Michael Addition Reaction Catalyzed by Imidazolium Chloride to Protect Amino Groups and Construct Medium Ring Heterocycles

Abstract
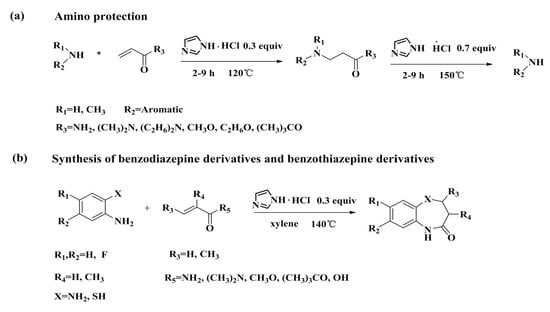
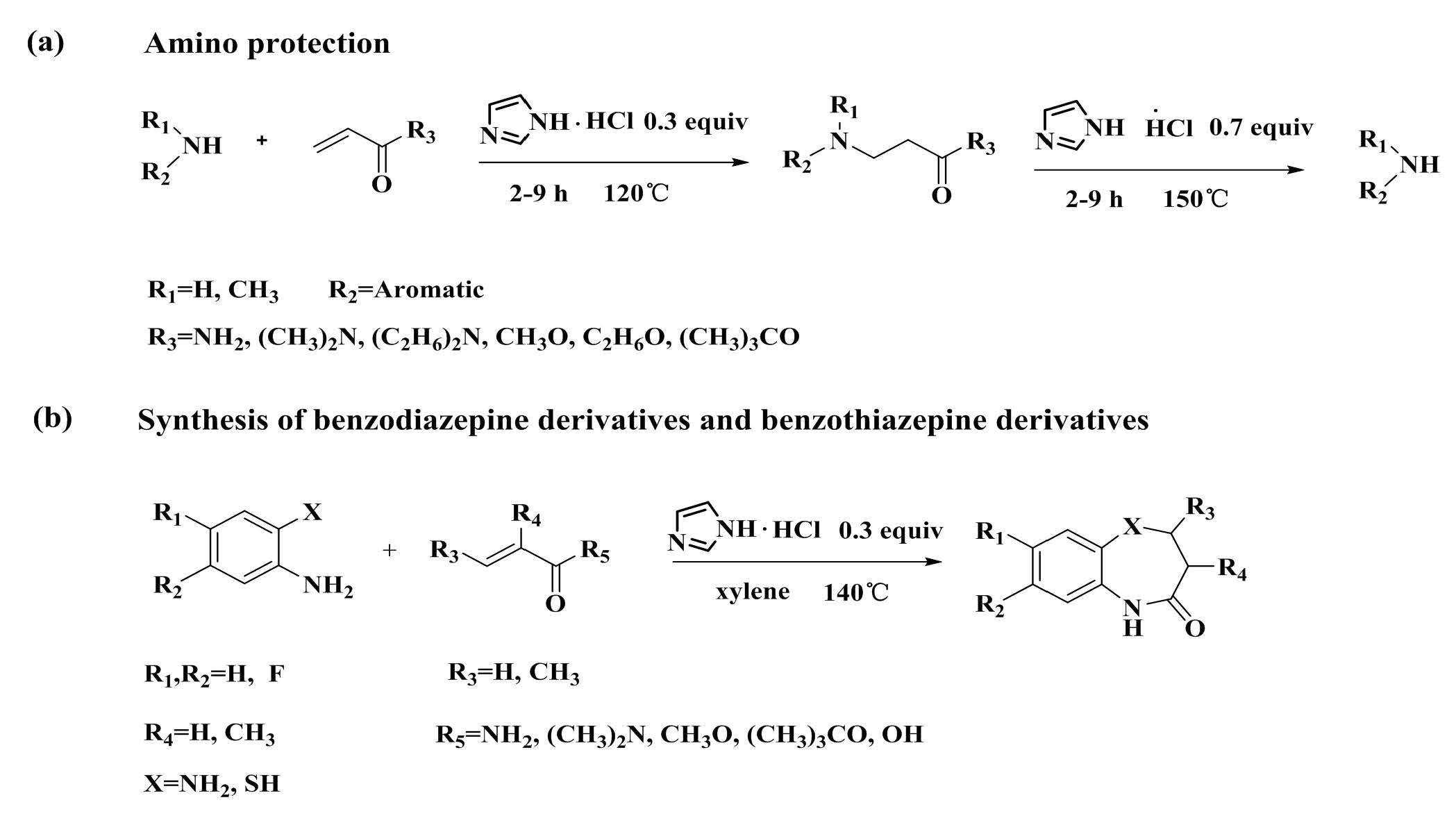
1. Introduction
2. Results and Discussion
2.1. Optimization of Reaction Conditions
2.2. Scope of Substituted Substrate
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Materials
4.2. General Procedures for Michael Addition Reaction
4.2.1. Synthesis of 3-(Pyrrolidin-1-yl)propanamide (3x)
4.2.2. Synthesis of 3-(4-Methylpiperidin-1-yl)propanamide (3y)
4.3. General Procedure for Amino Deprotection
4.4. General Methods for Synthesis of Substituted 1,3,4,5-Tetrahydro-2H-1,5-benzodiazepine-2-ones and Substituted 2,3-Dihydrobenzo[b][1,4]thiazepin-4(5H)-ones
Synthesis of 1,3,4,5-Tetrahydro-2H-benzo[b][1,4]diazepin-2-one (6c)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jianyong, Y.; Wen, L.; Zeshu, D. 一种咪唑盐酸盐催化氨基保护的方法. Patent CN110028422A, 17 July 2019. (In Chinese). [Google Scholar]
- Spaulding, A.; Takrouri, K.; Mahalingam, P.; Cleary, D.C.; Cooper, H.D.; Zucchi, P.; Tear, W.; Koleva, B.; Beuning, P.J.; Hirsch, E.B.; et al. Compound design guidelines for evading the efflux and permeation barriers of Escherichia coli with the oxazolidinone class of antibacterials: Test case for a general approach to improving whole cell Gram-negative activity. Bioorg. Med. Chem. Lett. 2017, 27, 5310–5321. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Shi, L.Y.; Wu, J.J.; Fang, X.; Yang, X.Y.; Wu, F.H. Microwave-assisted expeditious synthesis of 5-fluoroalkyl-3-(aryl/alkyl)-oxazolidin-2-ones. Tetrahedron 2013, 69, 3331–3337. [Google Scholar] [CrossRef]
- Pan, L.; Lei, D.Y.; Jin, L.; He, Y.; Yang, Q.Q. Promising Fungicides from Allelochemicals: Synthesis of Umbelliferone Derivatives and Their Structure (-) Activity Relationships. Molecules 2018, 23, 3002. [Google Scholar] [CrossRef] [PubMed]
- Samadi, S.; Jadidi, K.; Khanmohammadi, B.; Tavakoli, N. Heterogenization of chiral mono oxazoline ligands by grafting onto mesoporous silica MCM-41 and their application in copper-catalyzed asymmetric allylic oxidation of cyclic olefins. J. Catal. 2016, 340, 344–353. [Google Scholar] [CrossRef]
- Yang, E.G.; Mustafa, N.; Tan, E.C.; Poulsen, A.; Ramanujulu, P.M.; Chng, W.J.; Yen, J.J.; Dymock, B.W. Design and Synthesis of Janus Kinase 2 (JAK2) and Histone Deacetlyase (HDAC) Bispecific Inhibitors Based on Pacritinib and Evidence of Dual Pathway Inhibition in Hematological Cell Lines. J. Med. Chem. 2016, 59, 8233–8262. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Jang, D.O. Trifluoroacetylation of amines with trifluoroacetic acid in the presence of trichloroacetonitrile and triphenylphosphine. Tetrahedron Lett. 2010, 51, 683–685. [Google Scholar] [CrossRef]
