Green Synthesis and Electrochemical Properties of Mono- and Dimers Derived from Phenylaminoisoquinolinequinones

 and
and
Abstract
1. Introduction
2. Results
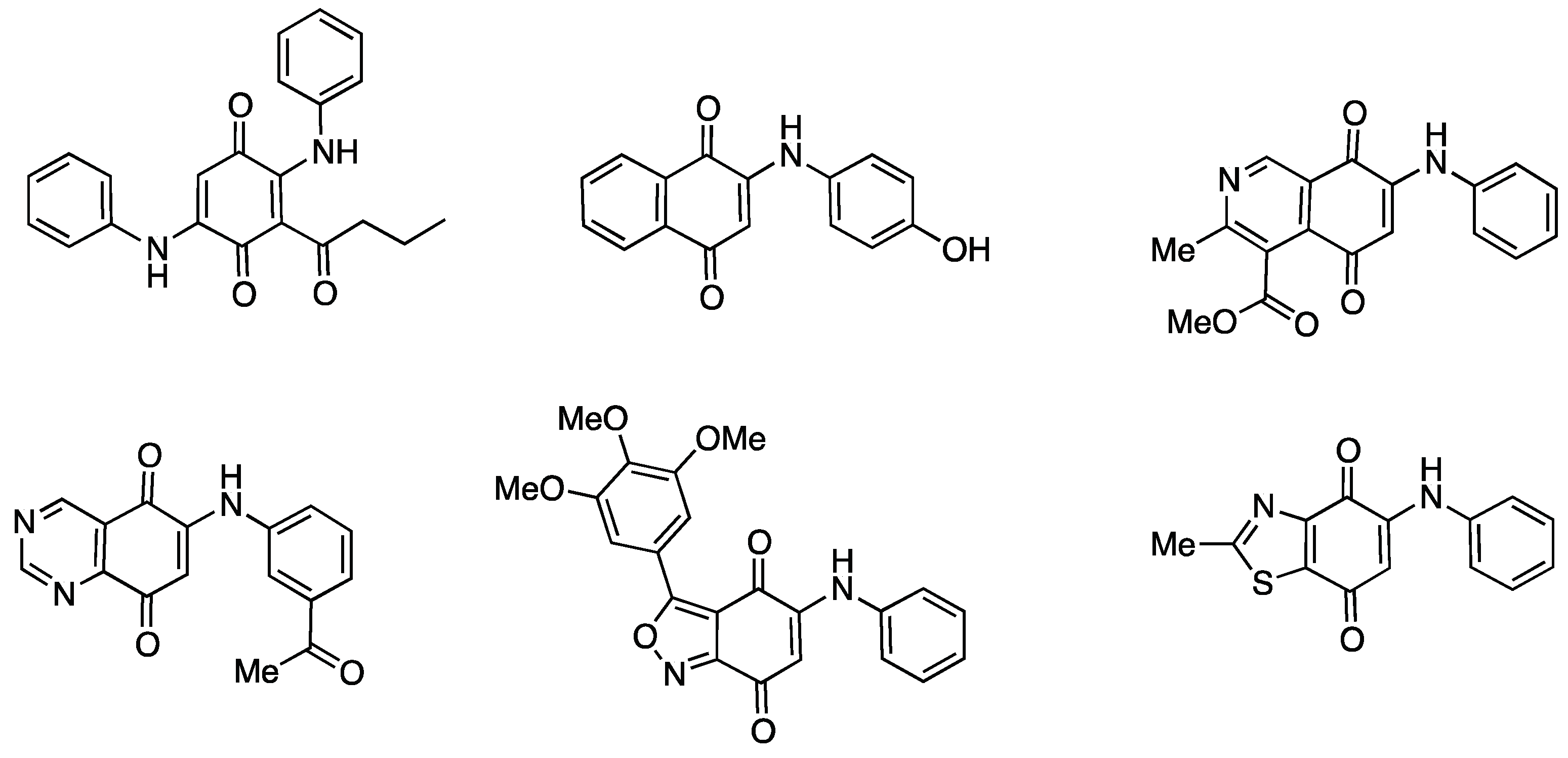
2.1. Chemistry
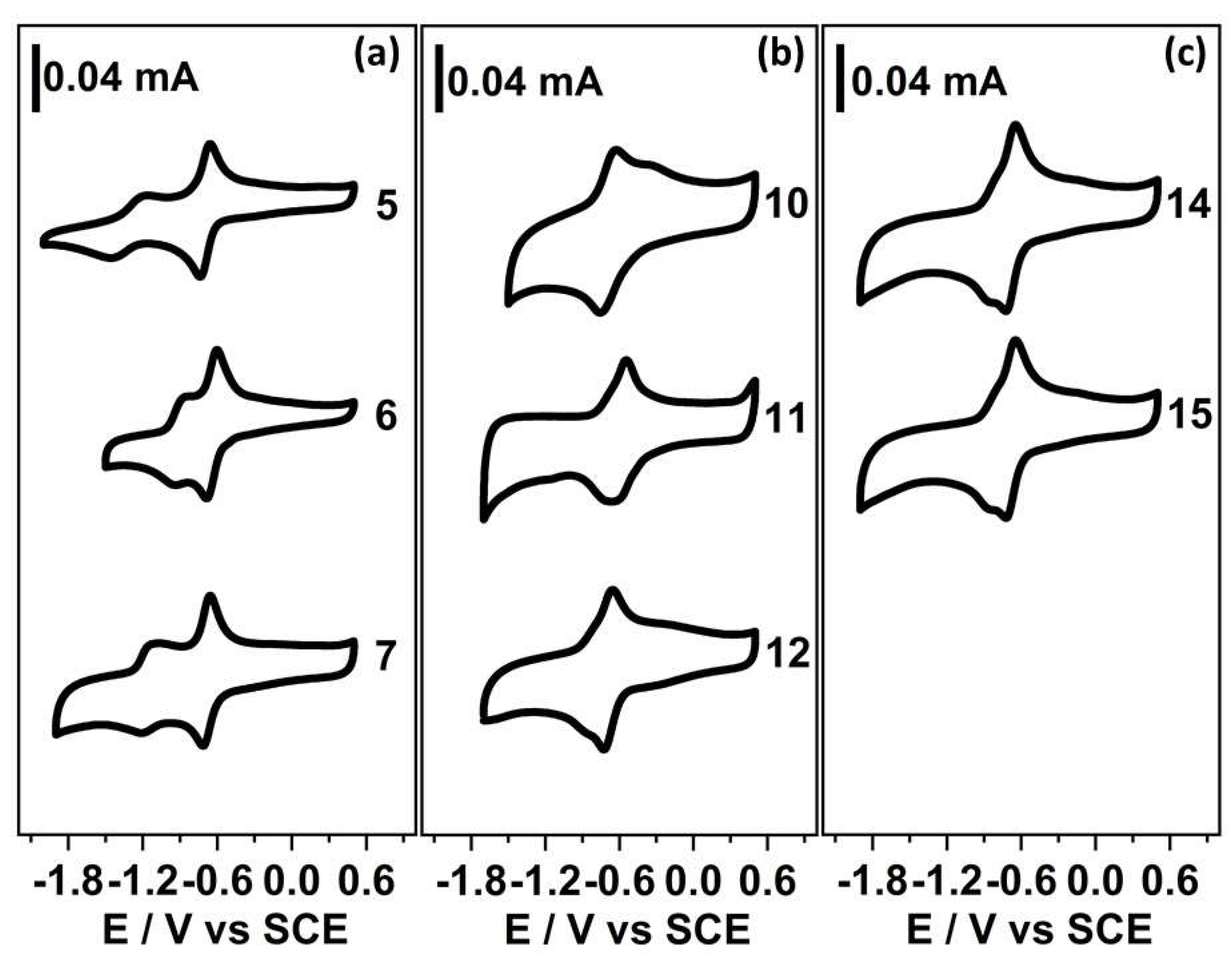
2.2. Electrochemistry
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. Preparation of Monoamination Compounds 5–7. General Procedure
3.2.2. Preparation of Heterodimers 10–12. General Procedure
3.2.3. Preparation of Homodimers 13–15. General Procedure
3.3. Electrochemical Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DDM | 4:4′-Diaminodiphenylmethane |
| IR | Infrared |
| 2D-NMR | Bidimensional Nuclear Magnetic Resonance |
| HRMS | High Resolution Mass Spectroscopy |
| SCE | Calomel Saturated Electrode |
| TBAP | Tetrabutylammonium perchlorate |
| MIC | Minimal Inhibitory Concentration |
References
- Thomson, R.H. Naturally Occurring Quinones III. Recent Advances, 3rd ed.; Chapman and Hall: Lincoln, UK, 1987. [Google Scholar]
- Sanchez-cruz, P.; Alegrıa, A.E. Quinone-Enhanced Reduction of Nitric Oxide by Xanthine/Xanthine Oxidase. Chem. Res. Toxicol. 2009, 22, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Esau, L.; Moosa, B.; Khashab, N.M.; Bajic, V.B.; Kaur, M. Cytotoxicity and Apoptosis Induced by a Plumbagin Derivative in Estrogen Positive MCF-7 Breast Cancer Cells. Anticancer. Agents Med. Chem. 2014, 14, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Simic, M.G.; Bergtold, D.S.; Karam, L.R. Generation of oxy radicals in biosystems. Mutat. Res. 1989, 214, 3–12. [Google Scholar] [CrossRef]
- Bolton, J.L.; Trush, M.A.; Penning, T.M.; Dryhurst, G.; Monks, T.J. Role of Quinones in Toxicology. Chem. Res. Toxicol. 2000, 13, 135–160. [Google Scholar] [CrossRef]
- Powis, G. Metabolism and reactions of quinoid anticancer agents. Pharmacol. Ther. 1987, 35, 57–162. [Google Scholar] [CrossRef]
- O’Brien, P.J. Molecular mechanisms of quinone cytotoxicity. Chem. Biol. Interact. 1991, 80, 1–41. [Google Scholar] [CrossRef]
- Paz, M.M.; Das, A.; Palom, Y.; He, Q.; Tomasz, M. Selective Activation of Mitomycin A by Thiols To Form DNA Cross-links and Monoadducts: Biochemical Basis for the Modulation of Mitomycin Cytotoxicity by the Quinone Redox Potential. J. Med. Chem. 2001, 44, 2834–2842. [Google Scholar] [CrossRef]
