
Identifying Promiscuous Compounds with Activity against Different Target Classes




Abstract
:1. Introduction
2. Results and Discussion
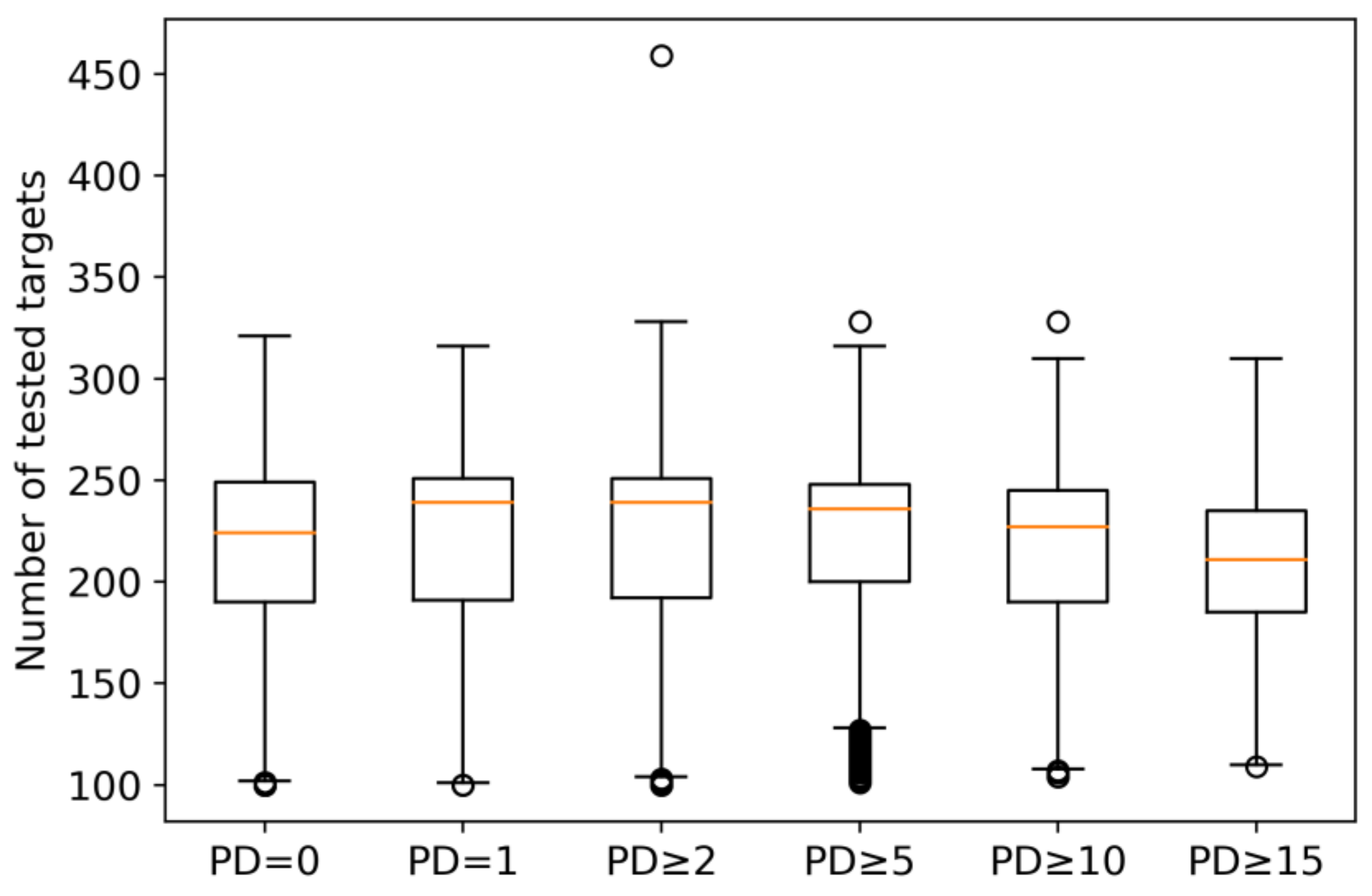
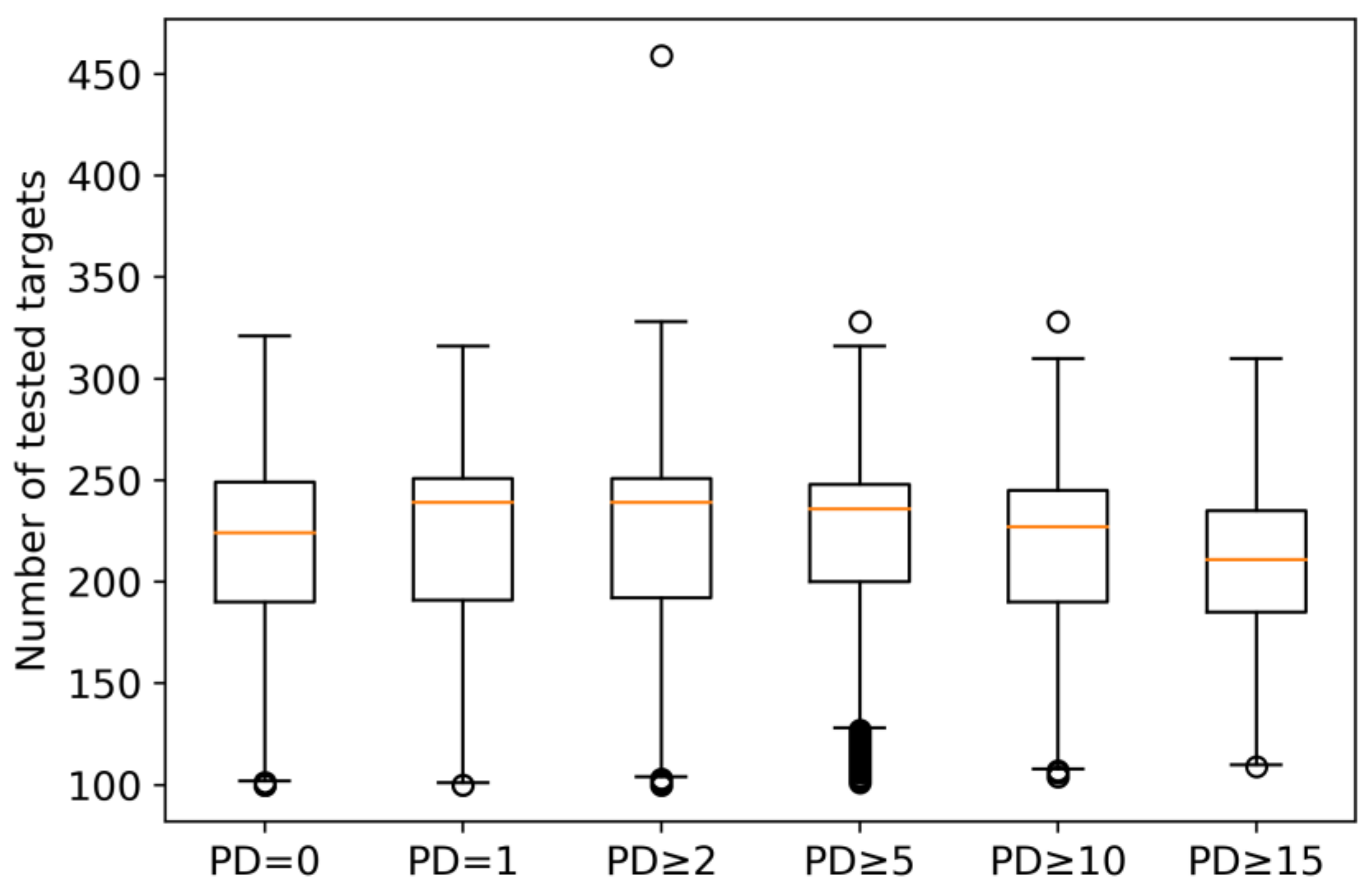
2.1. Qualifying Compounds and Promiscuity Degrees
2.2. Target Distribution and Classification
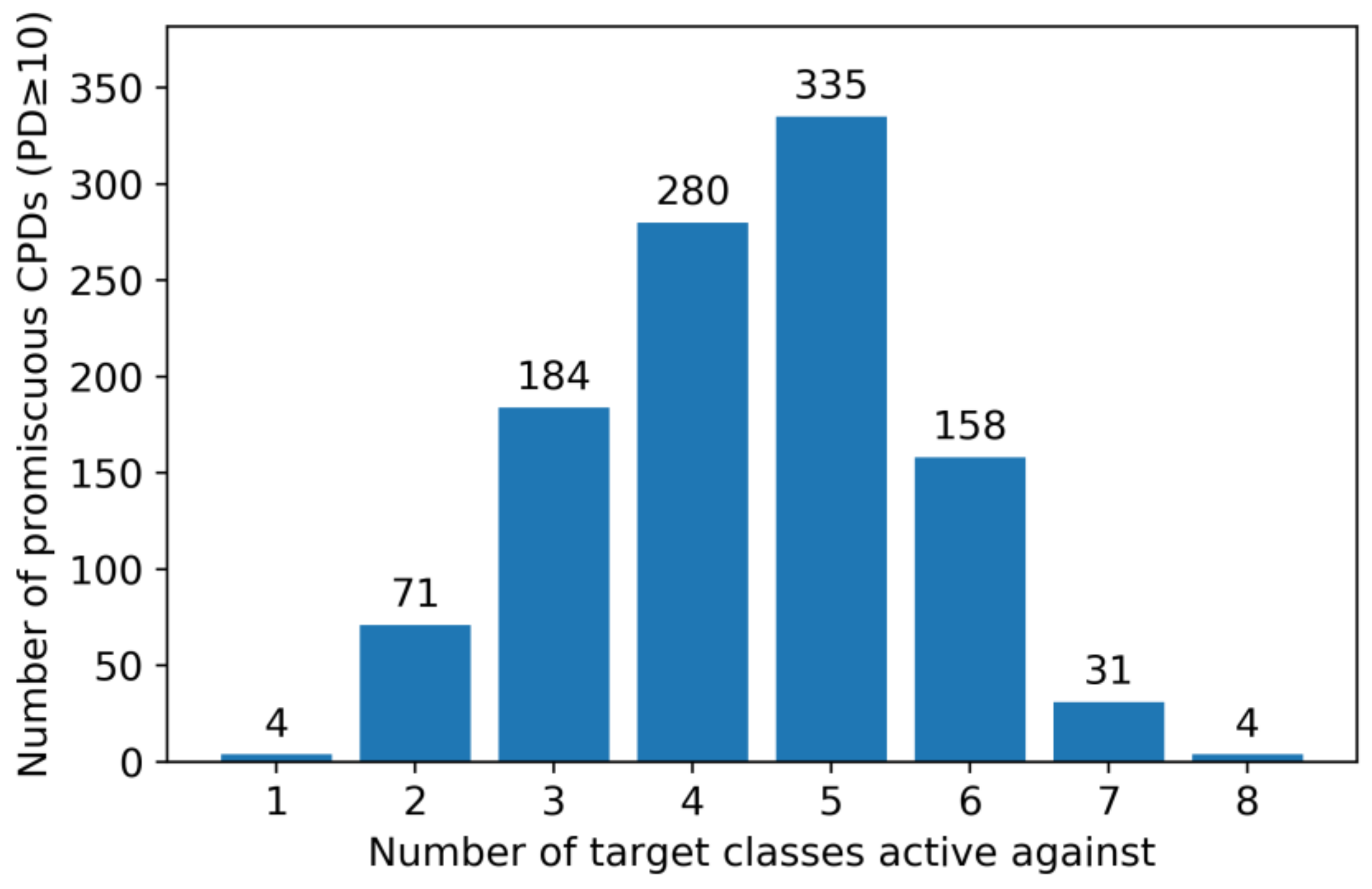
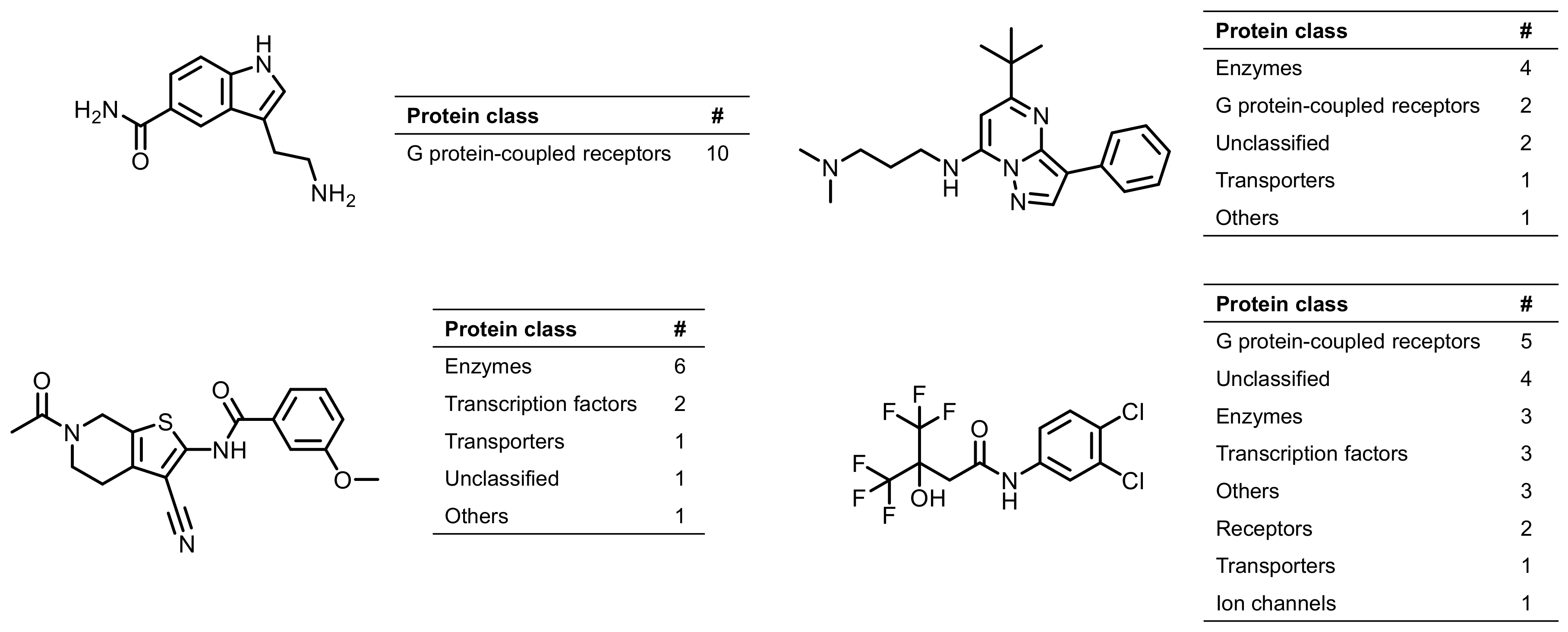
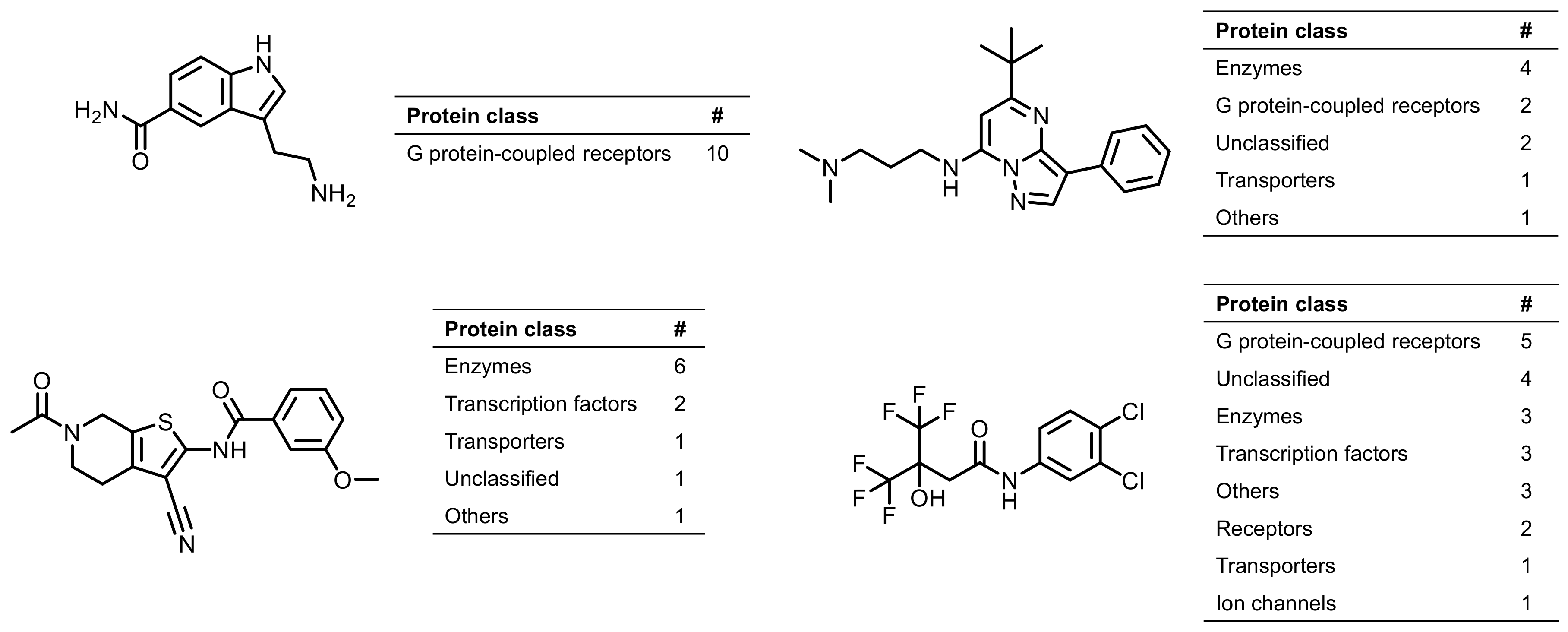
2.3. Promiscuous Compounds and Their Targets
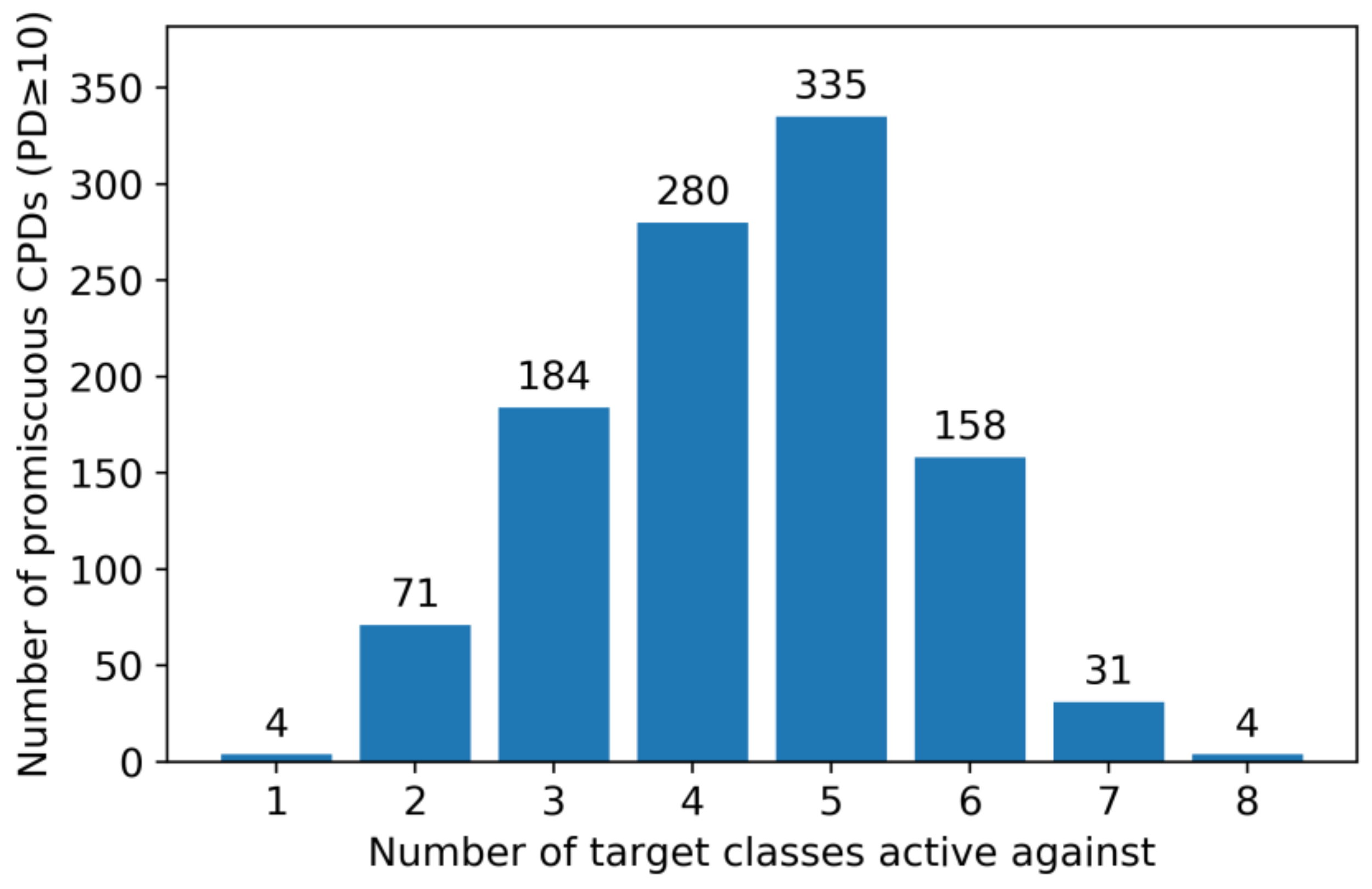
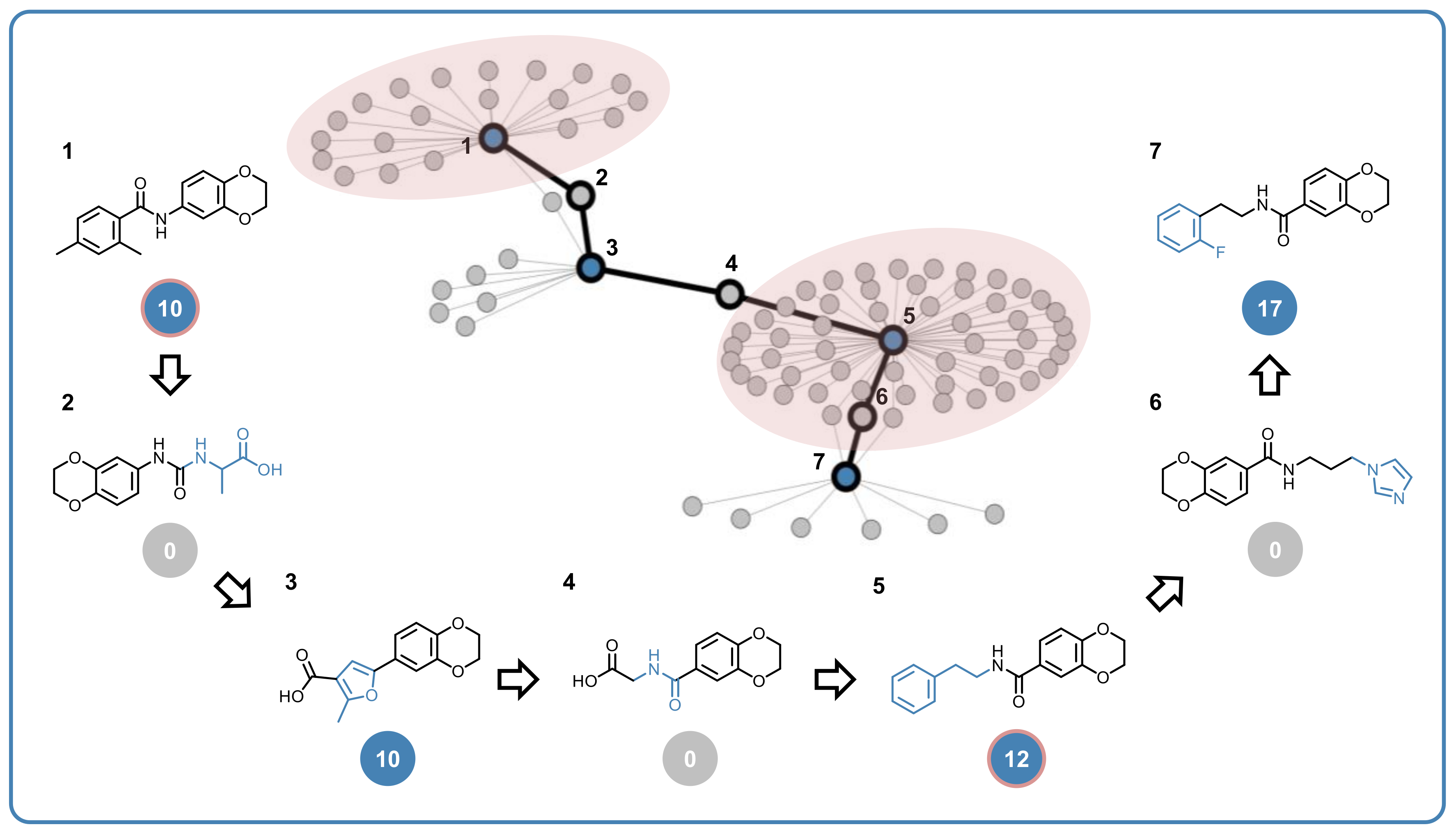
2.4. Multiclass Ligands
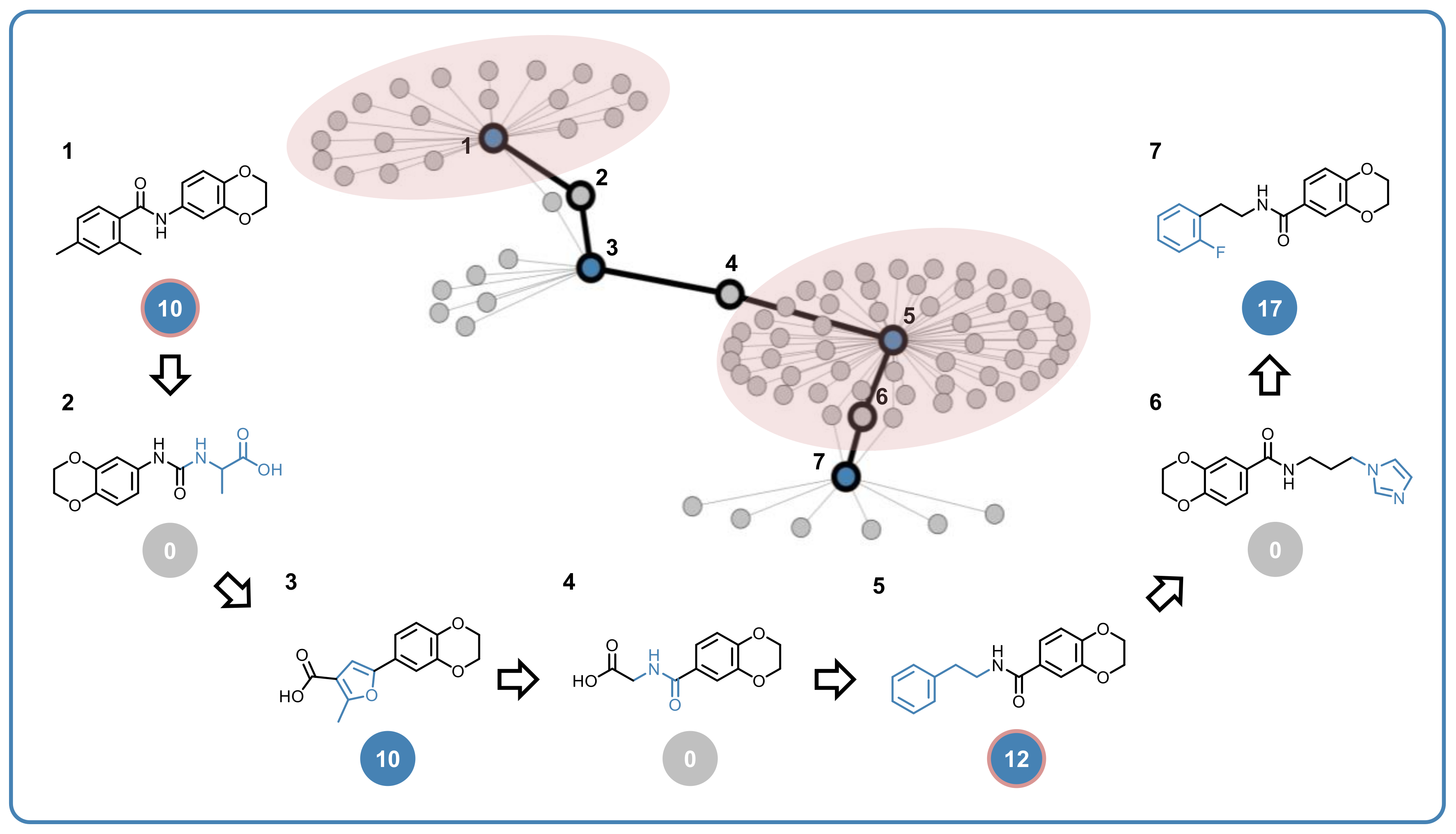
