
The Hydrophobicity of Lignocellulosic Fiber Network Can Be Enhanced with Suberin Fatty Acids

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
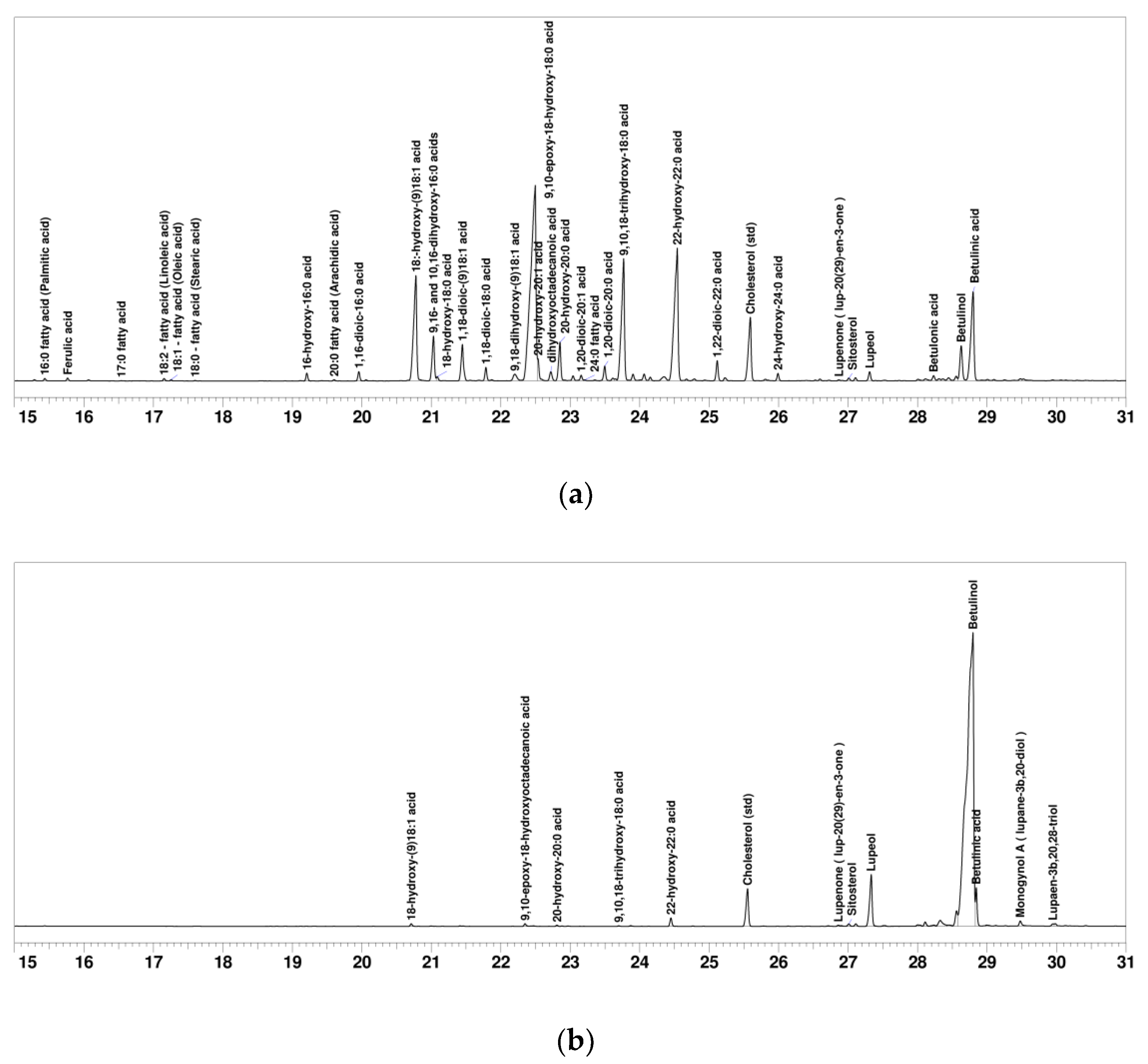
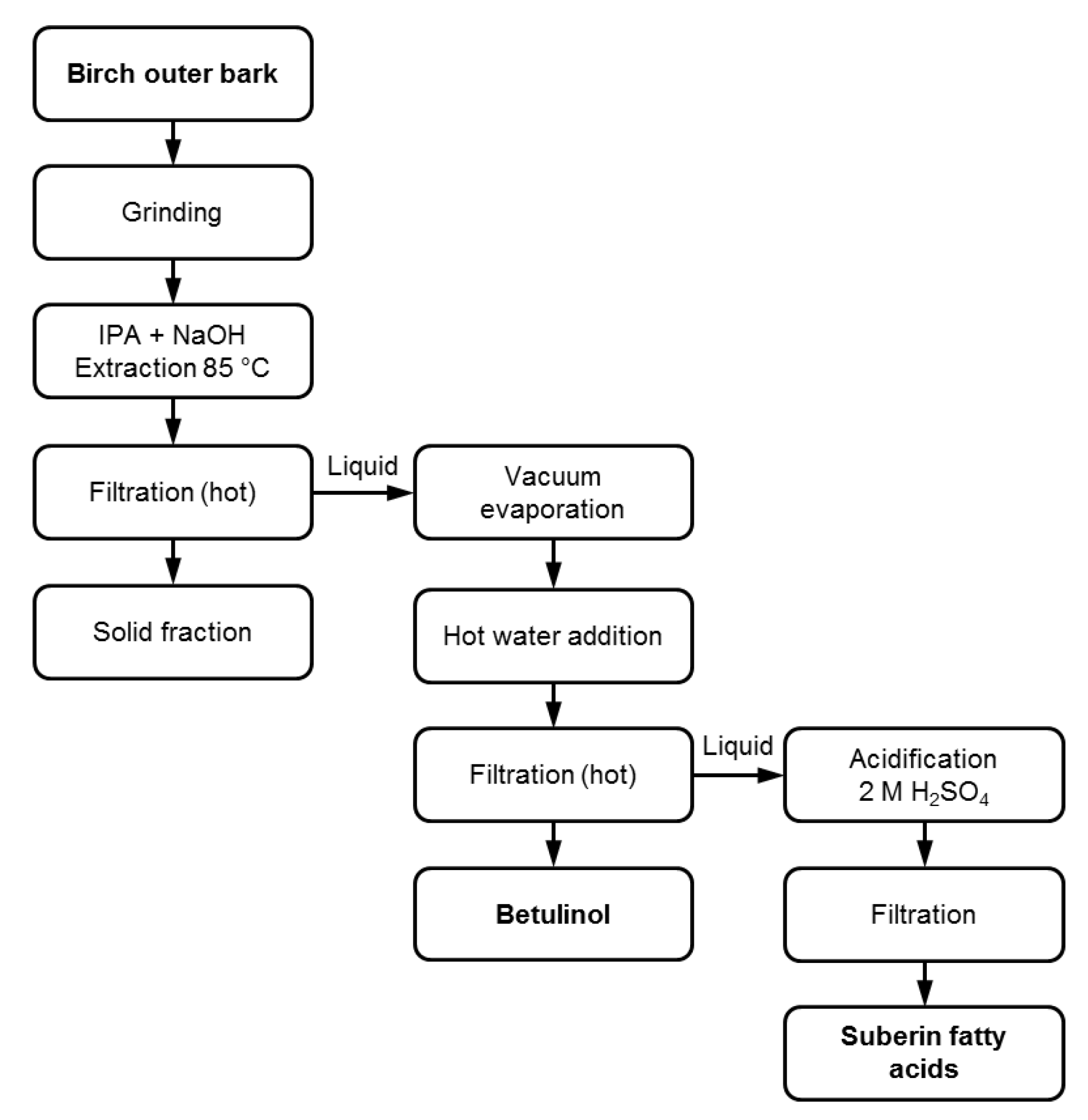
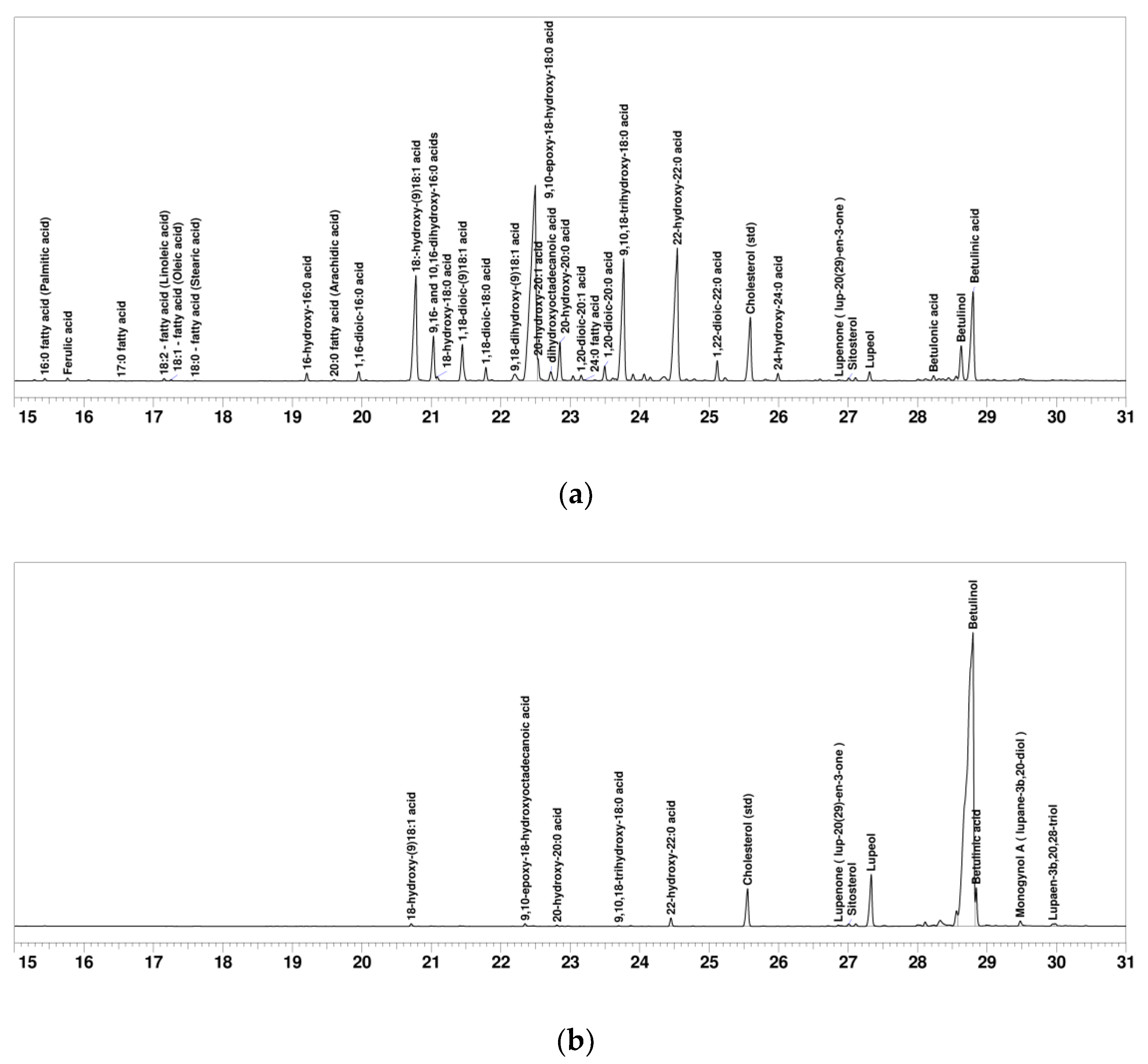
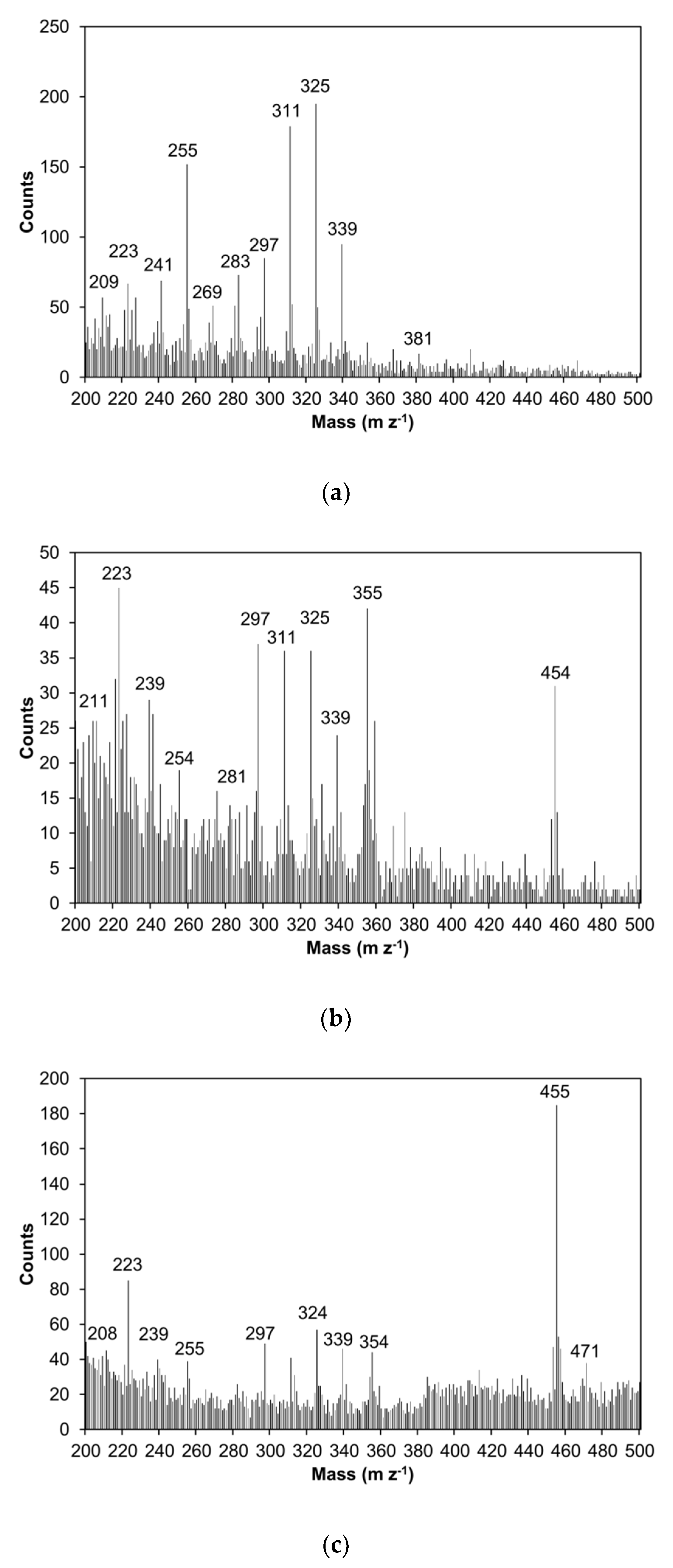
2.1. SFA and Betulinol Fractions
2.2. Elemental Analysis of SFA and Betulinol Fractions
2.3. The Effect of SFA Curing Agent Applied to the Laboratory Sheets (Lignocellulosic Fiber Network)
2.3.1. Basic Properties
2.3.2. Optical Properties
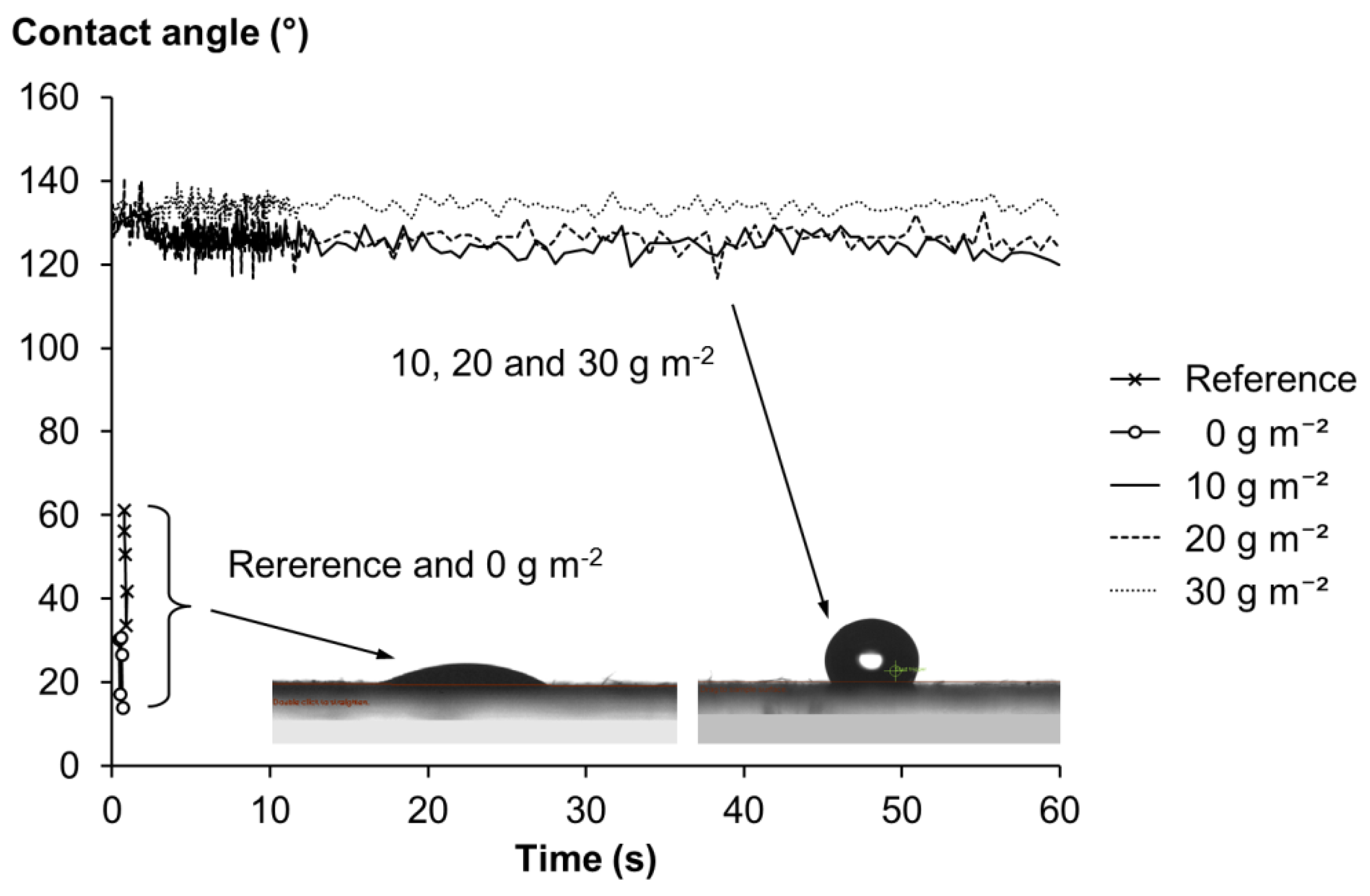
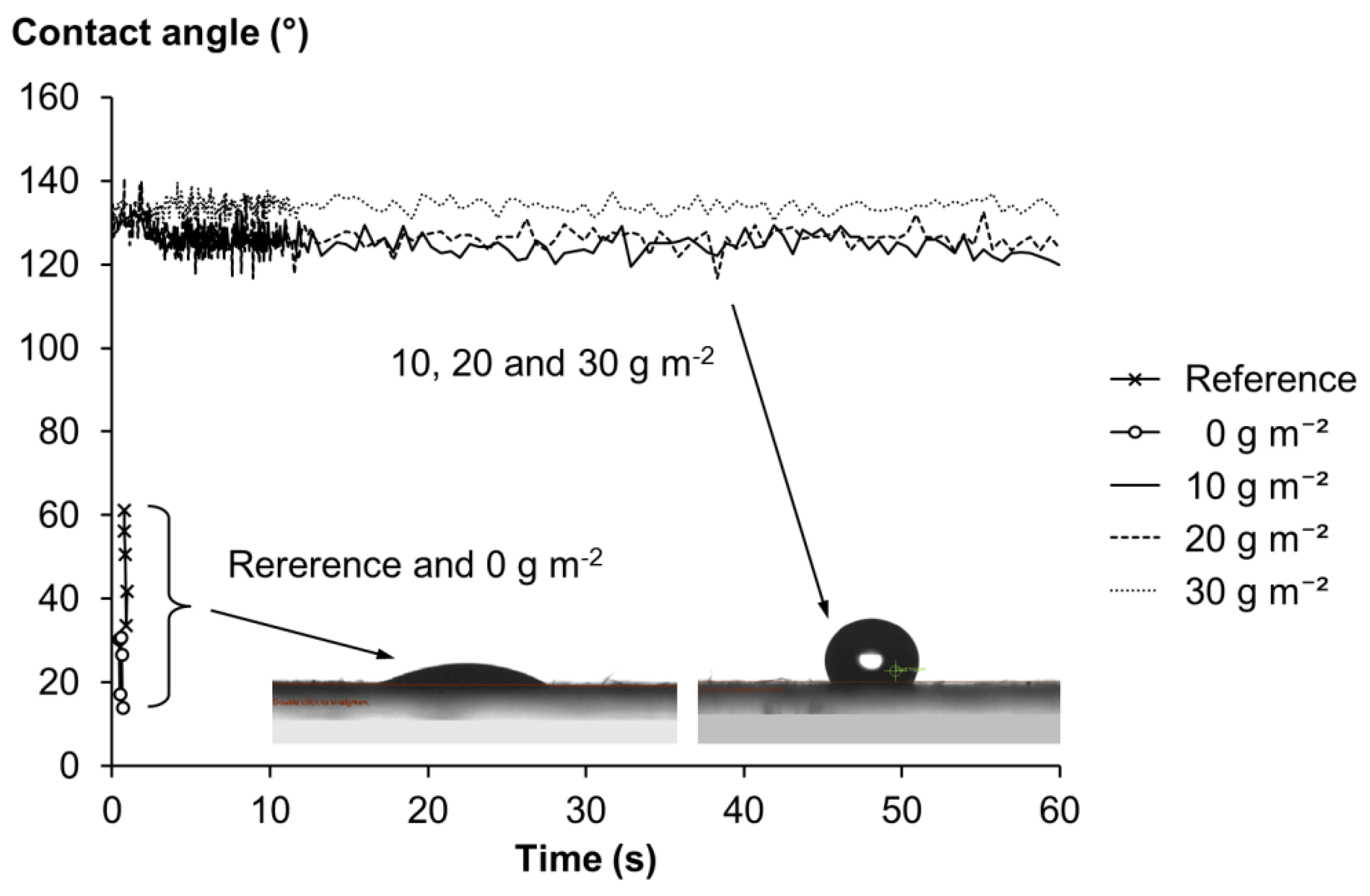
2.3.3. Contact Angle, Air Permeance and Water Vapor Transmission Rate (WVTR)
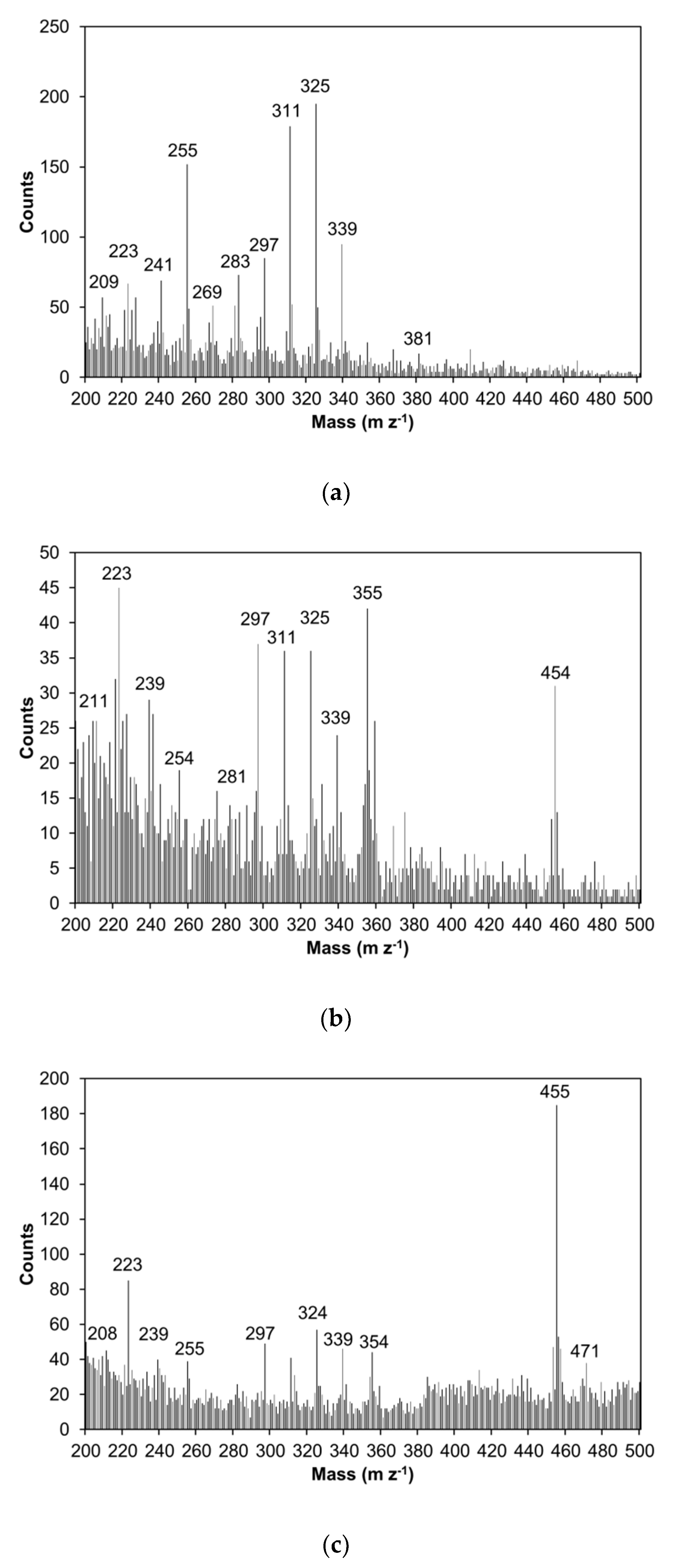
