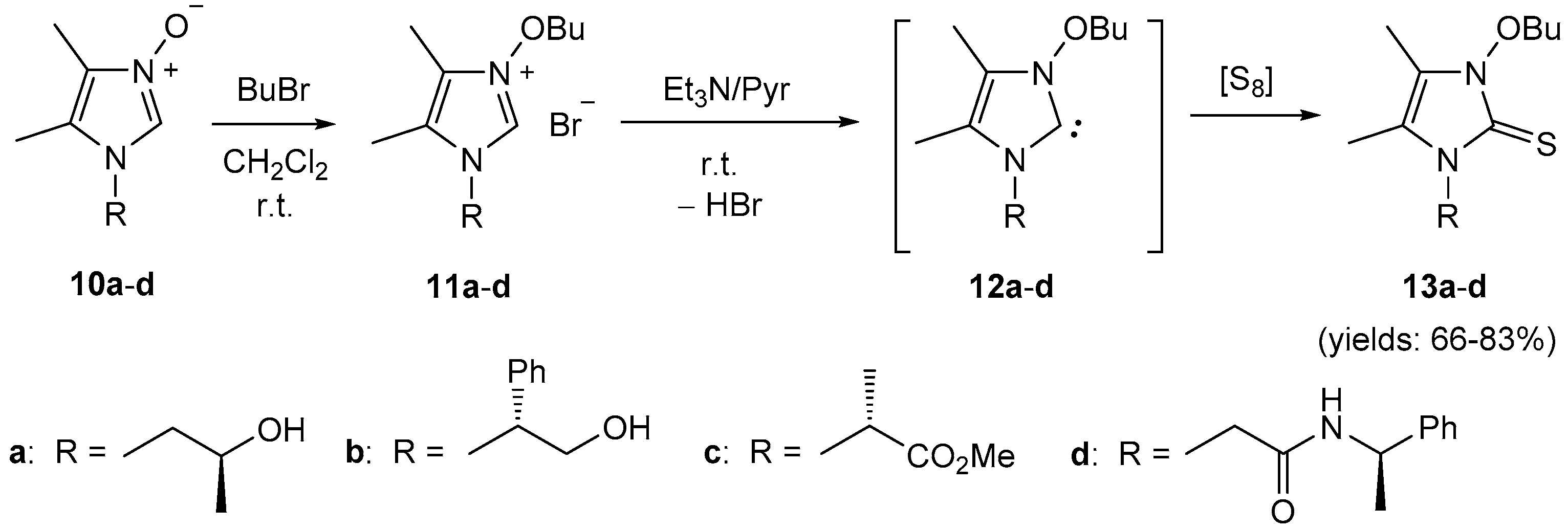
4.7. Synthesis of Imidazole N-Oxides 10a–d
A solution of diacetyl monooxime (
5a, 354 mg, 3.5 mmol) and the corresponding formaldimine
4 (3.0 mmol) in EtOH (10 mL) was refluxed for 3 h. The solvents were removed in vacuo, and the resulting oil was washed with Et
2O (3 × 20 mL). The crude product
10 was either purified by column chromatography on silica gel using AcOEt/MeOH mixtures as the eluent or by recrystallization from the appropriate solvents to give spectroscopically pure imidazole
N-oxides isolated as colorless materials. Compounds
10a [
21] and
10c [
22] were prepared following the analogous method, while imidazole
N-oxide
10d was obtained in two steps by condensation of
5a with methyl glycinate-derived formaldimine of type
4 followed by aminolysis of the resulting product with α-MBA as described [
22]; the NMR spectra of the obtained samples matches the data reported in the literature.