- Tudor, G.; Gutierrez, P.; Aguilera-Gutierrez, A.; Sausville, E.A. Cytotoxicity and apoptosis of benzoquinones: Redox cycling, cytochrome c release, and BAD protein expression. Biochem. Pharmacol. 2003, 65, 1061–1075. [Google Scholar] [CrossRef]
- Wellington, K.W. Understanding cancer and the anticancer activities of naphthoquinones—A review. RSC Adv. 2015, 26, 20309–20338. [Google Scholar] [CrossRef]
- Li, K.; Wang, B.; Zheng, L.; Yang, K.; Li, Y.; Hu, M.; He, D. Target ROS to induce apoptosis and cell cycle arrest by 5,7-dimethoxy-1,4-naphthoquinone derivative. Bioorg. Med. Chem. Lett. 2018, 28, 273–277. [Google Scholar] [CrossRef]
- Jiang, X.; Meng, L.; Wang, Y.; Zhang, Y. Novel 1,4-Naphthoquinone derivatives induce apoptosis via ROS-mediated p38/MAPK, Akt and STAT3 signaling in human hepatoma Hep3B cells. Int. J. Biochem. Cell. Biol. 2018, 96, 9–19. [Google Scholar] [CrossRef]
- Monks, T.; Jones, D. The Metabolism and Toxicity of Quinones, Quinonimines, Quinone Methides, and Quinone-Thioethers. Curr. Drug. Metab. 2002, 3, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Monks, T.J.; Hanzlik, R.P.; Cohen, G.M.; Ross, D.; Graham, D.G. Quinone chemistry and toxicity. Toxicol. Appl. Pharmacol. 1992, 112, 2–16. [Google Scholar] [CrossRef]
- Kim, H.J.; Mun, J.Y.; Chun, Y.J.; Choi, K.H.; Ham, S.W.; Kim, M.Y. Effects of a naphthoquinone analog on tumor growth and apoptosis induction. Arch. Pharm. Res. 2003, 26, 405–410. [Google Scholar] [CrossRef]
- Kondracki, M.-L.; Guyot, M. Smenospongine: A cytotoxic and antimicrobial aminoquinone isolated from sp. Tetrahedron Lett. 1987, 28, 5815–5818. [Google Scholar] [CrossRef]
- Kong, D.; Yamori, T.; Kobayashi, M.; Duan, H. Antiproliferative and Antiangiogenic Activities of Smenospongine, a Marine Sponge Sesquiterpene Aminoquinone Dexin. Mar. Drugs 2011, 9, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Dale, L.; Yasuda, M.; Steven, D. Streptonigrin and Lavendamycin Partial Structures. Probes for the Minimum, Potent Pharmacophore of Streptonigrin, Lavendamycin, and Synthetic Quinoline-5,8-diones. J. Med. Chem. 1987, 30, 1918–1928. [Google Scholar] [CrossRef]
- Rao, K.V.; Cullen, W.P. Streptonigrin, an antitumor substance. I. Isolation and characterization. In Antibiotics Annual 1959–1960; Welch, H., Martí-Ibáñez, F., Eds.; Medical Encyclopedia, Inc.: New York, NY, USA, 1960. [Google Scholar]