2.3.4. Scanning Electron Microscope Images and ToF-SIMS
3. Materials and Methods
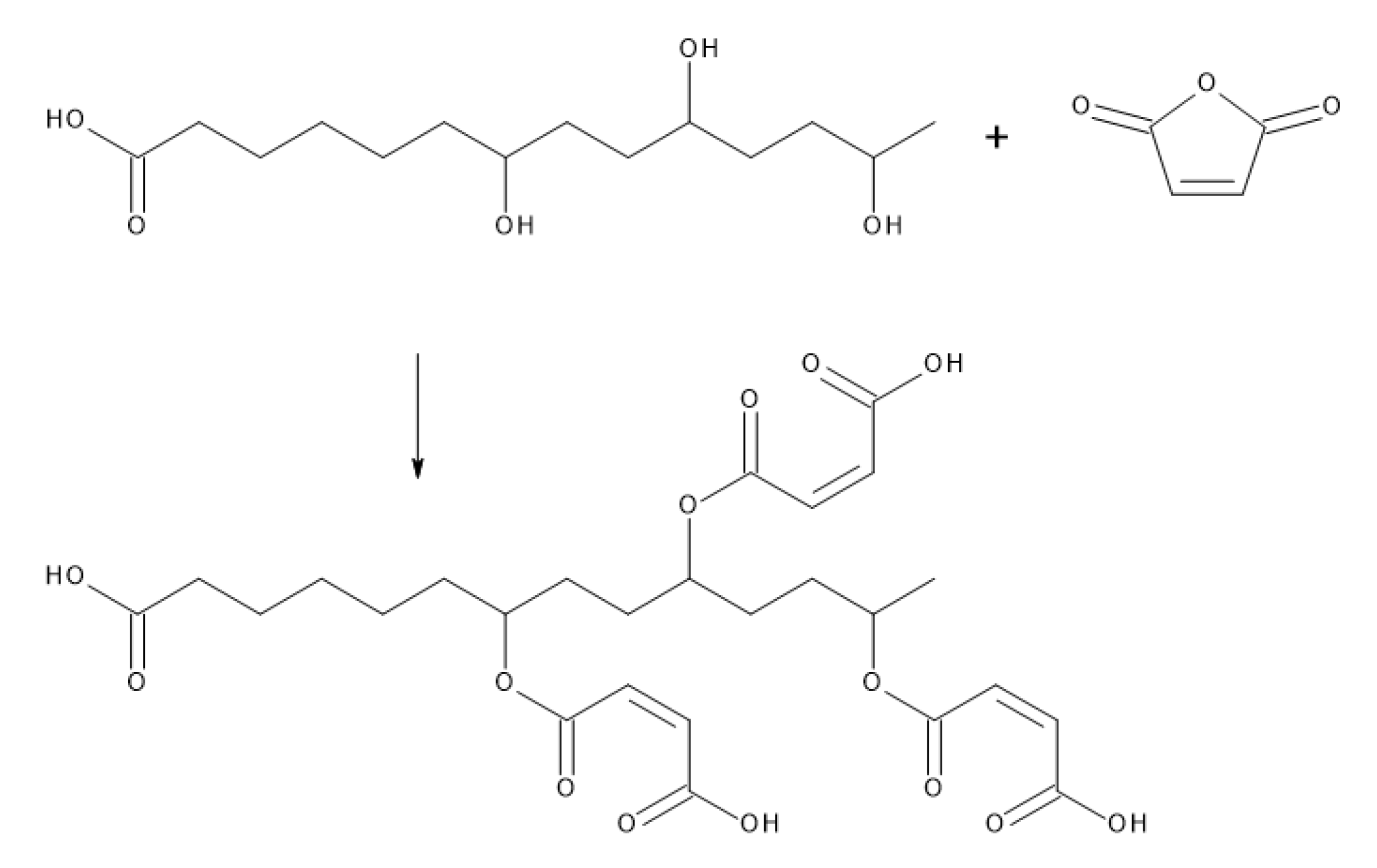
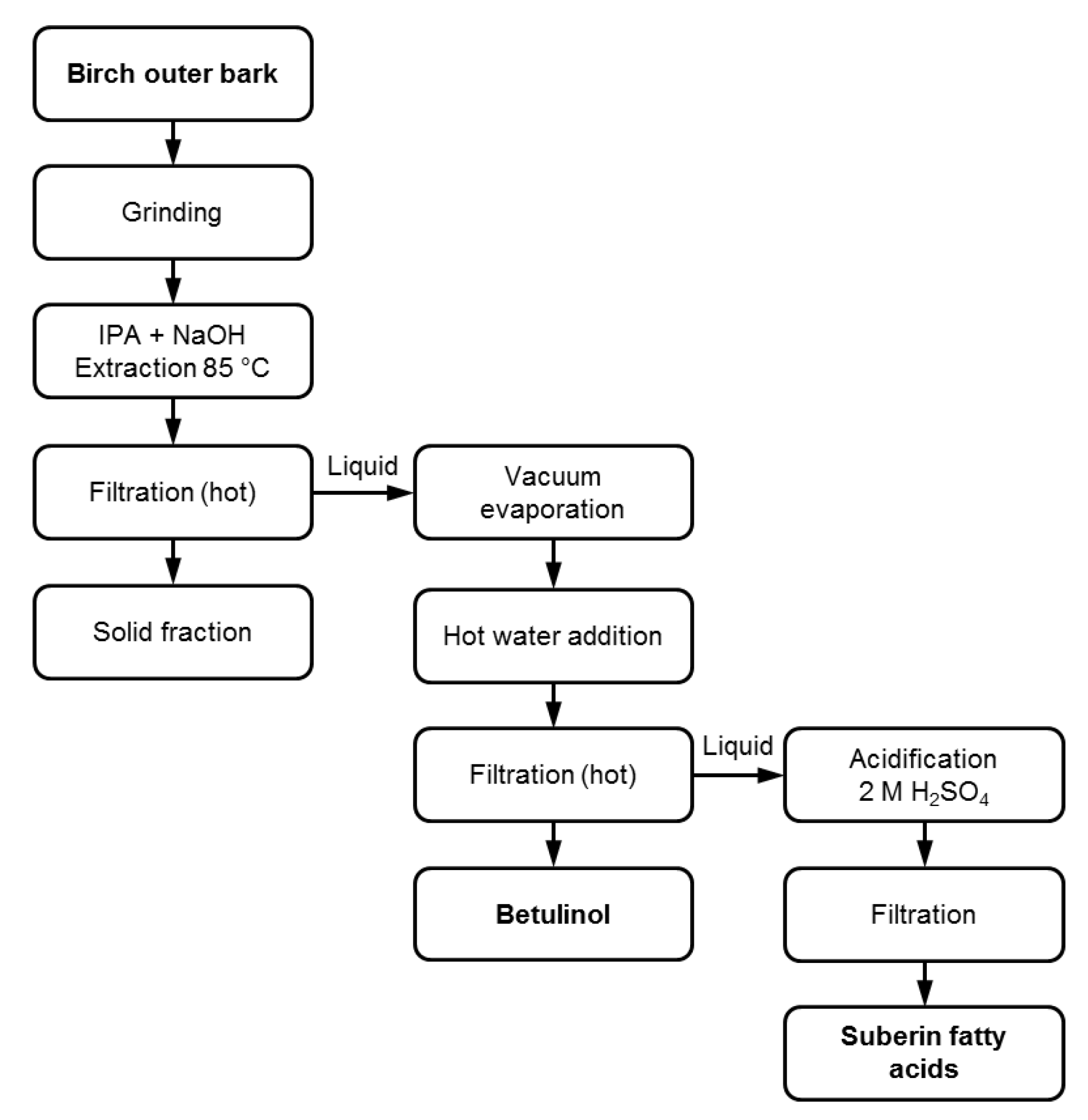
3.1. Suberin Fatty Acid Extraction and Isolation
3.2. Elemental Analysis
3.3. GC and GC-MS Analysis
3.4. Preparation of Laboratory Sheets (Lignocellulosic Fiber Network)
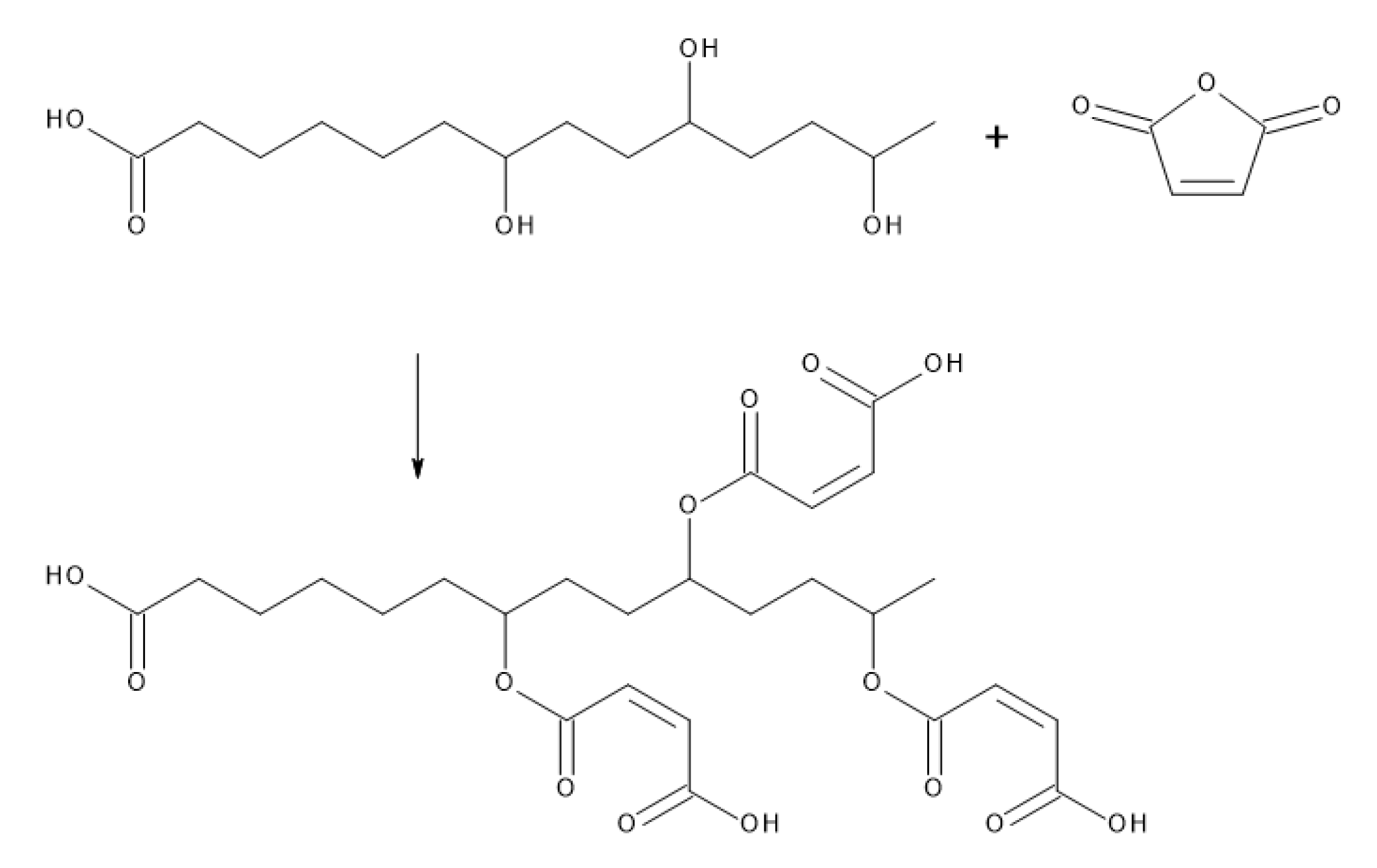
3.5. Suberin Fatty Acid Impregnation and Curing
3.6. Paper Technical Properties
- Thickness, density, grammage: ISO 534 Paper and board—Determination of thickness, density and specific volume.
- Tensile strength: ISO 1924 Paper and board—Determination of tensile strength.
- Tear strength: ISO 1974 Paper—Determination of tearing resistance.