(R)-1-(2-Hydroxy-1-phenylethyl)-4,5-dimethylimidazole 3-oxide (10b): Yield 474 mg (68%). Off-white solid. M.p. 198–200 °C (CH2Cl2/Et2O) (decomp.). = +49.1 (c 0.22, CHCl3). 1H-NMR (CDCl3): δ = 9.17 (s, 1H, C(2)H), 8.72 (br.s, 1H, OH), 7.01–7.04, 7.27–7.35 (2m, 2H, 3H, Ph), 5.14 (dd, J = 3.8, 10.7 Hz, 1H, CHPh), 4.21 (dd, J = 10.7, 13.4 Hz, 1H, CH2O), 3.95 (dd, J = 3.8, 13.4 Hz, 1H, CH2O), 1.84, 2.10 (2s, 3H each, 2Me) ppm. 13C-NMR (CDCl3): δ = 136.6 (s, Ph), 128.3, 129.0 (2d, 3CH, Ph), 126.9 (d, 2CH, Ph), 126.4 (s, C(4)), 124.9 (d, C(2)), 122.0 (s, C(5)), 63.9 (d, CHPh), 63.6 (t, CH2O), 7.0, 8.9 (2q, 2Me) ppm. IR (neat): ν 3110 (s), 3025–2926 (br, m), 1450 (m), 1405 (m), 1351 (s), 1325 (m), 1208 (m), 1064 (s) cm−1. ESI-MS (m/z): 465.4 (100, [2M + H]+), 233.3 (47, [M + H]+). C13H16N2O2 (232.28): calcd. C, 67.22; H, 6.94; N, 12.06; found: C 67.05, H 6.95, N 12.22.
4.8. Synthesis of Imidazolium Bromides 11a–d
To a solution of imidazole N-oxide 10 (1.0 mmol) in dry CH2Cl2 (1.0 mL) was added an excess of 1-bromobutane (548 mg, 4.0 mmol) and the resulting mixture was stirred until the starting material was fully consumed (TLC monitoring: SiO2, EtOAc/MeOH 7:1). The solvents were removed under reduced pressure to give the corresponding imidazolium bromide 11 quantitatively, which was used for the next step without further purification.
(
S)-3-Butoxy-1-(2-hydroxypropyl)-4,5-dimethylimidazolium bromide [
23] (
11a): Reaction time: 2 d. Pale yellow oil.
= +16.0 (
c 0.74, CHCl
3).
1H-NMR (CDCl
3): δ = 10.00 (s, 1H, C(2)H), 4.91 (br.d,
J ≈ 5.9 Hz, 1H, OH), 4.47 (td,
J = 2.1, 6.5 Hz, 2H, OCH
2, Bu), 4.34 (dd,
J = 9.1, 14.0 Hz, 1H, NCH
2), 4.22 (dd,
J = 2.6, 14.0 Hz, 1H, NCH
2), 4.16–4.22 (m, 1H, C
HCH
3), 2.26, 2.27 (2s, 3H each, 2Me), 1.77–1.81 (m, 2H, Bu), 1.47–1.43 (m, 2H, Bu), 1.34 (d,
J = 6.3 Hz, 3H, CHC
H3), 0.98 (t,
J = 7.4 Hz, 3H, CH
3, Bu) ppm.
13C-NMR (CDCl
3): δ = 131.4 (br.d, C(2)), 123.6, 124.7 (2s, C(4), C(5)), 82.7 (t, OCH
2, Bu), 64.8 (d,
CHCH
3), 53.5 (t, NCH
2), 29.6 (t, CH
2, Bu), 20.4 (q, CH
CH
3), 18.7 (t, CH
2, Bu), 13.6 (q, CH
3, Bu), 7.1, 8.9 (2q, 2Me) ppm. IR (neat): ν3308 (s), 2990–2876 (br, s), 1670 (m), 1634 (m), 1545 (m), 1457 (m), 1377 (m), 1139 (s), 1072 (s), 936 (s) cm
−1.
(R)-3-Butoxy-1-(2-hydroxy-1-phenylethyl)-4,5-dimethylimidazolium bromide (11b): Reaction time: 2 d. Yellow solid. M.p. 124–127 °C. = +83.5 (c 1.79, CHCl3). 1H-NMR (CDCl3): δ = 10.50 (s, 1H, C(2)H), 7.15–7.18, 7.32–7.38 (2m, 2H, 3H, Ph), 5.61 (dd, J = 3.8, 10.1 Hz, 1H, CHPh), 5.46 (br.s, 1H, OH), 4.80 (pseudo-q, J ≈ 6.6 Hz, 1H, OCH2, Bu), 4.52–4.59 (m, 2H, CH2OH (1H), OCH2 (1H)), 4.10 (dd, J = 3.8, 13.2 Hz, 1H, CH2OH), 2.06, 2.24 (2s, 3H each, 2Me), 1.79–1.84 (m, 2H, Bu), 1.49–1.55 (m, 2H, Bu), 0.98 (t, J = 7.4 Hz, 3H, CH3, Bu) ppm. 13C-NMR (CDCl3): δ = 134.2 (s, Ph), 131.4 (dbr, C(2)), 126.7, 129.3, 129.5 (3d, 5CH, Ph), 124.3, 125.0 (2s, C(4), C(5)), 83.2 (t, OCH2, Bu), 64.9 (d, CHPh), 62.4 (t, CH2OH), 29.8 (t, CH2, Bu), 18.8 (t, CH2, Bu), 13.7 (q, CH3, Bu), 7.1, 9.1 (2q, 2Me) ppm. IR (neat): ν 3248 (vs), 3027–2876 (br, s), 1636 (m), 1541 (m), 1448 (s), 1359 (m), 1066 (s), 935 (s) cm−1.