- Rao, K.V.; Biemann, K.; Woodward, R.B. The structure of streptonigrin. J. Am. Chem. Soc. 1963, 85, 2532–2533. [Google Scholar] [CrossRef]
- Harding, M.M.; Long, G.V.; Brown, C.L. Solution Conformation of the Antitumor Drug Streptonigrin. J. Med. Chem. 1993, 36, 3056–3060. [Google Scholar] [CrossRef]
- Hawas, U.W.; Shaaban, M.; Shaaban, K.A.; Speitling, M.; Maier, A.; Kelter, G.; Fiebig, H.H.; Meiners, M.; Helmke, E.; Laatsch, H. Mansouramycins A–D, Cytotoxic Isoquinolinequinones from a Marine Streptomycete. J. Nat. Prod. 2009, 72, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Liu, G.; Cao, R.; Zhang, L.; Yu, Q.; Sun, C. Mansouramycin C kills cancer cells through reactive oxygen species production mediated by opening of mitochondrial permeability transition pore. Oncotarget 2017, 8, 104057–104071. [Google Scholar] [CrossRef] [PubMed]
- Benites, J.; Valderrama, J.A.; Ramos, M.; Valenzuela, M.; Guerrero-castilla, A.; Muccioli, G.G.; Calderon, P.B. Half-Wave Potentials and In Vitro Cytotoxic Evaluation of 3-Acylated 2,5-Bis(phenylamino)-1,4- benzoquinones on Cancer Cells. Molecules 2019, 24, 1780. [Google Scholar] [CrossRef] [PubMed]
- Benites, J.; Valderrama, J.A.; Bettega, K.; Curi, R.; Buc, P.; Verrax, J. Biological evaluation of donor-acceptor aminonaphthoquinones as antitumor agents. Eur. J. Med. Chem. 2010, 45, 6052–6057. [Google Scholar] [CrossRef] [PubMed]
- Tandon, V.K.; Chhor, R.B.; Singh, R.V.; Yadav, D.B. Design, synthesis and evaluation of novel 1,4-naphthoquinone derivatives as antifungal and anticancer agents. Bioorg. Med. Chem. Lett. 2004, 14, 1079–1083. [Google Scholar] [CrossRef]
- Francisco, A.I.; Casellato, A.; Neves, A.P.; Carneiro, J.W.d.M.; Vargas, M.D.; Visentin, L.do.C.; Magalhães, A.; Câmara, C.A.; Pessoa, C.; Costa-Lotufo, L.V.; et al. Novel 2-(R-phenyl)amino-3-(2-methylpropenyl)-[1,4]-naphthoquinones: Synthesis, characterization, electrochemical behavior and antitumor activity. J. Braz. Chem. Soc. 2010, 21, 169–178. [Google Scholar] [CrossRef]
- Ibacache, J.A.; Delgado, V.; Benites, J.; Theoduloz, C.; Arancibia, V.; Muccioli, G.G.; Valderrama, J.A. Synthesis, Half-Wave Potentials and Antiproliferative Activity of 1-Aryl-substituted Aminoisoquinolinequinones. Molecules 2014, 19, 726–739. [Google Scholar] [CrossRef]