2.5. Structure–Promiscuity Relationships
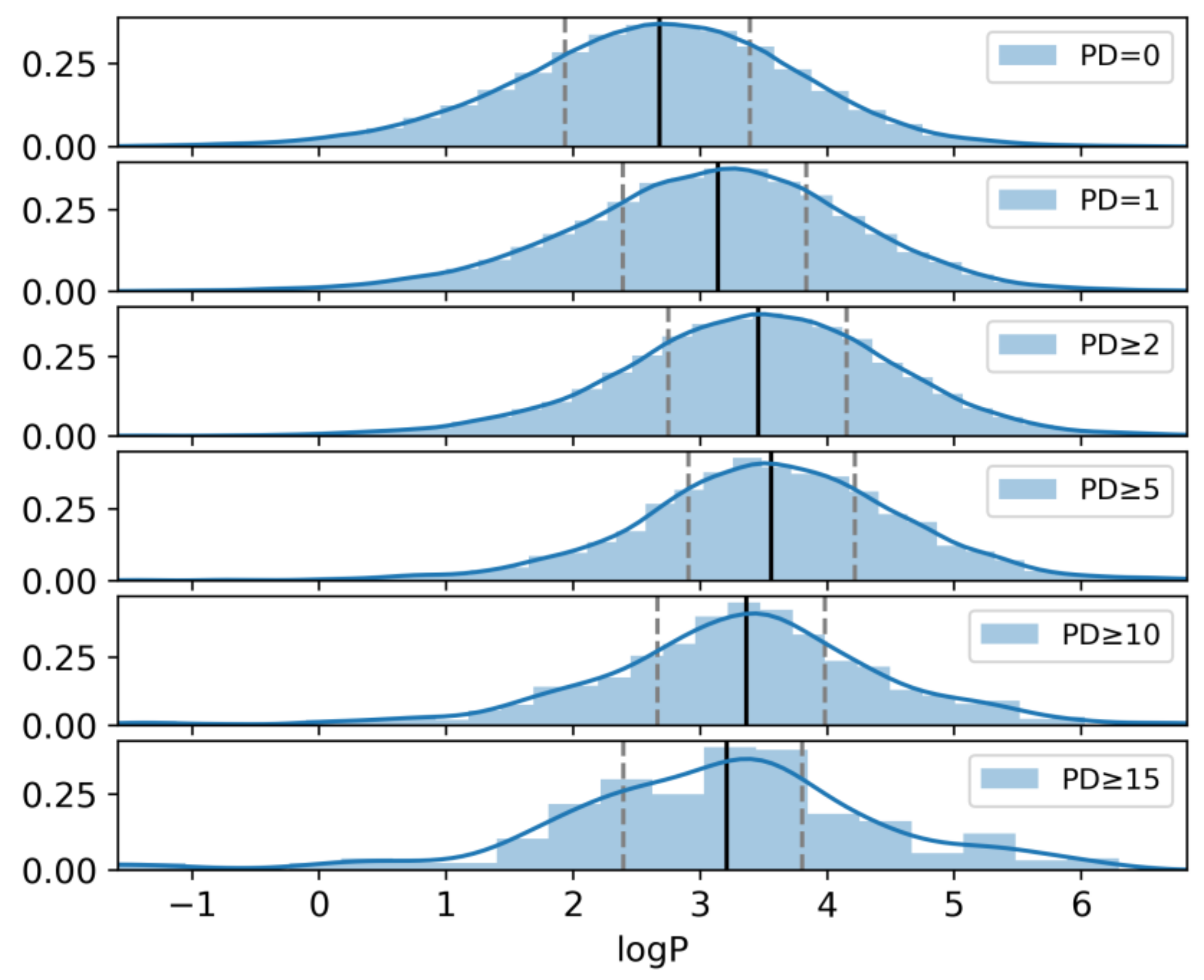
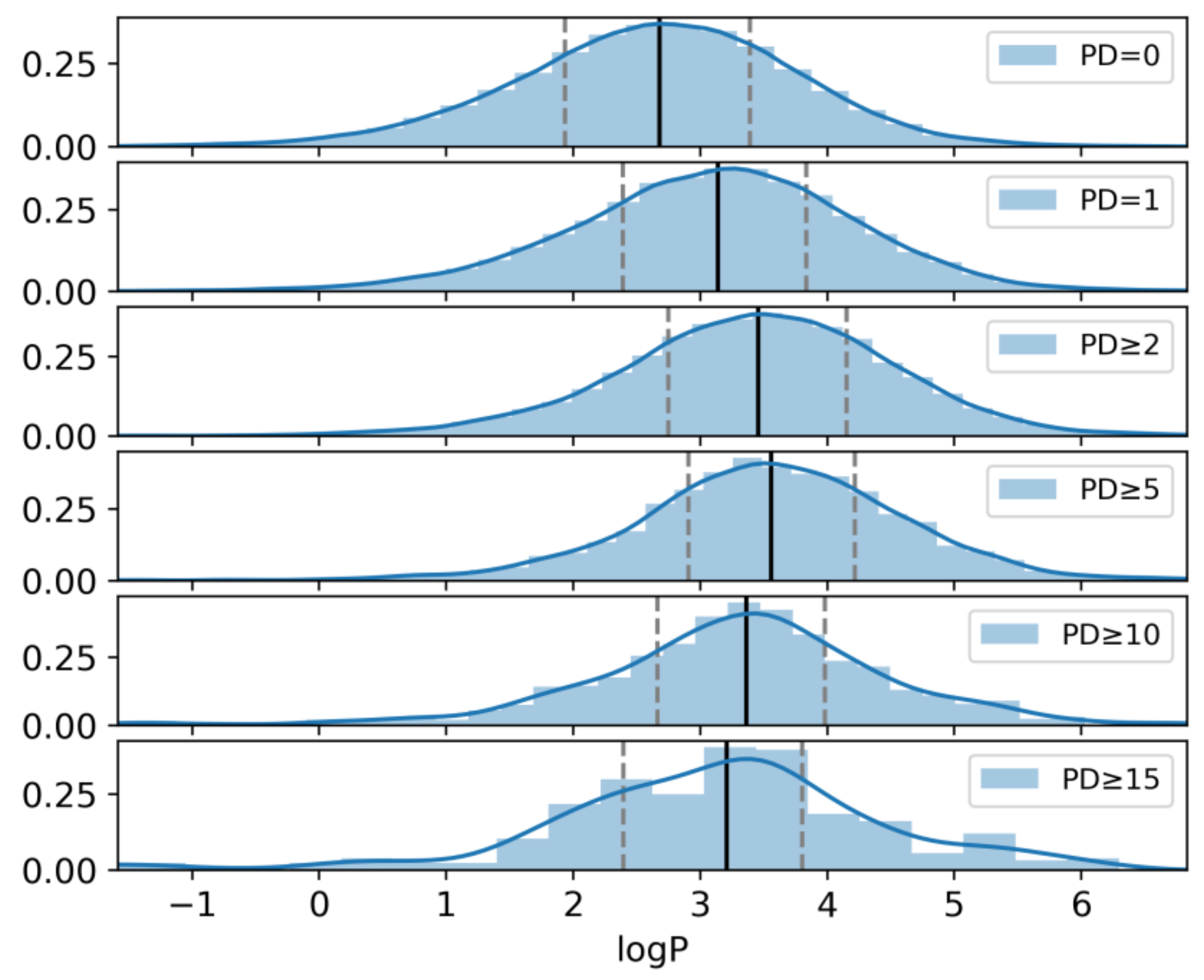
2.6. Hydrophobicity
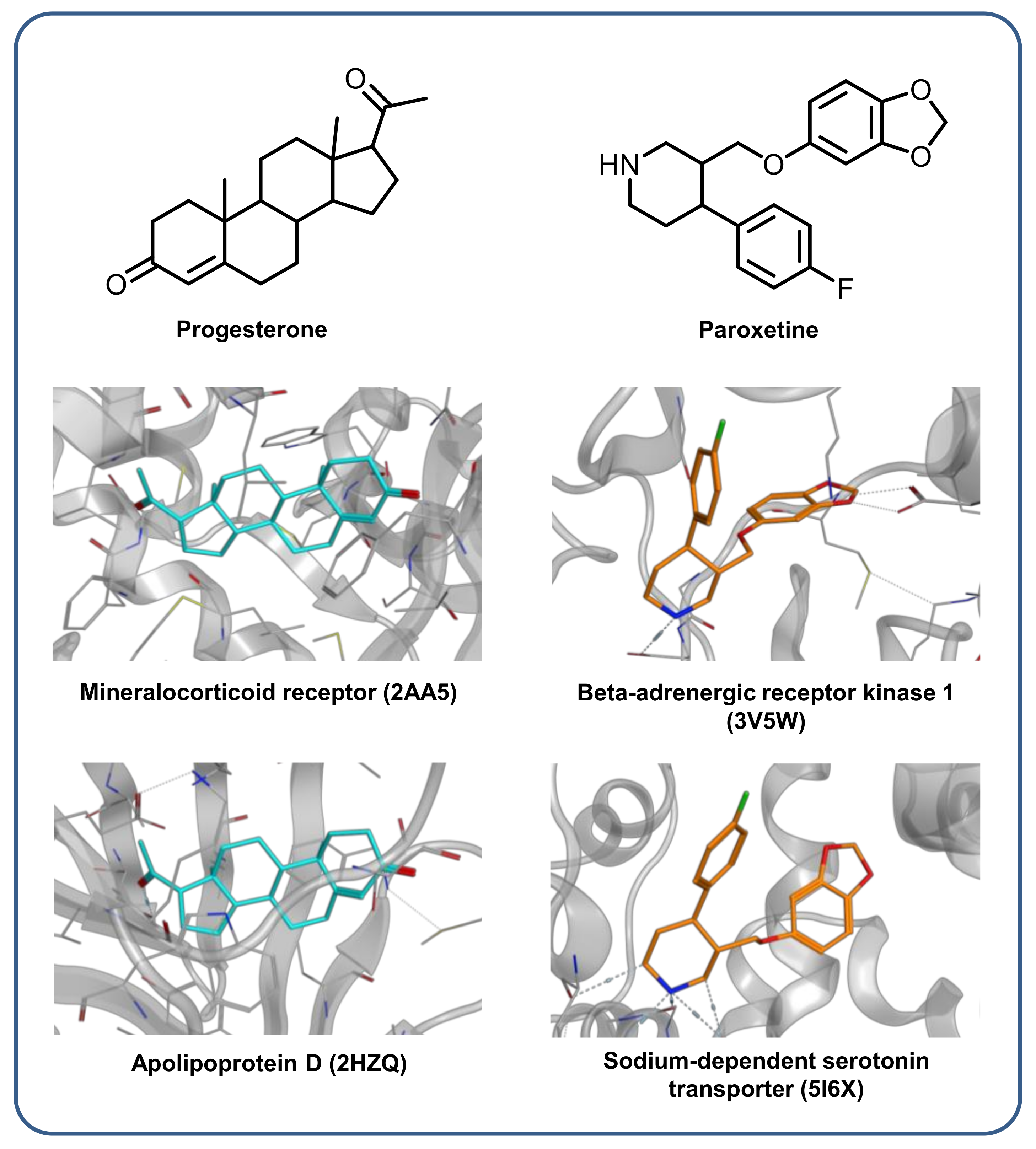
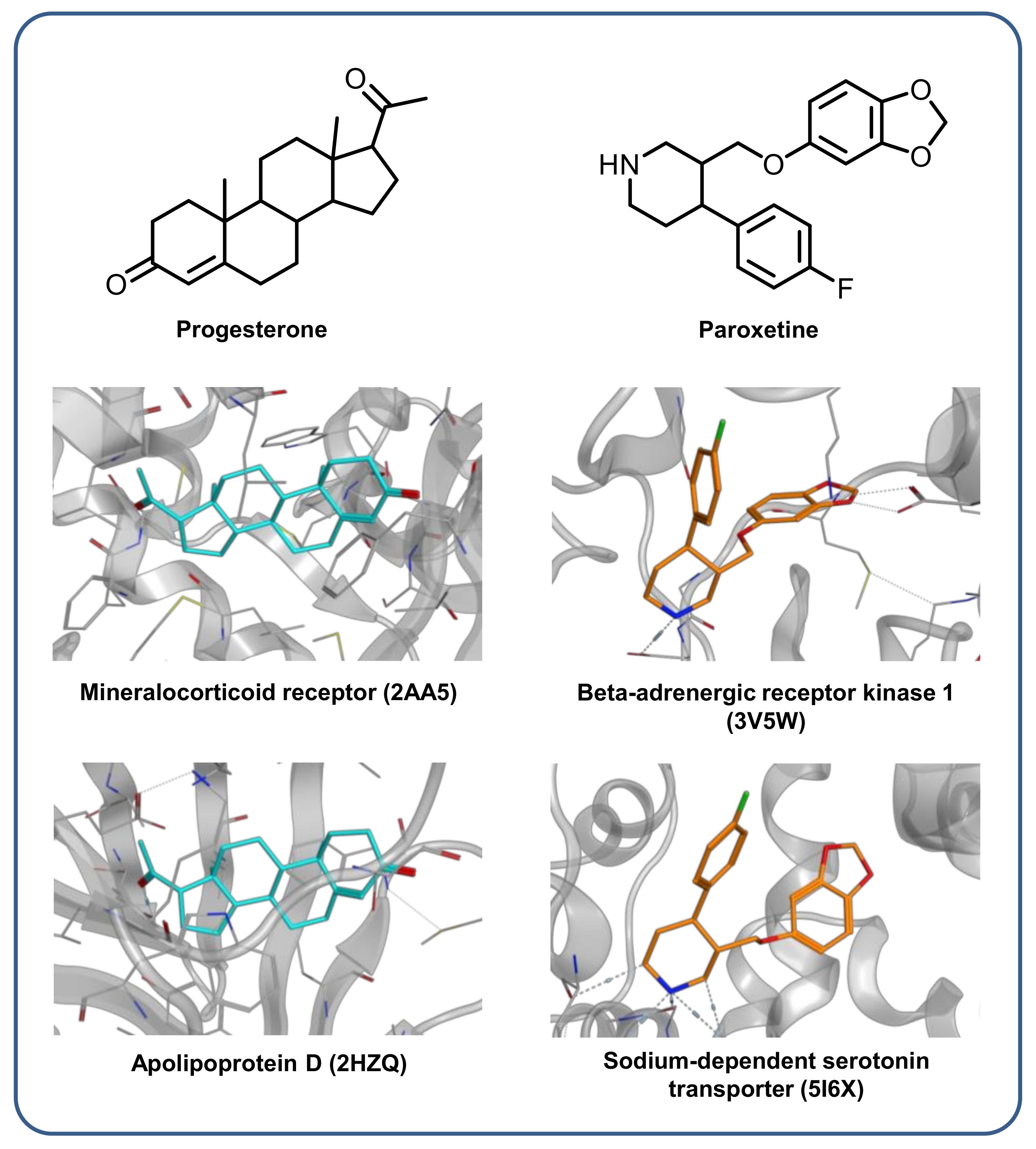
2.7. X-Ray Structures with Multiclass Ligands
3. Materials and Methods
3.1. Biological Screening Data
3.2. Compound Selection and Activity Assignment
3.3. Eliminating Compounds with Potential Chemical Liabilities
3.4. Target Classes
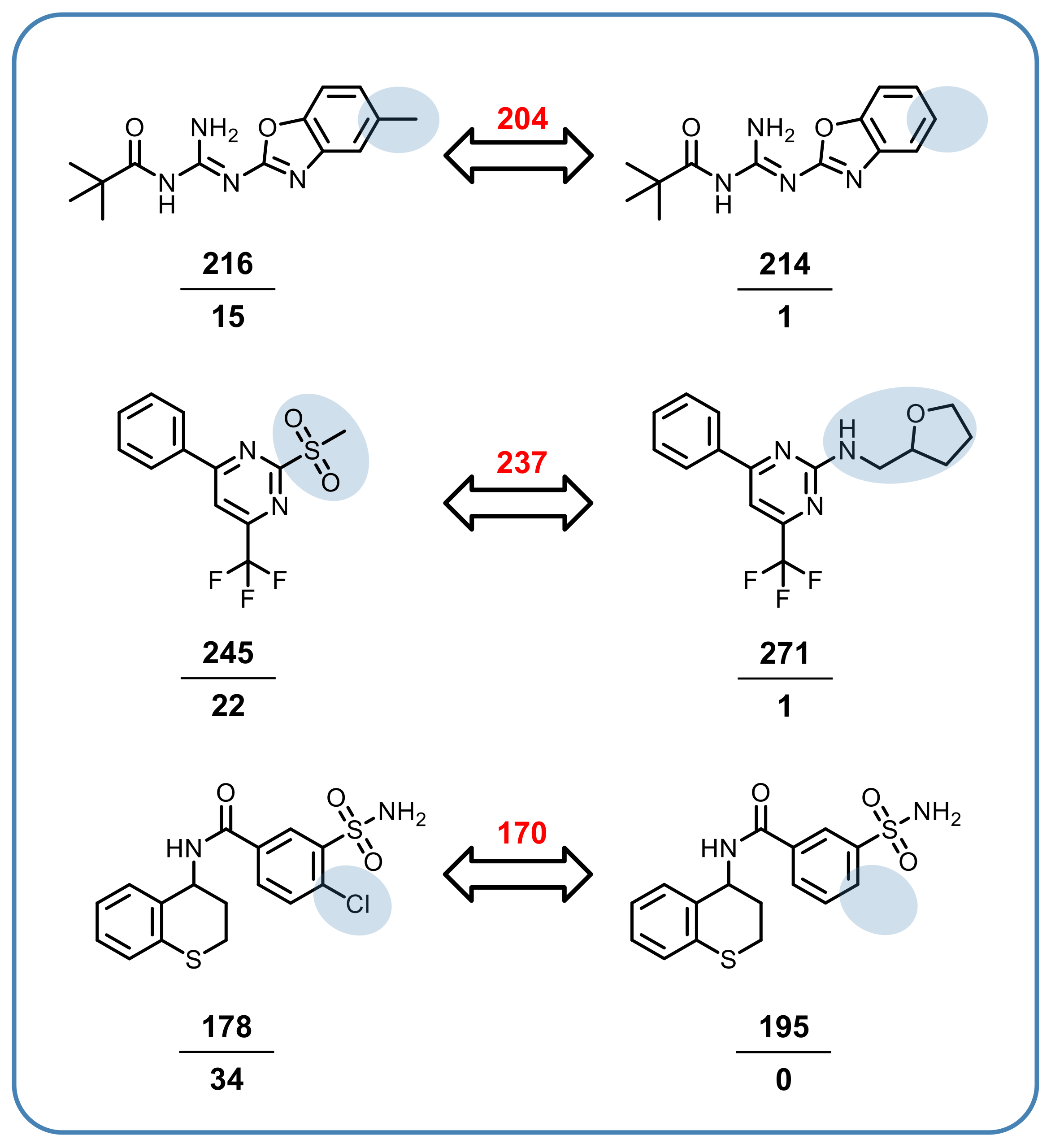
3.5. Promiscuity Cliffs
3.6. X-Ray Structures with Multiclass Ligands