(
R)-3-Butoxy-4,5-dimethyl-1-[1-(methoxycarbonyl)ethyl]imidazolium bromide [
23] (
11c): Reaction time: 2 d. Pale yellow oil.
= −16.6 (
c 0.51, CHCl
3). The NMR data in accordance with those reported in [
23].
(R)-3-Butoxy-4,5-dimethyl-1-[(N-phenylethyl)acetamido]imidazolium bromide (11d): Reaction time: 4 d. Pale yellow oil. = +51.2 (c 0.52, CHCl3). 1H-NMR (CDCl3): δ = 9.84 (s, 1H, C(2)H), 9.29 (d, J = 7.8 Hz, 1H, NH), 7.14–7.17, 7.23–7.26, 7.39–7.42 (3m, 1H, 2H, 2H, Ph), 5.35, 5.45 (AB system, J = 16.1 Hz, 2H, NCH2), 4.95 (dq, J = 7.1, 7.8 Hz, 1H, CH(Ph)CH3), 4.32 (td, J = 1.4, 6.5 Hz, 2H, OCH2, Bu), 2.14, 2.17 (2s, 3H each, 2Me), 1.71–1.76 (m, 2H, Bu), 1.53 (d, J = 7.1 Hz, 3H, CH(Ph)CH3), 1.42–1.48 (m, 2H, Bu), 0.94 (t, J = 7.4 Hz, 3H, CH3, Bu) ppm. 13C-NMR (CDCl3): δ = 163.6 (s, C=O), 143.6 (s, Ph), 131.4 (br.d, C(2)), 126.3, 126.9, 128.4 (3d, 5CH, Ph), 123.3, 125.9 (2s, C(4), C(5)), 82.6 (t, OCH2, Bu), 50.3 (d, CH(Ph)CH3), 49.7 (t, NCH2), 29.5 (t, CH2, Bu), 22.4 (q, CH(Ph)CH3), 18.6 (t, CH2, Bu), 13.6 (q, CH3, Bu), 7.0, 8.7 (2q, 2Me) ppm. IR (neat): ν 3200 (m), 3032–2872 (br, s), 1679 (vs, C=O), 1547 (s), 1448 (m), 1377 (m), 1250 (m) cm−1.
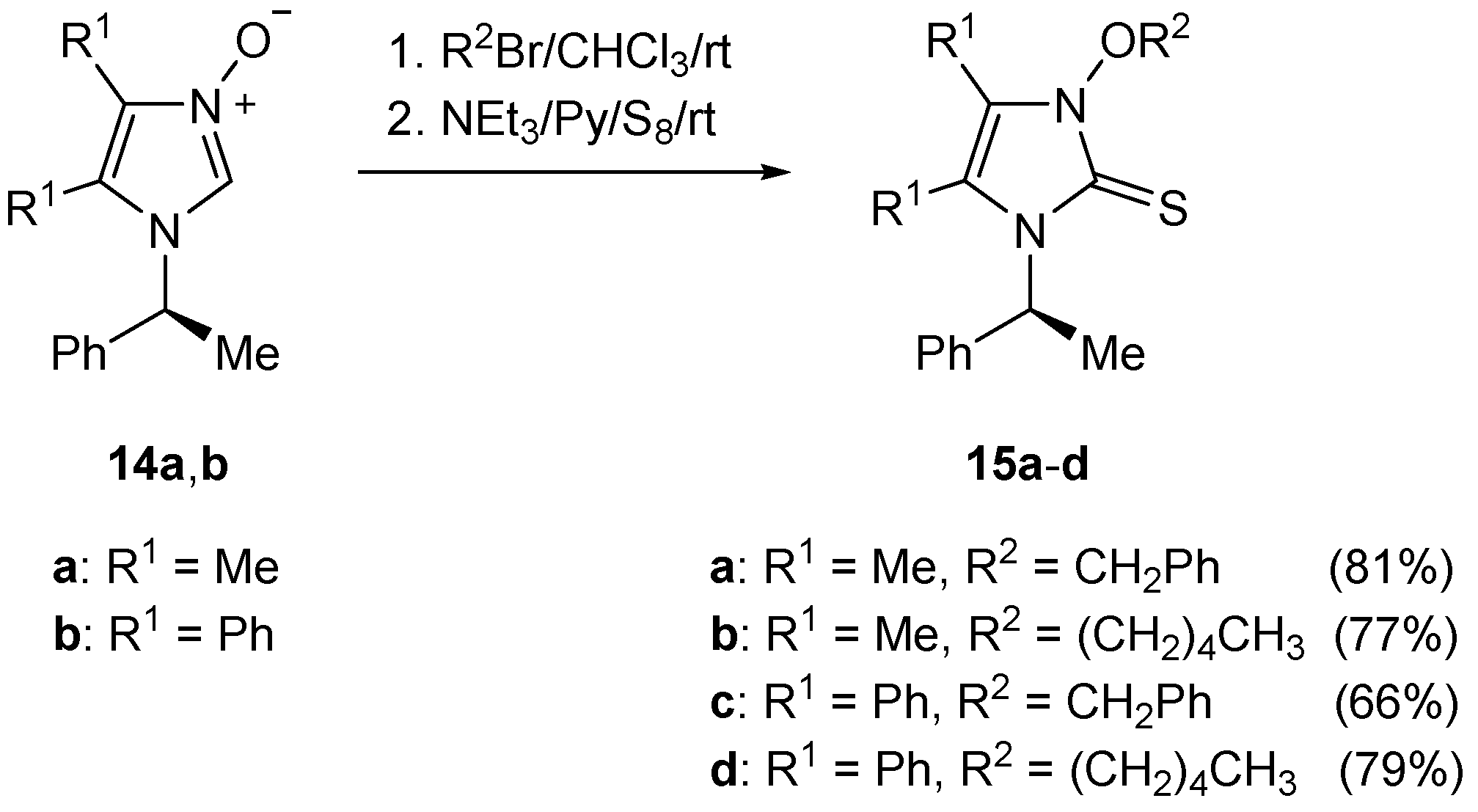
4.9. Synthesis of Imidazole-2-thiones 13a–d and 15a–d