- Pathania, D.; Kuang, Y.; Sechi, M.; Neamati, N. Mechanisms underlying the cytotoxicity of a novel quinazolinedione-based redox modulator, QD232, in pancreatic cancer cells. Br. J. Pharmacol. 2014, 172, 50–63. [Google Scholar] [CrossRef]
- Benites, J.; Valderrama, J.A.; Ramos, M.; Muccioli, G.G. Targeting Akt as strategy to kill cancer cells using 3-substituted 5- anilinobenzo[c]isoxazolequinones: A preliminary study. Biomed. Pharmacother. 2018, 97, 778–783. [Google Scholar] [CrossRef]
- Ryu, C.; Kang, H.; Lee, K.; Nam, A. 5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as Inhibitors of Cyclin-Dependent Kinase 4 and Cytotoxic Agents. Bioorg. Med. Chem. Lett. 2000, 10, 461–464. [Google Scholar] [CrossRef]
- Kongkathip, N.; Kongkathip, B.; Siripong, P.; Sangma, C.; Luangkamin, S.; Niyomdecha, M.; Pattanapa, S.; Piyaviriyagul, S.; Kongsaeree, P. Potent antitumor activity of synthetic 1,2-naphthoquinones and 1,4-naphthoquinones. Bioorganic. Med. Chem. 2003, 11, 3179–3191. [Google Scholar] [CrossRef]
- Bonifazi, E.L.; Ríos-Luci, C.; León, L.G.; Burton, G.; Padrón, J.M.; Misico, R.I. Antiproliferative activity of synthetic naphthoquinones related to lapachol. First synthesis of 5-hydroxylapachol. Bioorganic Med. Chem. 2010, 18, 2621–2630. [Google Scholar] [CrossRef] [PubMed]
- Duchowicz, P.R.; Bennardi, D.O.; Bacelo, D.E.; Bonifazi, E.L.; Rios-Luci, C.; Padrón, J.M.; Burton, G.; Misico, R.I. QSAR on antiproliferative naphthoquinones based on a conformation- independent approach. Eur. J. Med. Chem. 2014, 77, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Baiju, T.V.; Almeida, R.G.; Sivanandan, S.T.; de Simone, C.A.; Brito, L.M.; Cavalcanti, B.C.; Pessoa, C.; Namboothiri, I.N.N.; da Silva Júnior, E.N. Quinonoid compounds via reactions of lawsone and 2-aminonaphthoquinone with α- bromonitroalkenes and nitroallylic acetates: Structural diversity by C-ring modification and cytotoxic evaluation against cancer cells. Eur. J. Med. Chem. 2018, 151, 686–704. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, J.A.; Cabrera, M.; Benites, J.; Ríos, D.; Inostroza-Rivera, R.; Muccioli, G.G.; Calderon, P.B. Synthetic approaches and: In vitro cytotoxic evaluation of 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinones. RSC. Adv. 2017, 7, 24813–24821. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Andrea, I.J.; Arancibia, V.; Rodriguez, J.; Theoduloz, C. Studies on quinones. Part 45: Novel 7-aminoisoquinoline-5,8-quinone derivatives with antitumor properties on cancer cell lines. Bioorganic Med. Chem. 2009, 17, 2894–2901. [Google Scholar] [CrossRef]
