1. Introduction
The average number of new drugs approved by the US Food and Drug Administration (FDA) per year has declined since the 1990s, while costs have steadily increased over the years [
1]. Specifically, anti-cancer drugs show some of the highest attrition rates among all the therapeutic areas [
2]. Many authors argue that current in vitro models fail to mimic relevant cues in the microenvironment, which in turn impacts the results of drug-testing efficiency [
3]. Lately, the field has emphasized the need for improving traditional drug testing platforms to provide an increased correlation to clinical success [
4]. To address these concerns, researchers have speculated that a more physiologically relevant in vitro culture environment would encourage cells to behave more similarly to cells grown in vivo, therefore increasing the predictability of in vitro models [
5].
Breast cancer is the most common cancer in women, affecting one in eight women during their lifetime [
6]. Breast cancer originates in the mammary duct, and eventually tumor cells break the wall of the lumen and invade the surrounding tissue. Despite their common origin, breast tumors are highly heterogeneous from both a histopathological (e.g., ductal carcinoma in situ, invasive ductal carcinoma) and molecular (e.g., HER-2
+, PR
+, ER
+) perspective. Additionally, microenvironment alterations including hypoxia, presence of stromal cells critically influence cancer development, and progression are highlighted in breast cancer [
7,
8,
9,
10]. The vast number of parameters involved in breast cancer evolution requires new research tools that control costs yet allow for multiple replicates of microenvironmental conditions. In vitro models are well suited for this high-throughput analysis, but polystyrene-based Petri dishes that are traditionally used for in vitro studies do not reliably capture tissue complexity [
11]. In this context, microfluidic-based models offer great potential to mimic the tissue architecture. More specifically, the use of microfluidic platforms to generate breast ductal structures can help reproduce the breast cancer microenvironment [
12,
13]. Using such platforms, we have previously studied the metabolic adaptations undergone by tumor cells inside of the mammary duct; the effect of fibroblasts in breast cancer drug response; and the impact of obesity and adipocytes in hormonal therapy [
14,
15,
16]. However, the platform described in these studies, as well as most microdevices developed by other groups, are based on polydimethylsiloxane (PDMS) [