- Optical properties: ISO 2469 Paper, board and pulps—Measurement of diffuse radiance factor (diffuse reflectance factor) and ISO 2470-1 Paper, board and pulps—Measurement of diffuse blue reflectance factor—Part 1: Indoor daylight conditions (ISO brightness).
- Air permeance: ISO 5636 Paper and board—Determination of air permeance and air resistance (medium range).
3.7. Water Vapor Transmission Rate
3.8. Contact Angle Measurement
3.9. Scanning Electron Microscopy
3.10. Time-of-Flight Secondary-Ion Mass Spectrometry
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BSTFA | N,O-Bis(trimethylsilyl)trifluoroacetamide |
| EI | Electron ionization |
| FID | Flame ionization detection |
| GC | Gas chromatography |
| ICP | Inductively coupled plasma |
| LIMS | Liquid metal ion source |
| MS | Mass spectrometry |
| o.d. | Oven dried |
| RH | Relative humidity |
| SEM | Scanning electron microscopy |
| SFA | Suberin fatty acid |
| TMCS | Chlorotrimethylsilane |
| TMS | Trimethylsilyl |
| ToF-SIMS | Time-of-flight secondary ion mass spectrometry |
| WVTR | Water vapor transmission rate |
References
- Eriksen, M.; Lebreton, L.C.M.; Carson, H.S.; Thiel, M.; Moore, C.J.; Borerro, J.C.; Galgani, F.; Ryan, P.G.; Reisser, J. Plastic Pollution in the World’s Oceans: More than 5 Trillion Plastic Pieces Weighing over 250,000 Tons Afloat at Sea. PLoS ONE 2014, 9, e111913. [Google Scholar] [CrossRef] [PubMed]
- Teuten, E.L.; Saquing, J.M.; Knappe, D.R.U.; Barlaz, M.A.; Jonsson, S.; Björn, A.; Rowland, S.J.; Thompson, R.C.; Galloway, T.S.; Yamashita, R.; et al. Transport and release of chemicals from plastics to the environment and to