- Delgado, V.; Ibacache, A.; Theoduloz, C.; Valderrama, J.A. Synthesis and in vitro cytotoxic evaluation of aminoquinones structurally related to marine isoquinolinequinones. Molecules 2012, 17, 7042–7056. [Google Scholar] [CrossRef]
- Aguilar-Martinez, M.; Cuevas, G.; Jimenez-Estrada, M.; Gonzalez, I.; Lotina- Hennsen, B.; Macias-Ruvalcaba, N. An experimental and theoretical study of the substituent effects on the redox properties of 2-[(R-phenyl)-amine]-1,4-naphthalenediones in acetonitrile. J. Org. Chem. 1999, 64, 3684–3694. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Banzi, R.; Bartolini, M.; Cavalli, A.; Tarozzi, A.; Andrisano, V.; Minarini, A.; Rosini, M.; Tumiatti, V.; Bergamini, C.; et al. Novel class of quinone-bearing polyamines as multi-target-directed ligands to combat Alzheimer’s disease. J. Med. Chem. 2007, 50, 4882–4897. [Google Scholar] [CrossRef]
- Bernardo, P.H.; Khanijou, J.K.; Lam, T.H.; Tong, J.C.; Chai, C.L.L. Synthesis and potent cytotoxic activity of 8- and 9-anilinophenanthridine- 7,10-diones. Tetrahedron Lett. 2011, 52, 92–94. [Google Scholar] [CrossRef]
- Sanna, V.; Nurra, S.; Pala, N.; Marceddu, S.; Pathania, D.; Neamati, N.; Sechi, M. Targeted Nanoparticles for the Delivery of Novel Bioactive Molecules to Pancreatic Cancer Cells. J. Med. Chem. 2016, 59, 5209–5220. [Google Scholar] [CrossRef] [PubMed]
- Lisboa, C.D.S.; Santos, V.G.; Vaz, B.G.; De Lucas, N.C.; Eberlin, M.N.; Garden, S.J. C-H functionalization of 1,4-naphthoquinone by oxidative coupling with anilines in the presence of a catalytic quantity of copper(II) acetate. J. Org. Chem. 2011, 76, 5264–5273. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Verrax, J.; Gonze, T.; Zappone, M.; Curi, R.; Taper, H.; Feron, O.; Buc, P. Hsp90 cleavage by an oxidative stress leads to its client proteins degradation and cancer cell death. Biochem. Pharmacol. Ogy. 2009, 77, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Pathania, D.; Sechi, M.; Palomba, M.; Sanna, V.; Berrettini, F.; Sias, A.; Taheri, L.; Neamati, N. Design