17]. Although PDMS has excellent optical clarity, flexibility, and gas permeation properties, PDMS can nonspecifically absorb small molecules, including certain drugs [
17]. Polystyrene (PS) presents a more promising alternative due to its mass-production potential, impermeability to small hydrophobic molecules, and biocompatibility [
18].
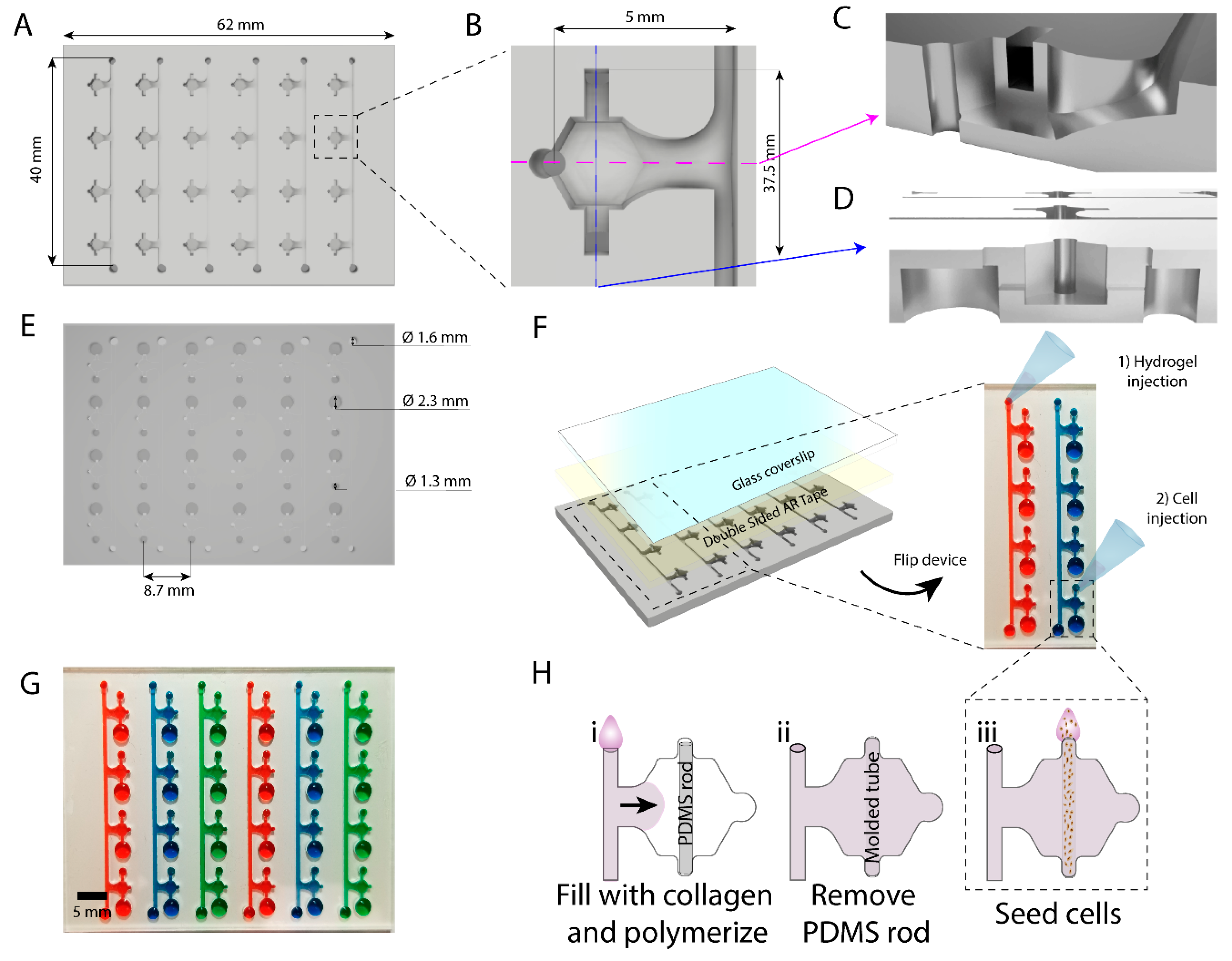
In this work we have developed an array of microdevices in polystyrene using micro-milling techniques for fabrication. This microfluidic array allows the generation of multiple luminal structures embedded in a 3D extracellular matrix for high-throughput screening. We demonstrated a lower diffusion of hydrophobic molecules into the PS device as compared to a similar comparable PDMS-based microdevice. As a proof of concept, we filled our device with collagen I and lined our resulting luminal structures with breast cancer cells within each device in the array. We monitored breast cancer invasion in the array by using co-cultures of cell lines with varying invasion potential. Additionally, we tested the model for use drug screening exposing the in vitro lumen models to the chemotherapy agent doxorubicin. Finally, we performed photodynamic therapy (PDT) in the lumen. PDT is based on the use of compounds that become toxic after light exposure (i.e., photosensitizers), allowing for a more localized delivery of active compounds, selectively targeting the tumor, and minimizing side effects in the normal tissue and other organs.
3. Methods
3.1. Cell Culture
MCF10A human mammary epithelial cells were obtained from ATCC (ATCC®CRL-10317™). MCF7 human mammary epithelial cells from metastatic site were obtained for ATCC as well (ATCC® HTB-22™) Human mammary adenocarcinoma cells, MDA-MB-231, both wild type and transfected to stably expressing green fluorescent protein (GFP), were a kind gift from Dr. Suzanne Ponik (University of Wisconsin, Madison, WI 53705, USA). All cells were maintained with RPMI base media (Gibco, 11875) with 10% fetal bovine serum (FBS, serum (VWR, 97068-085) and 1% penicillin/streptomycin (ThermoFisher, 15140-122, Grand Island, NY, USA) on cell culture-treated flasks (Corning, 156499, Oneonta, NY, USA). Supplemented RPMI media is referred to as relevant media in the rest of the paper. Cells were harvested via standard trypsinization. Briefly, cells were washed with PBS 1× (diluted from 10× with distilled water, Thermo Fisher BP3991), incubated with a 0.25% trypsin/EDTA solution (LifeTechnologies, 25200056, Fitchburg, WI, USA) for 5 min. Trypsin was inactivated with relevant media. Next, cells were pelleted at 300 g for 4 min and finally resuspended to the desired concentration for subsequent experiments.