To a solution of the respective imidazolium bromide (1.0 mmol) in dry pyridine (4.0 mL) was added triethylamine (150 μL) followed by elemental sulfur (33 mg, 1.1 mmol), and the resulting mixture was stirred overnight. The solvents were removed under reduced pressure, and the crude product was purified by standard column chromatography to give imidazole-2-thione of type 13 or 15, respectively.
(S)-3-Butoxy-1-(2-hydroxypropyl)-4,5-dimethylimidazole-2-thione (13a): (chromatographic separation, SiO2, CH2Cl2/EtOAc 5:1); 209 mg (81%). Pale yellow oil. = +1.3 (c 0.77, CHCl3). 1H-NMR (CDCl3): δ = 4.29–4.34 (m, 2H, OCH2, Bu), 4.18–4.23 (m, 1H, CHCH3), 4.03 (dd, J = 8.7, 14.4 Hz, 1H, NCH2), 3.97 (dd, J = 3.2, 14.4 Hz, 1H, NCH2), 3.28 (br.s, 1H, OH), 2.08, 2.12 (2s, 3H each, 2Me), 1.73–1.77 (m, 2H, Bu), 1.46–1.53 (m, 2H, Bu), 1.25 (d, J = 6.3 Hz, 3H, CHCH3), 0.96 (t, J = 7.4 Hz, 3H, CH3, Bu) ppm. 13C-NMR (CDCl3): δ = 156.2 (s, C=S), 118.5, 119.0 (2s, C(4), C(5)), 76.8 (t, OCH2, Bu), 67.3 (d, CHCH3), 51.3 (t, NCH2), 29.8 (t, CH2, Bu), 21.2 (q, CHCH3), 18.9 (t, CH2, Bu), 13.7 (q, CH3, Bu), 7.5, 9.1 (2q, 2Me) ppm. IR (neat): ν 3353 (s), 3040–2874 (br, m), 1433 (m), 1409 (s), 1375 (m), 1347 (m), 1127 (m), 1072 (m), 943 (m) cm−1. ESI-MS (m/z): 281.1 (53, [M + Na]+), 259.1 (100, [M + H]+), 208.1 (42). C12H22N2O2S (258.38): calcd. C 55.78, H 8.58, N 10.84, S 12.41; found: C 55.57, H 8.65, N 10.76, S 12.21.