and discovery of novel quinazolinedione-based redox modulators as therapies for pancreatic cancer. Biochim. Biophys. Acta 2014, 1840, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Ravichandiran, P.; Sheet, S.; Premnath, D.; Kim, A.R. 1,4-Naphthoquinone Analogues: Potent Antibacterial Agents and Mode of Action Evaluation. Molecules 2019, 24, 1437. [Google Scholar] [CrossRef]
- Ferraz, P.A.; de Abreu, F.C.; Pinto, A.V.; Glezer, V.; Tonholo, J.; Goulart, M.O. Electrochemical aspects of the reduction of biologically active 2-hydroxy-3-alkyl-1,4-naphthoquinones. J. Electroanal. Chem. 2001, 507, 275–286. [Google Scholar] [CrossRef]
- González, F.J.; Aceves, J.M.; Miranda, R.; González, I. The electrochemical reduction of perezone in the presence of benzoic acid in acetonitrile. J. Electroanal. Chem. Interfacial. Electrochem. 1991, 310, 293–303. [Google Scholar] [CrossRef]
- Hillard, E.A.; De Abreu, F.C.; Ferreira, D.C.M.; Jaouen, G.; Goulart, M.O.F.; Amatore, C. Electrochemical parameters and techniques in drug development, with an emphasis on quinones and related compounds. Chem. Commun. 2008, 2612–2628. [Google Scholar] [CrossRef]
- Armendáriz-Vidales, G.; Hernández-Muñoz, L.S.; González, F.J.; De Souza, A.A.; De Abreu, F.C.; Jardim, G.A.M.; Da Silva, E.N.; Goulart, M.O.F.; Frontana, C. Nature of electrogenerated intermediates in nitro-substituted nor-β-lapachones: The structure of radical species during successive electron transfer in multiredox centers. J. Org. Chem. 2014, 79, 5201–5208. [Google Scholar] [CrossRef]
- Hernández, D.M.; De Moura, M.A.B.F.; Valencia, D.P.; González, F.J.; González, I.; De Abreu, F.C.; Da Silva Júnior, E.N.; Ferreira, V.F.; Pinto, A.V.; Goulart, M.O.F.; et al. Inner reorganization during the radical-biradical transition in a nor-β-lapachone derivative possessing two redox centers. Org. Biomol. Chem. 2008, 6, 3414–3420. [Google Scholar] [CrossRef]
- Koyama, J.; Morita, I.; Kobayashi, N.; Osakai, T. Correlation of redox potentials and inhibitory effects on Epstein–Barr virus activation of naphthoquinones. Cancer Lett. 2003, 201, 25–30. [Google Scholar] [CrossRef]
- Ibacache, J.A.; Faundes, J.; Montoya, M.; Mejí, S.; Jaime, A. Preparation of Novel Homodimers Derived from Cytotoxic Isoquinolinequinones. A Twin Drug Approach. Molecules 2018, 23, 439. [Google Scholar] [CrossRef] [PubMed]