3.2. PDMS Device Fabrication
Standard SU-8 photolithography was used to create the device architecture as previously described [
13]. PDMS was mixed at a 10:1 polymer:curing agent ratio, poured onto SU-8 masters and used to fill 23G needles (BD, 305145). PDMS mixture was baked for 4 h at 80 °C. After baking, PDMS was extracted from the needles to form sacrificial PDMS rods. PDMS layers were detached from the SU-8 masters, ethanol bonded together with the sacrificial PDMS rod in place. Assembled devices were then plasma bonded to a 0.9 mm thick glass coverslip. To ensure collagen adhesion to PDMS surface, the devices were treated with a 0.2% aqueous poly(ethyleneimine) (PEI, Millipore-Sigma, 408727, St Louis, MO, USA) solution for 10 min, then with a 0.04% aqueous glutaraldehyde (GA, Millipore-Sigma, 354400, St Louis, MO, USA) solution for 30 min, then aspirated. To rinse residual PEI and GA, the devices were washed with deionized water three times.
3.3. Polystyrene Device Fabrication
Devices were micro-milled on a CNC mill out of sheets of 2 mm polystyrene (PS) with a 0.02 inch end-mill using a two-sided milling technique. We milled the hydrogel filling channel, the connection channels and all the devices into one side of the 2 mm thick PS sheet. Then, the platform was flipped upside-down and milled on the other surface. On this side of the PS, the ports for the devices and the filling channels were milled. Cell seeding ports (small and large) were milled at 1.5 mm-depth. The cell seeding ports and body of the device vertically overlapped by 0.5 mm, making room for the sacrificial PDMS rod to be suspended and therefore not in contact with the bottom surface of the platform. Milled devices were rinsed thoroughly with deionized water and dried with a high-pressure air stream. Sacrificial PDMS rods were loaded into individual lumen devices using fine tweezers. The flexibility of PDMS is required for the functionality of the rods, and therefore their fabrication with PS was not considered. The back of the PS devices was exposed to oxygen plasma to render the surface hydrophilic. Double-sided AR tape (AR care8890) was cut to the size of the PS device and adhered to a coverslip (50 × 75 mm, Corning 2947-75X50) to allow for a complete seal and optical transparency. Devices were flipped in order to access loading ports for further use.
3.4. Rhodamine B Diffusion
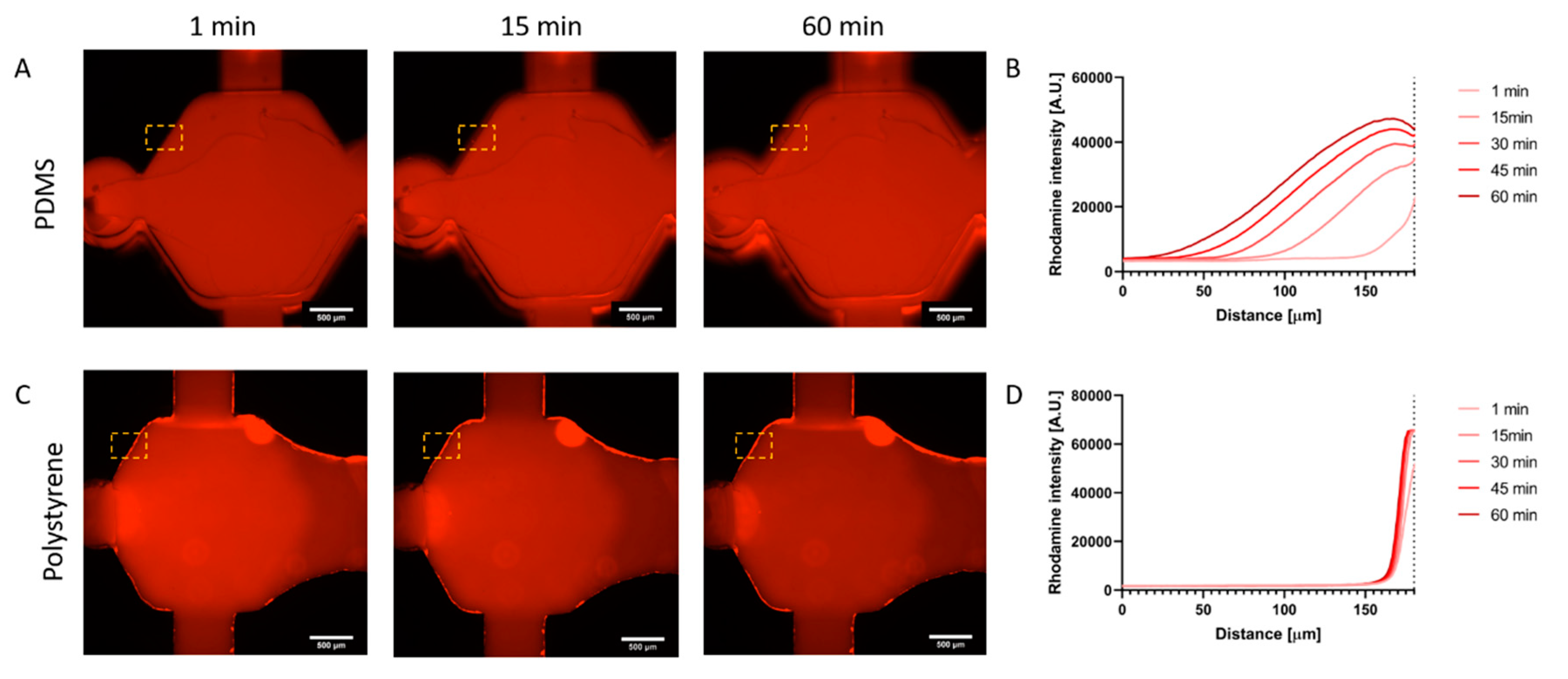
Rhodamine B (Millipore-Sigma, 83689, St Louis, MO, USA) was dissolved at 5 mg/mL in distilled water, and later diluted 1:100 in PBS. PDMS and PS fabricated devices were both filled with the Rhodamine B solution. Rhodamine B fluorescence was tracked via fluorescent microscopy for 1 h. The solution was then incubated at room temperature for 48 h and imaged again. Finally, the Rhodamine B solution was aspirated, and devices were washed thrice for 20 min each time before a final fluorescence image acquisition. ImageJ software was used to generate fluorescence plot profiles in the regions of interest (ROI) located at the edge of the chamber, and into the material.