- Hana, K.J.; Kim, M. A simple and efficient method for trifluoroacetylation of arylamines using trifluoroacetic acid and triphosgene. Lett. Org. Chem. 2011, 8, 559–561. [Google Scholar] [CrossRef]
- Ohtaka, J.; Sakamoto, T.; Kikugawa, Y. A one-pot procedure for trifluoroacetylation of arylamines using trifluoroacetic acid as a trifluoroacetylating reagent. Tetrahedron Lett. 2009, 50, 1681–1683. [Google Scholar] [CrossRef]
- Saitoh, T.; Shimada, C.; Takeiri, M.; Shiino, M.; Ohba, S.; Obata, R.; Ishikawa, Y.; Umezawa, K.; Nishiyama, S. A new NF-kB inhibitor based on the amino-epoxyquinol core of DHMEQ. Bioorg. Med. Chem. Lett. 2010, 20, 5638–5642. [Google Scholar] [CrossRef]
- Dighe, S.N.; Jadhav, H.R. Microwave assisted mild, rapid, solvent-less, and catalyst-free chemoselective N-tert-butyloxycarbonylation of amines. Tetrahedron Lett. 2012, 53, 5803–5806. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Gagliardi, A.; Leggio, A.; Leotta, V.; Romio, E.; Liguori, A. N-Urethane protection of amines and amino acids in an ionic liquid. RSC Adv. 2015, 5, 63407–63420. [Google Scholar] [CrossRef]
- Belsito, E.L.; Marco, R.D.; Gioia, M.L.D.; Liguori, A.; Perri, F.; Viscomi, M.C. N -(4-Nitrophenylsulfonyl)- and N-(Fluorenylmethoxycarbonyl)-N-ethyl Amino Acid Methyl Esters—A Practical Approach. Eur. J. Org. Chem. 2010, 4245–4252. [Google Scholar] [CrossRef]
- De Marco, R.; Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A new non-natural arginine-like amino acid derivative with a sulfamoyl group in the side-chain. Amino Acids 2010, 38, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Nardi, M.; Cano, N.H.; De Nino, A.; Di Gioia, M.L.; Maiuolo, L.; Oliverio, M.; Santiago, A.; Sorrentino, D.; Procopio, A. An eco-friendly tandem tosylation/Ferrier N -glycosylation of amines catalyzed by Er(OTf)3 in 2-MeTHF. Tetrahedron Lett. 2017, 58, 1721–1726. [Google Scholar] [CrossRef]
- Becerra-Figueroa, L.; Ojeda-Porras, A.; Gamba-Sanchez, D. Transamidation of carboxamides catalyzed by Fe(III) and water. J. Org. Chem. 2014, 79, 4544–4552. [Google Scholar] [CrossRef]
- Dai-Il, J.; Tae-wonchoi, C.; Yun-Young, K.; In-Shik, K.; You-Mi, P.; Yong-Gyun, L.; Doo-Hee, J. Synthesis Of 1,5-Benzodiazepine Derivatives. Synth. Commun. 1999, 29, 1941–1951. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Ruggiero, E.; Tabrizi, M.A. New Synthesis of Diazepino[3,2,1-ij]quinoline and Pyrido[1,2,3-de]quinoxalines via Addition-Elimination Followed by Cycloacylation. J. Heterocycl. Chem. 2014, 51, 101–105. [Google Scholar] [CrossRef]