- Delgado, V.; Ibacache, A.; Arancibia, V.; Theoduloz, C.; Valderrama, J.A. Synthesis and in Vitro Antiproliferative Activity of New Phenylaminoisoquinolinequinones against Cancer Cell Lines. Molecules 2013, 18, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Kaur, B.; Parmar, A.; Kumar, H. Applications of Ultrasound in Organic Synthesis - A Green Approach. Curr. Org. Chem. 2013, 17, 1790–1828. [Google Scholar] [CrossRef]
- Guin, P.S.; Das, S.; Mandal, P.C. Electrochemical Reduction of Quinones in Different Media: A Review. Int. J. Electrochem. 2011, 2011, 1–22. [Google Scholar] [CrossRef]
- He, P.; Crooks, R.M.; Faulkner, L.R. Adsorption and electrode reactions of disulfonated anthraquinones at mercury electrodes. J. Phys. Chem. 2005, 94, 1135–1141. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R.; Swain, E.; Robey, C. Electrochemical Method—Fundamentals and Applications, 2nd ed.; Harris, D., Swain, E., Robey, C., Aiello, E., Eds.; John Wiley & Sons: New York, NY, USA, 2001; ISBN 0471043729. [Google Scholar]
- Patai, S.; Rappoport, Z. (Eds.) The Chemistry of the Quinonoid Compounds; John Wiley & Sons: New York, NY, USA, 1988; Chapter 12; Volume 2, ISBN 9780470772119. [Google Scholar]
- Skancke, A.; Skancke, P.N. General and Theoretical Aspects of Quinones; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons: New York, NY, USA, 1988; Chapter 14; Volume 1, ISBN 9780470772119. [Google Scholar]
- Wu, D.; Huang, Y.; Hu, X. A sulfurization-based oligomeric sodium salt as a high-performance organic anode for sodium ion batteries. Chem. Commun. 2016, 52, 11207–11210. [Google Scholar] [CrossRef]
- Han, C.; Li, H.; Shi, R.; Zhang, T.; Tong, J.; Li, J.; Li, B. Organic quinones towards advanced electrochemical energy storage: Recent advances and challenges. J. Mater. Chem. A 2019, 1–38. [Google Scholar] [CrossRef]
- Mirkhalaf, F.; Tammeveski, K.; Schiffrin, D.J. Substituent effects on the electrocatalytic reduction of oxygen on quinone-modified glassy carbon electrodes. Phys. Chem. Chem. Phys. 2004, 6, 1321–1327. [Google Scholar] [CrossRef]
- Jardim, G.A.M.; Reis, W.J.; Ribeiro, M.F.; Ottoni, F.M.; Alves, R.J.; Silva, T.L.; Goulart, M.O.F.; Braga, A.L.; Menna-Barreto, R.F.S.; Salomão, K.; et al. On the investigation of hybrid quinones: Synthesis, electrochemical studies and evaluation of trypanocidal activity. RSC Adv. 2015, 5, 78047–78060. [Google Scholar] [CrossRef]