(R)-3-Butoxy-1-(2-hydroxy-1-phenylethyl)-4,5-dimethylimidazole-2-thione (13b): (chromatographic separation, SiO2, CH2Cl2/EtOAc 5:1); 240 mg (75%). Yellow oil. = −10.8 (c 0.73, CHCl3). 1H-NMR (CDCl3): δ = 7.24–7.30, 7.32–7.35 (2m, 3H, 2H, Ph), 6.26 (m, 1H, CHPh), 4.60–4.65 (m, 1H, CH2OH), 4.34–4.42 (m, 3H, OCH2(Bu) (2H), CH2OH (1H)), 3.09 (br.s, 1H, OH), 2.09 (s, 3H, Me), 1.79–1.84 (m, 2H, Bu), 1.72 (s, 3H, Me), 1.52–1.58 (m, 2H, Bu), 1.00 (t, J = 7.4 Hz, 3H, CH3, Bu) ppm. 13C-NMR (CDCl3): δ = 157.4 (s, C=S), 136.7 (s, Ph), 126.9, 127.8, 128.7 (3d, 5CH, Ph), 118.8, 120.1 (2s, C(4), C(5)), 76.8 (t, OCH2, Bu), 62.8 (t, CH2OH), 60.9 (d, CHPh), 29.9 (t, CH2, Bu), 19.0 (t, CH2, Bu), 13.8 (q, CH3, Bu), 7.4, 9.9 (2q, 2Me) ppm. IR (neat): ν 3347 (s), 3016–2874 (br, s), 1407 (s), 1333 (m), 1156 (m), 1060 (m), 1031 (m) cm–1. ESI-MS (m/z): 343.1 (54, [M + Na]+), 321.2 (100, [M + H]+), 270.1 (45). HRMS (ESI-TOF): calcd for C17H25N2O2S: 321.1637; found: 321.1640.
(R)-3-Butoxy-4,5-dimethyl-1-[1-(methoxycarbonyl)ethyl]imidazole-2-thione (13c): (chromatographic separation, SiO2, CH2Cl2/EtOAc 95:5); 236 mg (83%). Yellow oil. = −45.4 (c 0.20, CHCl3). 1H-NMR (CDCl3): δ = 6.03 (d, J = 7.4 Hz, 1H, CHCH3), 4.31–4.39 (m, 2H, OCH2, Bu), 3.74 (s, 3H, OCH3), 2.02, 2.12 (2s, 3H each, 2Me), 1.75–1.80 (m, 2H, Bu), 1.62 (d, J = 7.4 Hz, 3H, CHCH3), 1.49–1.55 (m, 2H, Bu), 0.98 (t, J = 7.4 Hz, 3H, CH3, Bu) ppm. 13C-NMR (CDCl3): δ = 170.8 (s, C=O), 157.6 (s, C=S), 117.4, 119.8 (2s, C(4), C(5)), 76.7 (t, OCH2, Bu), 53.2 (d, CHCH3), 52.6 (q, OCH3), 29.9 (t, CH2, Bu), 19.0 (t, CH2, Bu), 16.0 (q, CHCH3), 13.8 (q, CH3, Bu), 7.4, 9.8 (2q, 2Me) ppm. IR (neat): ν 2932–2872 (m), 1743 (vs, C=O), 1407 (s), 1344 (m), 1224 (s), 1183 (s), 1109 (m), 1066 (s), 963 (m) cm−1. ESI-MS (m/z): 287.3 (100, [M + H]+). C13H22N2O3S (286.39): calcd. C 54.52, H 7.74, N, 9.78, S 11.19; found: C 54.58, H 7.79, N 9.77, S 11.18.