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Bolognesi, L.M. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Rosini, M. Polypharmacology: The rise of multitarget drugs over combination therapies. Future Med. Chem. 2014, 6, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A. Multitarget drug discovery and polypharmacology. Chem. Med. Chem. 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Bajorath, J. Compound promiscuity — what can we learn from current data. Drug Discov. Today 2013, 18, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Gilberg, E.; Gütschow, M.; Bajorath, J. Promiscuous ligands from experimental structures, binding conformations, and protein family dependent interaction hotspots. ACS Omega 2019, 4, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An aggregation advisor for ligand discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef]
- Baell, J.B.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M., Jr.; Schepartz, A.; Wang, S. The ecstasy and agony of assay interference compounds. J. Chem. Inf. Model. 2017, 57, 387–390. [Google Scholar] [CrossRef]
- Stumpfe, D.; Tinivella, A.; Rastelli, G.; Bajorath, J. Promiscuity of inhibitors of human protein kinases at varying data confidence levels and test frequencies. RSC Adv. 2017, 7, 41265–41271. [Google Scholar] [CrossRef]
- Hu, Y.; Bajorath, J. Entering the ‘big data’ era in medicinal chemistry: Molecular promiscuity analysis revisited. Future Sci. OA 2017, 3, FSO179. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.B.; Shoemaker, A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017, 45, D955–D963. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Jasial, S.; Gilberg, E.; Bajorath, J. Structure-Promiscuity relationship puzzles - extensively assayed analogs with large differences in target annotations. AAPS J. 2017, 19, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed]