- Tang, X.J.; Yan, Z.L.; Chen, W.L.; Gao, Y.R.; Mao, S.; Zhang, Y.L.; Wang, Y.Q. Aza-Michael reaction promoted by aqueous sodium carbonate solution. Tetrahedron Lett. 2013, 54, 2669–2673. [Google Scholar] [CrossRef]
- Payra, S.; Saha, A.; Banerjee, S. On-water magnetic NiFe2O4 nanoparticle-catalyzed Michael additions of active methylene compounds, aromatic/aliphatic amines, alcohols and thiols to conjugated alkenes. RSC Adv. 2016, 6, 95951–95956. [Google Scholar] [CrossRef]
- Dabiri, M.; Salehi, P.; Bahramnejad, M.; Baghbanzadeh, M. Ecofriendly and efficient procedure for hetero-Michael addition reactions with an acidic ionic liquid as catalyst and reaction medium. Mon. Chem. 2011, 143, 109–112. [Google Scholar] [CrossRef]
- Vijender, M.; Kishore, P.; Satyanarayana, B. Cadmium chloride (CdCl2): An effificient catalyst for conjugate addition of amines to electron-poor alkenes. Synth. Commun. 2007, 37, 591–594. [Google Scholar] [CrossRef]
- Duan, Z.; Xuan, X.; Li, T.; Yang, C.; Wu, Y. Cerium (IV) ammonium nitrate (CAN) catalyzed aza-Michael addition of amines to α,β-unsaturated electrophiles. Tetrahedron Lett. 2006, 47, 5433–5436. [Google Scholar] [CrossRef]
- Meshram, H.M.; Lakshindra, C.; Reddy, P.N.; Sadashiv, K.; Yadav, J.S. Zirconium(IV) chloride-mediated chemoselective conjugate addition of aliphatic amines to α,β-ethylenic compounds. Synth. Commun. 2006, 36, 795–801. [Google Scholar] [CrossRef]
- Saidi, M.R.; Pourshojaei, Y.; Aryanasab, F. Highly Efficient Michael Addition Reaction of Amines Catalyzed by Silica-Supported Aluminum Chloride. Synth. Commun. 2009, 39, 1109–1119. [Google Scholar] [CrossRef]
- Lin, Y.D.; Kao, J.Q.; Chen, C.T. Catalytic conjugate additions of nitrogen-, phosphorus-, and carbon-containing nucleophiles by amphoteric vanadyl triflate. Org. Lett. 2007, 9, 5195–5198. [Google Scholar] [CrossRef]
- Yadav, J.S.; Ramesh Reddy, A.; Gopal Rao, Y.; Narsaiah, A.V.; Reddy, B.V.S. Lanthanum trichloride (LaCl3): An efficient catalyst for conjugate addition of amines to electron-deficient olefins. Lett. Org. Chem. 2007, 4, 462–464. [Google Scholar]
- Nguyen, L.T.L.; Nguyen, T.T.; Nguyen, K.D.; Phan, N.T.S. Metal-organic framework MOF-199 as an efficient heterogeneous catalyst for aza-Michael reaction. Appl. Catal. A Gen. 2012, 425–426, 44–52. [Google Scholar] [CrossRef]
- Ai, X.; Wang, X.; Liu, J.; Ge, Z.; Cheng, T.; Li, R. An effective aza-Michael addition of aromatic amines to electron-deficient alkenes in alkaline Al2O3. Tetrahedron 2010, 66, 5373–5377. [Google Scholar] [CrossRef]