3.5. Collagen Loading in Devices
200 µl of Rat Tail Collagen I, High Concentration (Corning, 354249, Bedford, MA, USA, stock at 8.28 mg/mL) was neutralized with 5 µl of 0.5 M NaOH (Sigma, S5881, St Louis, MO, USA) and diluted to 4.5 mg/mL. Relevant osmolarity was achieved using 40 µl PBS10× (Fisher Scientific, BP3991, Fair Lawn, NJ, 07410, USA) with 155 µL of sterile distilled water. The mixture was kept on ice for 20 min prior to loading the devices. pH was measured using pH Test Strips (Whatman, 2629-990, Little Chalfont, UK), and only mixtures with pH values of 7–7.4 were used for experiments. PS devices were thoroughly rinsed with deionized water twice and dried using vacuum aspiration. During collagen loading, the devices were placed on a metal block on top of ice to prevent the collagen solution from polymerizing until the devices were fully filled. Approximately 140 µL of collagen solution were injected in each line port to fill the connected line of lumen devices, whereas 10 µL were used per PDMS lumen device. Once all devices were filled, they were left to polymerize at room temperature for 10 min. After that, the devices were transferred to an omni-tray plate (Thermo, 242811, Rochester, NY, USA) and incubated at 37 °C for 30 min prior to cell loading.
3.6. Vybrant Staining
MCF10a cells were stained in red with the fluorescent lipid Dil (Thermo Fisher, V22889, Eugene, OR, USA) according to manufacturer’s instructions. Briefly, MFC10a cells were resuspended at 5 cells million/mL in PBS. DiI was added at a 1:200 dilution from the stock and incubated at 37 °C protected from light for 10 min. Next, cells were pelleted and washed twice with PBS before resuspending in relevant media at the desired concentration (20 million cells/mL).
3.7. Cell Loading into Lumen Devices
Sacrificial PDMS rods were removed from each device and the resulting hollow lumen was filled with relevant media. Cells were resuspended at the desired concentration of 6 million/mL. Three microliters of the cell solution were pipetted into the small port connected to the luminal geometry and passively pumped through the lumen. Cultures were incubated at 37 °C for 15 min at a time and flipped 3 times (a total of 1 h of incubation) so that the cells adhered to the entire surface of luminal geometry. After, excess media was gently aspirated to remove unattached cells. Devices were cultured overnight at 37 °C, and media was replenished immediately the following morning. Experimental conditions were introduced 24 h after seeding, replenishing every 24 h.