(R)-3-Butoxy-4,5-dimethyl-1-[(N-phenylethyl)acetamido]imidazole-2-thione (13d): (chromatographic separation, SiO2, CH2Cl2/EtOAc 4:1); 238 mg (66%). Pale orange solid. M.p. 118–119 °C. = +2.7 (c 1.89, CHCl3). 1H-NMR (CDCl3): δ = 7.71 (br.d, J ≈ 8.1 Hz, 1H, NH), 7.20–7.23, 7.25–7.30 (2m, 1H, 4H, Ph), 5.00 (pseudo-p, J ≈ 7.1 Hz, 1H, CH(Ph)CH3), 4.65, 4.70 (AB system, J = 14.9 Hz, 2H, NCH2), 4.35 (dt, J ≈ 6.6, 8.1 Hz, 1H, OCH2, Bu), 4.28 (dt, J ≈ 6.6, 8.1 Hz, 1H, OCH2, Bu), 2.11, 2.13 (2s, 3H each, 2Me), 1.76–1.81 (m, 2H, Bu), 1.50–1.56 (m, 2H, Bu), 1.43 (d, J = 7.0 Hz, 3H, CH(Ph)CH3), 0.99 (t, J = 7.4 Hz, 3H, CH3, Bu) ppm. 13C-NMR (CDCl3): δ = 166.2 (s, C=O), 156.3 (s, C=S), 143.1 (s, Ph), 125.8, 127.0, 128.4 (3d, 5CH, Ph), 118.5, 119.5 (2s, C(4), C(5)), 76.9 (t, OCH2, Bu), 49.3 (d, CH(Ph)CH3), 48.6 (t, NCH2), 29.8 (t, CH2, Bu), 22.5 (q, CH(Ph)CH3), 18.9 (t, CH2, Bu), 13.7 (q, CH3, Bu), 7.4, 9.1 (2q, 2Me) ppm. IR (neat): ν 3282 (m), 2956–2871 (br, m), 1662 (vs, C=O), 1549 (s), 1407 (s), 1372 (m), 1247 (m), 1003 (m) cm−1. ESI-MS (m/z): 384.2 (100, [M + Na]+), 362.2 (21, [M + H]+), 311.1 (48), 241.1 (69). C19H27N3O2S (361.50): calcd. C 63.13, H 7.53, N 11.62, S 8.87; found: C 63.04, H 7.80, N 11.70, S 8.76.
(S)-3-Benzyloxy-4,5-dimethyl-1-(1-phenylethyl)imidazole-2-thione (15a): Yield: 275 mg (81%). Pale yellow crystals. M.p. 102–104 °C (petroleum ether/CH2Cl2). = +110.5 (c 0.32, CHCl3). 1H-NMR (CDCl3): δ = 7.58–7.53 (m, 2H), 7.43–7.40 (m, 3H), 7.37–7.34 (m, 2H), 7.30–7.26 (m, 3H), 6.80 (q, J = 7.3 Hz, 1H), 5.58, 5.45 (AB system, J = 10.0 Hz, 2H, OCH2), 1.83 (d, J = 7.3 Hz, 3H), 1.78, 1.63 (2s, 6H, 2Me) ppm. 13C-NMR (CDCl3): δ = 157.3 (s, C=S), 126.4, 127.4, 128.5, 128.6, 129.3, 130.4 (6d, 10CH), 117.5, 120.7 134.0, 139 (4s), 77.9 (t, OCH2), 53.4 (d, CH), 7.2, 10.2, 17.3 (3q, 3Me) ppm. IR (neat): ν 2971 (m), 2924 (m), 1449 (m), 1403 (s), 1375 (m), 1332 (s), 1299 (m), 1151 (m), 1025 (m), 956 (m), 909 (m), 747 (s), 697 (vs) cm−1. HRMS (ESI-TOF) calcd for C20H23N2OS: 339.1531; found: 339.1535.