- Takamura, K.; Shioya, A.; Minamoto, K.; Asada, N.; Takaku, S.; Yoshimitsu, A.; Nitta, Y. Studies on analgesics of aniline series. I. Preparation and properties of beta-alaninamide series. Chem. Pharm. Bull. 1965, 13, 198–204. [Google Scholar] [CrossRef][Green Version]
- Bosica, G.; Abdilla, R. Aza-Michael Mono-addition Using Acidic Alumina under Solventless Conditions. Molecules 2016, 21, 815. [Google Scholar] [CrossRef]
- You, L.; Song, F.; Rui, A.; Wang, X.; Bai, D. ChemInform Abstract: Silica Gel Accelerated Aza-Michael Addition of Amines to α,β-Unsaturated Amides. Tetrahedron Lett. 2008, 49, 5147–5149. [Google Scholar] [CrossRef]
- Rostamnia, S.; Alamgholiloo, H. Synthesis and Catalytic Application of Mixed Valence Iron (FeII/FeIII)-Based OMS-MIL-100(Fe) as an Efficient Green Catalyst for the aza-Michael Reaction. Catal. Lett. 2018, 148, 2918–2928. [Google Scholar] [CrossRef]
- Neogi, S.; Naskar, D. ChemInform Abstract: One-Pot Reductive Mono-N-Alkylation of Aromatic Nitro Compounds Using Nitriles as Alkylating Reagents. Synth. Commun. 2011, 41, 1901–1915. [Google Scholar] [CrossRef]
- Xu, W.Q.; Ren, Y.J.; Wang, Q.W.; Sun, Y.X. Triflic Acid as Efficient Catalyst for the Hydroamination of Ethyl Acrylate with 2-Aminopyridines. Lett. Org. Chem. 2015, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.S.; Engel-Andreasen, J.; Fristrup, P.; Harris, P.; Olsen, C.A. Cis-trans amide bond rotamers in beta-peptoids and peptoids: Evaluation of stereoelectronic effects in backbone and side chains. J. Am. Chem. Soc. 2013, 135, 2835–2844. [Google Scholar] [CrossRef]
- Ying, A.; Li, Z.; Yang, J.; Liu, S.; Xu, S.; Yan, H.; Wu, C. DABCO-based ionic liquids: Recyclable catalysts for aza-Michael addition of α, β-unsaturated Amides under Solvent-Free Conditions. J. Org. Chem. 2014, 79, 6510–6516. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Liu, Y.; Ling, C.; Lai, Z.; Fang, X.; Liu, B.; Zhang, W.; Lu, M.; Xu, Y.; Hao, X. Nmp-based ionic liquids: Recyclable catalysts for both hetero-Michael addition and Knoevenagel condensation in water. Synth. Commun. 2018, 48, 1060–1067. [Google Scholar] [CrossRef]
- Kreye, O.; Wald, S.; Meier, M.A.R. ChemInform Abstract: Introducing Catalytic Lossen Rearrangements: Sustainable Access to Carbamates and Amines. Adv. Synth. Catal. 2013, 355, 81–86. [Google Scholar] [CrossRef]
- Cantillo, D.; Baghbanzadeh, M.; Kappe, C.O. In situ generated iron oxide nanocrystals as efficient and selective catalysts for the reduction of nitroarenes using a continuous flow method. Angew. Chem. Int. Ed. 2012, 51, 10190–10193. [Google Scholar] [CrossRef]