3.8. Cell Viability
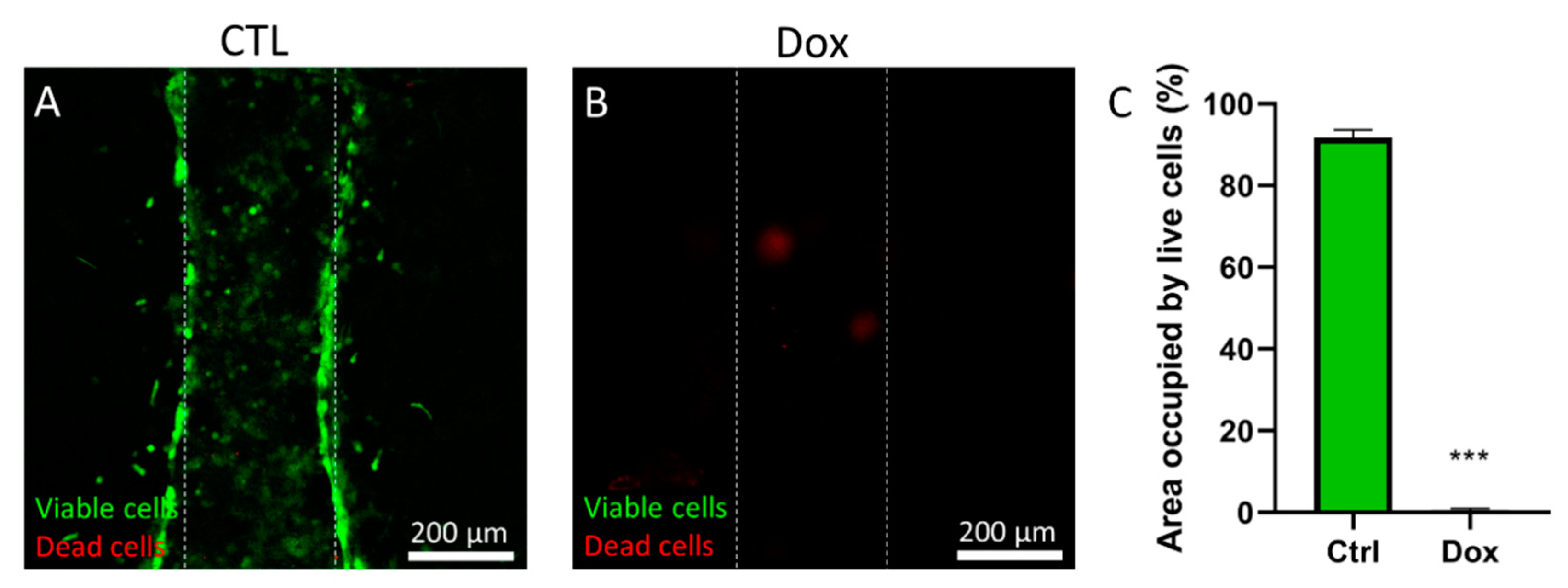
Calcein AM (Thermo Fisher, C1430, Eugene, OR, USA) and propidium iodide (Millipore Sigma, P4170, St Louis, MO, USA) were resuspended in DMSO at 5 mM and 2 mg/mL respectively, and then diluted in PBS to final concentrations of 5 µM and 2 µg/mL respectively. The cells were stained in this solution for 30 min at 37 °C prior to confocal imaging with an SP8 Leica microscope with stimulated emission depletion microscopy (STED) module. Green fluorescence was detected using an excitation laser 485 nm, emission filter 495–525 nm. Red fluorescence was detected using an excitation laser, 532 nm, 540–600 nm. The number of propidium iodide positive (dead, red) cells were counted and divided by the total number of cells (calcein AM positive, green; and PI positive cells, red) to calculate the percentage of viable cells.