- Elhabiri, M.; Sidorov, P.; Cesar-Rodo, E.; Marcou, G.; Lanfranchi, D.A.; Davioud-Charvet, E.; Horvath, D.; Varnek, A. Electrochemical properties of substituted 2-methyl-1,4-naphthoquinones: Redox behavior predictions. Chem.-A Eur. J. 2015, 21, 3415–3424. [Google Scholar] [CrossRef] [PubMed]
- Turek, A.K.; Hardee, D.J.; Ullman, A.M.; Nocera, D.G.; Jacobsen, E.N. Activation of Electron-Deficient Quinones through Hydrogen-Bond-Donor-Coupled Electron Transfer. Angew. Chemie.-Int. Ed. 2016, 55, 539–544. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the synthesized compounds 5, 6, 13 and 15 are available from the corresponding authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound N° | Structure | −EI1/2 (mV) a | −EII1/2 (mV) a | n b |
|---|---|---|---|---|
| 5 |  | 697 | 1318 | 1.1 |
| 6 |  | 681 | 897 | 1.2 |
| 7 |  | 688 | 1174 | 1.2 |
| 10 |  | 661 | - | 2.0 |
| 11 |  | 580 | - | 1.7 |
| 12 |  | 667 | - | 1.9 |
| 14 |  | 652 | - | 1.8 |
| 15 |  | 689 | - | 1.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibacache, J.A.; Valderrama, J.A.; Faúndes, J.; Danimann, A.; Recio, F.J.; Zúñiga, C.A. Green Synthesis and Electrochemical Properties of Mono- and Dimers Derived from Phenylaminoisoquinolinequinones. Molecules 2019, 24, 4378. https://doi.org/10.3390/molecules24234378
Ibacache JA, Valderrama JA, Faúndes J, Danimann A, Recio FJ, Zúñiga CA. Green Synthesis and Electrochemical Properties of Mono- and Dimers Derived from Phenylaminoisoquinolinequinones. Molecules. 2019; 24(23):4378. https://doi.org/10.3390/molecules24234378
Chicago/Turabian StyleIbacache, Juana Andrea, Jaime A. Valderrama, Judith Faúndes, Alex Danimann, Francisco J. Recio, and César A. Zúñiga. 2019. "Green Synthesis and Electrochemical Properties of Mono- and Dimers Derived from Phenylaminoisoquinolinequinones" Molecules 24, no. 23: 4378. https://doi.org/10.3390/molecules24234378
APA StyleIbacache, J. A., Valderrama, J. A., Faúndes, J., Danimann, A., Recio, F. J., & Zúñiga, C. A. (2019). Green Synthesis and Electrochemical Properties of Mono- and Dimers Derived from Phenylaminoisoquinolinequinones. Molecules, 24(23), 4378. https://doi.org/10.3390/molecules24234378