- Ji, P.; Manna, K.; Lin, Z.; Feng, X.; Urban, A.; Song, Y.; Lin, W. Single-Site Cobalt Catalysts at New Zr12(μ3-O)8(μ3-OH)8(μ2-OH)6 Metal-Organic Framework Nodes for Highly Active Hydrogenation of Nitroarenes, Nitriles, and Isocyanides. J. Am. Chem. Soc. 2017, 139, 7004–7011. [Google Scholar] [CrossRef]
- Ding, Z.C.; Li, C.Y.; Chen, J.J.; Zeng, J.H.; Tang, H.T.; Ding, Y.J.; Zhan, Z.P. Palladium/Phosphorus-Doped Porous Organic Polymer as Recyclable Chemoselective and Efficient Hydrogenation Catalyst under Ambient Conditions. Adv. Synth. Catal. 2017, 359, 2280–2287. [Google Scholar] [CrossRef]
- Cohen, S.; Bilyachenko, A.N.; Gelman, D. Bifunctional Pincer Catalysts for Chemoselective Transfer Hydrogenation and Related Reactions. Eur. J. Inorg. Chem. 2019, 2019, 3203–3209. [Google Scholar] [CrossRef]
- Gayakwad, E.M.; Patel, K.P.; Shankarling, G.S. Sodium sulfate–hydrogen peroxide–sodium chloride adduct: Selective protocol for the oxidative bromination, iodination and temperature dependent oxidation of sulfides to sulfoxides and sulfones. New J. Chem. 2019, 43, 6001–6009. [Google Scholar] [CrossRef]
- Duan, Z.; Ma, G.; Zhang, W. Preparation of Copper Nanoparticles and Catalytic Properties for the Reduction of Aromatic Nitro Compounds. Bull. Korean Chem. Soc. 2012, 33, 4003–4006. [Google Scholar] [CrossRef]
- Portada, T.; Margetic, D.; Strukil, V. Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives. Molecules 2018, 23, 3163. [Google Scholar] [CrossRef]
- Jiang, Z.; Wu, Z.; Wang, L.; Wu, D.; Zhou, X. Preparation of aromatic amines by copper-catalyzed coupling of boronic acids with aqueous ammonia. Can. J. Chem. 2010, 88, 964–968. [Google Scholar] [CrossRef]
- Doebelin, C.; Schmitt, M.; Antheaume, C.; Bourguignon, J.J.; Bihel, F. Nucleophilic Substitutionof Azide Acting as a Pseudo Leaving Group: One-Step Synthesis of VariousAza Heterocycles. J. Org. Chem. 2013, 78, 11335–11341. [Google Scholar] [CrossRef]
- Taylor, A.M.; Vaswani, R.G.; Gehling, V.S.; Hewitt, M.C.; Leblanc, Y.; Audia, J.E.; Bellon, S.; Cummings, R.T.; Cote, A.; Harmange, J.C.; et al. Discovery of Benzotriazolo[4,3-d][1,4]diazepines as orally active Inhibitors of BET bromodomains. ACS Med. Chem. Lett. 2015, 7, 145–150. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Zhu, X.; Mao, H.; Zou, X.; Kong, L.; Li, X. Features and applications of reactions of α,β-unsaturated -acylbenzotriazoles with amino compounds. Tetrahedron 2008, 64, 6510–6521. [Google Scholar] [CrossRef]
- Pan, Y.; Chen, C.; Xu, X.; Zhao, H.; Han, J.; Li, H.; Xu, L.; Fan, Q.; Xiao, J. Metal-free tandem cyclization/hydrosilylation to construct tetrahydroquinoxalines. Green Chem. 2018, 20, 403–411. [Google Scholar] [CrossRef]