3.9. Invasion Assay
MCF10a cells were stained with DiI as described in previous sections and resuspended at 20 million cells/mL. GFP-fluorescent MDA-MB-231 cells were resuspended at 6 million/mL. Both cell suspensions were mixed 1:1 and loaded through the lumens in PS devices prepared as previously described. Devices were imaged with a confocal microscope (Leica SP8 with STED module) at 24 and 48 h. Cell invasion was quantified as the area occupied by cells in different ROIs defined in the captured field with ImageJ, both for green and red fluorescence.
3.10. Photodynamic Therapy Experiment
PS lumen devices were prepared as described above to create tubular void structures embedded in hydrogel. GFP fluorescent MDA-MB-231 cells were resuspended at 5 million cells/mL and loaded into the lumen structure as described above and left to adhere for 24 h. Verteporfin (Selleckchem, S1786, Pittsburgh, PA, USA), was dissolved at 25 mg/mL in DMSO, and later diluted 1:50,000 (to a final concentration of 500 ng/mL) in relevant media. Verteporfin solution was perfused through the lumen and incubated at 37 °C for 1 h. Thereafter, cells in the lumen were exposed to fluorescent light using a Nikon Eclipse Ti microscope and a 485/35 nm filter for 45 s. After the light activation of verteporfin, cells were left in the incubator overnight. Viability was assessed via propidium iodide staining as described in subsequent sections, since cells were already fluorescently green, calcein was not required to stain live cells. Images were analyzed using ImageJ by plotting the fluorescence profile in three different region of interests (ROIs) in the image within the lumen (at the beginning, center and end of the lumen along the axis). The area under the curve was calculated and normalized to the highest value (among green and red) to demonstrate which of the fluorescence channels was more prevalent in each section (and ROI) of the lumen.
3.11. Doxorubicin Cytotoxicity Assay
Doxorubicin was dissolved in DMSO at 100 mM, and then dissolved 1:1000 in culture media. PS microarrays were prepared as previously described and MDA-MB-231 were loaded in the lumen at 5 million cells/mL. Cells were left to adhere for 24 h and then the doxorubicin solution was added and incubated for 72 h. Then, viability was measured using calcein AM and propidium iodide as described in the following section.
3.12. Statistical Analysis
All the experiments were repeated at least three times as independent biological replicates. All results are presented as the mean ± standard deviation. Data were analyzed using GraphPad Prism v7 and statistical significance was set at p < 0.05. First, normal distribution was assessed by the Shapiro-Wilk test. If the normality test was passed, a Student’s T-test was performed. Multiple comparisons by ANOVA were corrected using Tukey’s tests.
4. Conclusions
The pharmaceutical industry now relies on high throughput systems to test thousands of compounds a year. However, most used systems have shown to produce confounding results that do not fully recapitulate in vivo effects of drugs. On the other hand, microfluidics can provide very useful models that recapitulate functions of in vivo systems more accurately, therefore providing with great opportunities for more relevant in vitro drug testing. However, most microfluidic devices rely on PDMS, which have important drawbacks for drug testing. Among these drawbacks, the use of highly hydrophobic materials, which could impact the effective concentration of molecules (e.g., drugs) in the cell culture system; and the labor-intensive protocols are important limitations for potential users to adopt microfluidic drug testing. In an effort to increase the throughput of microfluidic organotypic models of breast cancer, as well as decrease user handling, we have developed a lumen array platform made of polystyrene. We have leveraged microfluidic physical principles (i.e., capillarity, passive pumping), to decrease user operation compared to traditional PDMS-based microfluidic devices and increase throughput by incorporating a side channel to fill several microdevices with hydrogel solution at the same time. Compared to PDMS, PS has low retention capabilities of lipophilic molecules and is widely accepted among in vitro-focused researchers. We have illustrated this premise by tracking Rhodamine B diffusion in our microfluidic lumen array, revealing minimal Rhodamine B penetration in the PS-based device. Likewise, we have demonstrated three cell-based applications for breast cancer in this platform. First, we have assessed cancer cell invasion in a model of both normal (MCF10a) and invasive breast cancer cells (MDA-MB-231) and demonstrated a higher invasiveness of MDA-MB-231 cells in the surrounding matrix. Next, we have used doxorubicin, a common chemotherapeutic, and assessed its cytotoxicity in a lumen model of breast cancer cells. Finally, we have leveraged our model to test the cytotoxicity and reach of photodynamic therapy (PDT) with verteporfin, showing a gradient of cell cytotoxicity along the lumen.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