wildlife. Philos. T. R. Soc. B 2009, 364, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Hubbe, M.; Venditti, R.; Rojas, O. What happens to cellulosic fibers during papermaking and recycling? A review. Bioresources 2007, 2, 739–788. [Google Scholar]
- Kumar, H.; Christopher, L. Recent trends and developments in dissolving pulp production and application. Cellulose 2017, 24, 2347–2365. [Google Scholar] [CrossRef]
- Measuring Sustainability in Cotton Farming Systems: Towards a Guidance Framework. Available online: http://www.fao.org/3/a-i4170e.pdf (accessed on 26 November 2019).
- Sixta, H.; Iakolev, M.; Testova, L.; Roselli, A.; Hummel, M.; Borrega, M.; van Heiningen, A.; Froschauer, C.; Schottenberger, H. Novel concepts of dissolving pulp production. Cellulose 2013, 20, 1547–1561. [Google Scholar] [CrossRef]
- Hauru, L.K.J.; Hummel, L.; Nieminen, K.; Michud, A.; Sixta, H. Cellulose regeneration and spinnability from ionic liquids. Soft Matter 2016, 12, 1487–1495. [Google Scholar] [CrossRef]
- Holmbom, T.; Holmbom, B. A novel natural hydrophobisation technology utilising birch bark extractives. In Proceedings of the 5th Nordic Wood Biorefinery Conference (NWBC), Stockholm, Sweden, 25–27 March 2014; pp. 305–306. [Google Scholar]
- Huang, T.; Li, D.; Ek, M. Water repellency improvement of cellulosic textile fibers by betulin and a betulin-based copolymer. Cellulose 2018, 25, 2115–2128. [Google Scholar] [CrossRef]
- Forest Industries’ Wood Consumption. Available online: https://stat.luke.fi/en/wood-consumption (accessed on 28 October 2019).