- Nageswara Rao, S.; Chandra Mohan, D.; Adimurthy, S. ChemInform Abstract: Chitosan: An Efficient Recyclable Catalyst for Transamidation of Carboxamides with Amines under Neat Conditions. Green Chem. 2014, 16, 4122–4126. [Google Scholar] [CrossRef]
- Rana, N.K.; Singh, V.K. ChemInform Abstract: Enantioselective Enolate Protonation in Sulfa—Michael Addition to α-Substituted N-Acryloyloxazolidin-2-ones with Bifunctional Organocatalyst. Org. Lett. 2011, 13, 6520–6523. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}

| Entry | Cat (eq) | Solvent | Temperature (°C) | Time (h) | Yield (%) c |
|---|---|---|---|---|---|
| 1 | - | - | 120 | 4 | trace |
| 2 | Imidazolium chloride (0.3) | - | 120 | 4 | 69 |
| 3 | Imidazole (0.3) | - | 120 | 6 | 10 |
| 4 | HCl (0.3) | - | 120 | 4 | 60 |
| 5 | Imidazolium chloride (0.3) | - | 90 | 6 | 50 |
| 6 | Imidazolium chloride (0.3) | - | 120 | 4 | 75 |
| 7 | Imidazolium chloride (0.3) | - | 130 | 4 | 65 |
| 8 | Imidazolium chloride (0.3) | - | 140 | 4 | 40 |
| 9 | Imidazolium chloride (0.1) | - | 120 | 4 | 32 |
| 10 | Imidazolium chloride (0.2) | - | 120 | 4 | 61 |
| 11 | Imidazolium chloride (0.5) | - | 120 | 4 | 73 |
| 12 b | Imidazolium chloride (0.3) | H2O | 100 | 6 | 18 |
| 13 b | Imidazolium chloride (0.3) | CH3CN | 70 | 6 | NO |
| 14 b | Imidazolium chloride (0.3) | Toluene | 120 | 4 | 63 |
| 15 b | Imidazolium chloride (0.3) | Xylene | 120 | 4 | 66 |

| Entry | Cat (eq) | Solvent | Temperature (°C) | Time (h) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | - | - | 150 | 6 | Trace |
| 2 | Imidazole (0.7) | - | 150 | 6 | Trace |
| 3 | HCl (0.7) | - | 150 | 6 | 8 |
| 4 | Imidazolium chloride (0.7) | - | 120 | 6 | Trace |
| 5 | Imidazolium chloride (0.7) | - | 150 | 6 | 61 |
| 6 | Imidazolium chloride (0.7) | - | 160 | 6 | 63 |
| 7 | Imidazolium chloride (0.7) | - | 180 | 6 | 65 |
| 8 | Imidazolium chloride (0.1) | - | 150 | 6 | 23 |
| 9 | Imidazolium chloride (0.5) | - | 150 | 6 | 35 |
| 10 | Imidazolium chloride (1.0) | - | 150 | 6 | 65 |
| 11 c | Imidazolium chloride (0.7) | H2O | 100 | 6 | NO |
| 12 c | Imidazolium chloride (0.7) | benzene | 90 | 6 | NO |
| 13 c | Imidazolium chloride (0.7) | xylene | 150 | 6 | 57 |

| Entry | Cat(eq) | Solvent | Temp(°C) | Time(h) | Yield(%) b |
|---|---|---|---|---|---|
| 1 | Imidazolium chloride(0.3) | xylene | 120 | 5 | 45 |
| 2 | - | xylene | 120 | 5 | NO |
| 3 | HCl(0.3eq) | xylene | 120 | 5 | 34 |
| 4 | Imidazolium(0.3) | xylene | 120 | 5 | trace |
| 5 | Imidazolium chloride(0.3) | xylene | 80 | 5 | 13 |
| 6 | Imidazolium chloride(0.3) | xylene | 100 | 5 | 25 |