(S)-4,5-Dimethyl-3-pentyloxy-1-(1-phenylethyl)imidazole-2-thione (15b): Yield: 245 mg (77%). Pale yellow crystals. M.p. 70–73 °C (petroleum ether/CH2Cl2). = +125.9 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.28–7.39 (m, 5H), 6.76 (q, J = 7.3 Hz, 1H), 4.43–4.46 (pseudo q, J = 7.2 Hz, 1H, OCH2), 4.37–4.40 (pseudo q, J = 7.2 Hz, 1H, OCH2), 2.09 (s, 3H, Me), 1.83–1.88 (m, 2H, CH2), 1.80 (d, J = 7.3 Hz, 3H), 1.68 (s, 3H, Me), 1.49–1.53 (m, 2H, CH2), 1.41–1.45 (m, 2H, CH2), 0.96 (t, J = 7.3 Hz, 3H, CH3) ppm. 13C-NMR (CDCl3): δ = 157.5 (C=S), 126.5, 127.3, 128.5 (3d, 5CH), 117.8, 119.9, 139.9 (3s), 76.9 (t, OCH2), 53.4 (d, CH), 22.5, 27.7, 27.9 (3t, 3CH2), 7.3, 10.1, 13.9, 16.8 (4q, 4Me) ppm. IR (neat): ν 2954 (m), 2927 (m), 1450 (m), 1410 (s), 1334 (s), 1299 (s), 1157 (m), 1027 (m), 1001 (s), 998 (m), 876 (m), 700 (vs), 687 (s) cm−1. C18H26N2OS (318.48): calcd. C 67.88, H 8.23, N 8.80, S 10.07; found: C 67.61, H 8.28, N 8.70, S 10.06.
(S)-3-Benzyloxy-4,5-diphenyl-1-(1-phenylethyl)imidazole-2-thione (15c): Yield: 305 mg (66%). Pale yellow oil. = +188.5 (c 0.40, CHCl3). 1H-NMR (CDCl3): δ = 7.30–7.32 (m, 2H), 7.20–7.25 (m, 10H), 7.14–7.18 (m, 2H), 7.05–7.12 (m, 6H), 6.75 (br.s, 1H, CH3-CH), 6.67 (br.s, 2H), 5.30, 5.32 (AB system, J = 12.5 Hz, 2H, OCH2), 1.62 (d, J = 6.5 Hz, 3H, CH3-CH) ppm. 13C-NMR (CDCl3): δ = 158.4 (s, C=S), 124.1, 125.9, 126.2, 128.6, 129.8, 132.9 (6s), 126.9, 127.3, 127.9, 128.0, 128.1, 128.2, 128.3, 128.9, 129.1, 129.4, 130.6, 132.0 (12d, 20CH), 77.7 (t, OCH2), 54.6 (d, CH), 17.5 (q, CH3) ppm. IR (neat): ν 3032 (m), 2927 (m), 1496 (m), 1446 (m), 1395 (s), 1325 (m), 1316s, 1215 (m), 1187 (m), 1073 (m), 907 (m), 730 (s), 695 (vs) cm−1. HRMS (ESI-TOF): calcd for C30H27N2O: 463.1844; found: 463.1849.
(S)-3-Pentyloxy-4,5-diphenyl-1-(1-phenylethyl)imidazole-2-thione (15d): Yield: 350 mg (79%). Beige crystals. M.p. 78–81 °C (petroleum ether/CH2Cl2). = +130.1 (c 0.30, CHCl3). 1H-NMR (CDCl3): δ = 7.20–7.25 (m, 9H), 7.06–7.09 (m, 4H), 6.67–6.73 (m, 3H, 2CHarom, CH-CH3), 4.31 (pseudo q, J = 6.8 Hz, 1H, OCH2), 4.14 (pseudo q, J = 6.8 Hz, 1H, OCH2), 1.63–1.66 (m, 2H, CH2), 1.59 (d, J = 6.8 Hz, 3H, CH3-CH), 1.24–1.27 (m, 2H, CH2), 1.19–1.23 (m, 2H, CH2), 0.83 (t, J = 7.3 Hz, 3H, CH3) ppm. 13C-NMR (CDCl3): δ = 158.6 (s, C=S), 126.9, 127.2, 127.9, 128.1, 128.2, 128.3, 128.9, 129.3, 132.0 (9d, 15CH), 124.4, 125.2, 126.0, 140.3 (4s), 76.9 (t, OCH2), 54.7 (d, CH), 22.2, 27.4, 27.8 (3t, 3CH2), 13.9, 17.5 (2q, 2CH3) ppm. IR (neat): ν 2968 (m), 2953 (m), 1448 (m), 1392 (s), 1321 (s), 1187 (m), 1026 (m), 1002 (m), 963 (m), 702 (vs), 695 (vs) cm−1. C28H30N2OS (442.62): C 75.98; H 6.83; N 6.33; S 7.24. Found: C 75.78; H 6.88; N 6.58; S 7.50.




 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