- Rižikovs, J.; Zandersons, J.; Paže, A.; Tardenaka, A.; Spince, B. Isolation of Suberinic Acids from Extracted Outer Birch Bark Depending on the Application Purposes. Balt. For. 2014, 20, 98–105. [Google Scholar]
- Ekman, R. The Suberin Monomers and Triterpenoids from the Outer Bark of Betula verrucosa Ehrh. Holzforschung 1983, 37, 205–211. [Google Scholar] [CrossRef]
- Pinto, P.C.R.O.; Sousa, A.F.; Silvestre, A.J.D.; Neto, C.P.; Gandini, A.; Eckerman, C.; Holmbom, B. Quercus suber and Betula pendula outer barks as renewable sources of oleochemicals: A comparative study. Ind. Crop. Prod. 2009, 29, 126–132. [Google Scholar] [CrossRef]
- Li, D.; Iversen, T.; Ek, M. Hydrophobic materials based on cotton linter cellulose and anepoxy-activated polyester derived from a suberin monomer. Holzforschung 2015, 69, 721–730. [Google Scholar] [CrossRef]
- Karnaouri, A.; Rova, U.; Christakopoulos, P. Effect of Different Pretreatment Methods on Birch Outer Bark: New Biorefinery Routes. Molecules 2016, 21, 427. [Google Scholar] [CrossRef] [PubMed]
- Heinämäki, J.; Pirttimaa, M.M.; Alakurtti, S.; Pitkänen, H.P.; Kanerva, H.; Hulkko, J.; Paaver, U.; Aruväli, J.; Yliruusi, J.; Kogermann, K. Suberin Fatty Acids from Outer Birch Bark: Isolation and Physical Material Characterization. J. Nat. Prod. 2017, 80, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Garcia, H.; Sousa, A.F.; Freire, C.S.R.; Silvestre, A.J.D.; Rebeloa, L.P.N.; Pereira, C.S. Isolation of suberin from birch outer bark and cork using ionic liquids: A new source of macromonomers. Ind. Crop. Prod. 2013, 44, 520–527. [Google Scholar] [CrossRef]
- Miranda, I.; Gominho, J.; Mirra, I.; Pereira, H. Fractioning and chemical characterization of bark of Betula pendula and Eucalyptus globulus. Ind. Crop. Prod. 2013, 41, 299–305. [Google Scholar] [CrossRef]
- Graça, J. Suberin: The biopolyester at the frontier of plants. Front. Chem. 2015, 3, 1–11. [Google Scholar] [CrossRef]
- Graça, J.; Pereira, H. Methanolysis of Bark Suberins: Analysis of Glycerol and Acid Monomers. Phytochem. Anal. 2000, 11, 45–51. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Gaëtan, V.; Xu, L.; Beisson, F. Cutin and Suberin Polyesters. In Essential for Life Science (eLS); John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 1–12. [Google Scholar]
- Bernards, M. Demystifying suberin. Can. J. Bot. 2002, 80, 227–240. [Google Scholar] [CrossRef]
- Ekman, R.; Eckerman, C. Aliphatic carboxylic acids from suberin in birch outer bark by hydrolysis, methanolysis, and alkali fusion. Pap. Puu 1985, 67, 255–273. [Google Scholar]
- Gandini, A.; Neto, C.P.; Silvestre, A.J.D. Suberin: A promising renewable resource for novel macromolecular materials. Prog. Polym. Sci. 2006, 31, 878–892. [Google Scholar] [CrossRef]
- Sudakova, I.G.; Kuznetsov, B.N.; Garyntseva, N.V.; Pavlenko, N.I.; Ivanchenko, N.M. Functional and Thermal Analysis of Suberin Isolated from Birch Bark. J. Sib. Fed. Univ. Chem. 2008, 4, 355–362. [Google Scholar]
- Garcia, H.; Ferreira, R.; Petkovic, M.; Ferguson, J.L.; Leitão, M.C.; Gunaratne, H.Q.; Seddon, K.; Rebelo, L.; Silva Pereira, C. Dissolution of cork biopolymers in biocompatible ionic liquids. Green Chem. 2010, 12, 367–369. [Google Scholar] [CrossRef]
- Ferreira, R.; Garcia, H.; Sousa, A.F.; Petkovic, M.; Lamosa, P.; Freire, C.S.R.; Silvestre, A.J.D.; Rebelo, L.P.N.; Silva Pereira, C.S. Suberin isolation from cork using ionic liquids: Characterisation of ensuing products. New J. Chem. 2012, 36, 2014–2024. [Google Scholar] [CrossRef]
- Ferreira, R.; Garcia, H.; Sousa, A.F.; Guerreiro, M.; Duarte, F.J.S.; Freire, C.S.R.; Calhorda, M.J.; Silvestre, A.J.D.; Rebelo, L.P.N.; Silva Pereira, C. Unveiling the dual role of the cholinium hexanoate ionic liquid as solvent and catalyst in suberin depolymerisation. RSC Adv. 2014, 4, 2993–3002. [Google Scholar]
- Garcia, H.; Ferreira, R.; Martins, C.; Sousa, A.F.; Freire, C.S.R.; Silvestre, A.J.D.; Kunz, W.; Rebelo, L.P.N.; Silva Pereira, C. Ex Situ Reconstitution of the Plant Biopolyester Suberin as a Film. Biomacromolecules 2014, 15, 1806–1813. [Google Scholar] [CrossRef]
- Flynn, A. Biodegradable chewing gum bases and uses thereof. U.S. Patent Application US13/127,737, 3 November 2009. [Google Scholar]
- Mazo, P.C.; Estenoz, D.; Ríos, L.A. Kinetics of the esterification of maleic anhydride with castor oil. Lat. Am. Appl. Res. 2011, 41, 11–15. [Google Scholar]
- Miller, R.O. Microwave Digestion of Plant Tissue in a Closed Vessel. In Handbook of Reference Methods for Plant Analysis; Kalra, Y.P., Ed.; CRC Press: Boka Raton, FL, USA, 1998; pp. 69–73. [Google Scholar]
- Invercote GP + PET Coatings. Available online: https://www.iggesund.com/globalassets/iggesund/products/plastic-coatings/product-specifications/pet-coating-en-pdf/ (accessed on 30 November 2019).
- Invercote GP + PE Coatings. Available online: https://www.iggesund.com/globalassets/iggesund/products/plastic-coatings/product-specifications/invercote-pe-coating-en-pdf/ (accessed on 30 November 2019).