| 7 | Imidazolium chloride(0.3) | xylene | 110 | 5 | 31 |
| 8 | Imidazolium chloride(0.3) | xylene | 140 | 5 | 75 |
| 9 | Imidazolium chloride(0.3) | xylene | 150 | 5 | 76 |
| 10 | Imidazolium chloride(0.3) | CH3CN | 70 | 9 | 15 |
| 11 | Imidazolium chloride(0.3) | H2O | 100 | 9 | 10 |
| 12 | Imidazolium chloride(0.3) | dioxane | 100 | 9 | 27 |
| 13 | Imidazolium chloride(0.3) | toluene | 110 | 9 | 10 |
| 14 | Imidazolium chloride(0.3) | ethylene glycol diethyl ether | 120 | 5 | 39 |
| 15 | Imidazolium chloride(0.3) | 2-methoxyethanol | 125 | 5 | 43 |
| 16 | Imidazolium chloride(0.3) | 2-ethoxyethanol | 140 | 5 | trace |
| 17 | Imidazolium chloride(0.1) | xylene | 140 | 5 | 19 |
| 18 | Imidazolium chloride(0.5) | xylene | 140 | 5 | 49 |
 |
 |
 | ||||
|---|---|---|---|---|
| Entry | Substance | Time (h) | Product | Yield b (%) |
| 1 |  | 6 |  | 63 |
| 2 |  | 4 |  | 82 |
| 3 |  | 9 |  | 85 |
| 4 |  | 5 |  | 72 |
| 5 |  | 12 |  | 40 |
| 6 |  | 11 |  | 70 |
| 7 |  | 10 |  | 73 |
| 8 |  | 6 |  | 70 |
| 9 |  | 4 |  | 81 |
| 10 |  | 10 |  | 35 |
| 11 |  | 9 |  | 72 |
| 12 |  | 6 |  | 76 |
| 13 |  | 6 |  | 72 |
| 14 |  | 6 |  | 82 |
 | |||||
|---|---|---|---|---|---|
| Entry | Substance | Time (h) | Product | Yield b (%) | |
| 1 |  |  | 5 |  | 74 |
| 2 |  |  | 5 |  | 68 |
| 3 c |  |  | 13 |  | 34 |
| 4 |  |  | 0.5 |  | 93 |
| 5 |  |  | 4 |  | 90 |
| 6 |  |  | 2 |  | 34 |
| 7 |  |  | 2 |  | 31 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Z.; Tian, Q.; Li, Y.; Shang, S.; Luo, W.; Wang, X.; Li, D.; Zhang, Y.; Li, Z.; Yuan, J. Michael Addition Reaction Catalyzed by Imidazolium Chloride to Protect Amino Groups and Construct Medium Ring Heterocycles. Molecules 2019, 24, 4224. https://doi.org/10.3390/molecules24234224
Dai Z, Tian Q, Li Y, Shang S, Luo W, Wang X, Li D, Zhang Y, Li Z, Yuan J. Michael Addition Reaction Catalyzed by Imidazolium Chloride to Protect Amino Groups and Construct Medium Ring Heterocycles. Molecules. 2019; 24(23):4224. https://doi.org/10.3390/molecules24234224
Chicago/Turabian StyleDai, Zeshu, Qingqiang Tian, Yanwu Li, Suqin Shang, Wen Luo, Xuetong Wang, Dan Li, Ying Zhang, Zhiyao Li, and Jianyong Yuan. 2019. "Michael Addition Reaction Catalyzed by Imidazolium Chloride to Protect Amino Groups and Construct Medium Ring Heterocycles" Molecules 24, no. 23: 4224. https://doi.org/10.3390/molecules24234224
APA StyleDai, Z., Tian, Q., Li, Y., Shang, S., Luo, W., Wang, X., Li, D., Zhang, Y., Li, Z., & Yuan, J. (2019). Michael Addition Reaction Catalyzed by Imidazolium Chloride to Protect Amino Groups and Construct Medium Ring Heterocycles. Molecules, 24(23), 4224. https://doi.org/10.3390/molecules24234224