Sample Availability: No samples are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | SFA Fraction (mg g−1) | Betulinol Fraction (mg g−1) | Retention Time (min) | Kovats’ RI |
|---|---|---|---|---|
| Ferulic acid | 1.0 | 15.765 | 2073 | |
| 17:0 fatty acid (margaric acid) | 0.1 | 16.537 | 2141 | |
| 18:2 fatty acid (linoleic acid) | 1.0 | 17.153 | 2200 | |
| 18:1 fatty acid (oleic acid) | 0.4 | 17.254 | 2208 | |
| 18:0 fatty acid (stearic acid) | 0.2 | 17.600 | 2239 | |
| 16-hydroxy-16:0 acid | 2.7 | 19.601 | 2395 | |
| 20:0 - fatty acid (arachidic acid) | 0.5 | 19.208 | 2437 | |
| 1,16-dioic-16:0 acid | 3.5 | 19.957 | 2472 | |
| 18:-hydroxy-(9)18:1 acid | 62.4 | 2.0 | 20.778 | 2558 |
| 9,16- and 10,16-dihydroxy-16:0 acids | 19.7 | 21.032 | 2580 | |
| 18-hydroxy-18:0 acid | 1.5 | 21.087 | 2591 | |
| 1,18-dioic-(9)18:1 acid | 16.8 | 21.448 | 2633 | |
| 1,18-dioic-18:0 acid | 5.0 | 21.786 | 2669 | |
| 9,18-dihydroxy-(9)18:1 acid | 5.0 | 22.202 | 2713 | |
| 9,10-epoxy-18-hydroxy-18:0 acid | 198.0 | 2.3 | 22.496 | 2746 |
| 20-hydroxy-20:1 acid | 6.2 | 22.536 | 2752 | |
| Dihydroxyoctadecanoic acid | 4.4 | 22.721 | 2769 | |
| 20-hydroxy-20:0 acid | 16.5 | 0.9 | 22.852 | 2787 |
| 1,20-dioic-20:1 acid | 2.4 | 23.157 | 2828 | |
| 24:0 fatty acid (lignoceric acid) | 0.4 | 23.210 | 2834 | |
| 1,20-dioic-20:0 acid | 6.0 | 23.497 | 2866 | |
| 9,10,18-trihydroxy-18:0 acid | 70.2 | 0.6 | 23.769 | 2888 |
| 22-hydroxy-22:0 acid | 94.1 | 6.6 | 24.541 | 2988 |
| 1,22-dioic-22:0 acid | 7.7 | 25.116 | 3064 | |
| Cholesterol (standard) | - | - | 25.592 | 3151 |
| 24-hydroxy-24:0 acid | 2.8 | 25.990 | 3178 | |
| Lupenone (lup-20(29)-en-3-one) | 0.5 | 1.1 | 26.866 | 3330 |
| Sitosterol | 1.3 | 2.3 | 27.010 | 3347 |
| Lupeol | 3.8 | 50.8 | 27.310 | 3397 |
| Betulonic acid | 2.4 | 28.231 | 3529 | |
| Betulinol | 16.8 | 802.4 | 28.628 | 3574 |
| Betulinic acid | 53.1 | 23.8 | 28.799 | 3599 |
| Monogynol A (lupane-3b,20-diol) | 0.0 | 6.1 | 29.478 | 3706 |
| Lupane-3b,20,28-triol | 0.0 | 2.3 | 29.950 | 3766 |
| Total identified | 607.2 | 901.2 | ||
| Total eluted | 743.7 | 971.8 |
| SFA Fraction | Betulinol Fraction | ||
|---|---|---|---|
| Element | Content (mg kg−1) | Element | Content (mg kg−1) |
| S | 210 | Na | 4910 |
| Na | 77.2 | Mn | 42.3 |
| P | 29.8 | Mg | 22.7 |
| K | <10.2 | S | 22.1 |
| Ca | 3.83 | Fe | 18.8 |
| Pb | <1.02 | Ca | 17.7 |
| Cu | 0.782 | K | 17 |
| Al | 0.671 | P | 12.8 |
| B | 0.508 | Zn | 2.56 |
| Fe | 0.335 | Al | 2.38 |
| Zn | 0.305 | Pb | <1.02 |
| Mg | 0.213 | B | 0.969 |
| Cr | <0.203 | Cu | 0.948 |
| Ni | <0.203 | Cr | <0.204 |
| Cd | <0.07 | Ni | <0.204 |
| Mn | 0.061 | Cd | <0.07 |
| Grammage | Thickness | Density | Tear Index | Tensile Index | Brightness | Yellowness | WVTR | Air Permeance | |
|---|---|---|---|---|---|---|---|---|---|
| (g m−2) | (µm) | (kg m−3) | (mNm2 g−1) | (Nm g−1) | (%-ISO) | (%-ISO) | (g m−2d−2) | (ml min−1) | |
| Reference | 56.3 | 183.5 (8.1) | 306.7 (13.5) | 5.7 (0.4) | 11.5 (0.9) | 26.1 (0.3) | 51.4 (0.2) | 2523 (113) | 8820 |
| 0 g m−2 | 57.9 | 181.0 (6.0) | 319.9 (10.4) | 5.7 (0.2) | 14.0 (0.6) | 24.8 (0.1) | 56.6 (0.2) | 2577 (11) | 8820 |
| 10 g m−2 | 62.3 | 190.9 (8.4) | 326.4 (15.0) | 4.6 (0.2) | 16.6 (0.8) | 24.4 (0.1) | 60.5 (0.2) | 2576 (78) | 8820 |
| 20 g m−2 | 66.9 | 204.8 (4.6) | 326.8 (7.5) | 4.2 (0.1) | 16.7 (1.8) | 23.9 (0.3) | 61.6 (0.6) | 2755 (29) | 8820 |
| 30 g m−2 | 69.8 | 207.4 (2.8) | 336.7 (4.6) | 4.1 (0.4) | 16.0 (4.1) | 22.5 (1.9) | 63.2 (3.5) | 2847 (47) | 8820 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korpinen, R.I.; Kilpeläinen, P.; Sarjala, T.; Nurmi, M.; Saloranta, P.; Holmbom, T.; Koivula, H.; Mikkonen, K.S.; Willför, S.; Saranpää, P.T. The Hydrophobicity of Lignocellulosic Fiber Network Can Be Enhanced with Suberin Fatty Acids. Molecules 2019, 24, 4391. https://doi.org/10.3390/molecules24234391
Korpinen RI, Kilpeläinen P, Sarjala T, Nurmi M, Saloranta P, Holmbom T, Koivula H, Mikkonen KS, Willför S, Saranpää PT. The Hydrophobicity of Lignocellulosic Fiber Network Can Be Enhanced with Suberin Fatty Acids. Molecules. 2019; 24(23):4391. https://doi.org/10.3390/molecules24234391
Chicago/Turabian StyleKorpinen, Risto I., Petri Kilpeläinen, Tytti Sarjala, Maristiina Nurmi, Pauliina Saloranta, Thomas Holmbom, Hanna Koivula, Kirsi S. Mikkonen, Stefan Willför, and Pekka T. Saranpää. 2019. "The Hydrophobicity of Lignocellulosic Fiber Network Can Be Enhanced with Suberin Fatty Acids" Molecules 24, no. 23: 4391. https://doi.org/10.3390/molecules24234391
APA StyleKorpinen, R. I., Kilpeläinen, P., Sarjala, T., Nurmi, M., Saloranta, P., Holmbom, T., Koivula, H., Mikkonen, K. S., Willför, S., & Saranpää, P. T. (2019). The Hydrophobicity of Lignocellulosic Fiber Network Can Be Enhanced with Suberin Fatty Acids. Molecules, 24(23), 4391. https://doi.org/10.3390/molecules24234391