- OEChem TK. OpenEye Scientific Software, Inc., Santa Fe, NM, US. Available online: www.eyesopen.com/oechem-tk (accessed on 1 November 2019).
- Baell, B.J.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Cheminformatics and Machine Learning Software. Available online: http://www.rdkit.org (accessed on 1 November 2019).
- Sterling, T.; Irwin, J. ZINC 15 – Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the utility of alerts for pan-assay interference compounds. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef]
- Jasial, S.; Hu, Y.; Bajorath, J. How frequently are pan-assay interference compounds active? Large-Scale analysis of screening data reveals diverse activity profiles, low global hit frequency, and many consistently inactive compounds. J. Med. Chem. 2017, 60, 3879–3886. [Google Scholar] [CrossRef]
- Bruns, R.F.; Watson, I.A. Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.A.; Crippen, G.M. Prediction of physiochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Dimova, D.; Bajorath, J. Rationalizing promiscuity cliffs. ChemMedChem 2018, 13, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Hu, Y.; Vogt, M.; Stumpfe, D.; Bajorath, J. MMP-Cliffs: Systematic identification of activity cliffs on the basis of matched molecular pairs. J. Chem. Inf. Model. 2012, 52, 1138–1145. [Google Scholar] [CrossRef]
- Kenny, P.W.; Sadowski, J. Structure Modification in chemical databases. In Chemoinformatics in Drug Discovery; Oprea, T.I., Ed.; Wiley-VCH: Weinheim, Germany, 2005; pp. 271–285. [Google Scholar] [CrossRef]
- Hussain, J.; Rea, C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J. Chem. Inf. Model. 2010, 50, 339–348. [Google Scholar] [CrossRef]
- Miljković, F.; Vogt, M.; Bajorath, J. Systematic computational identification of promiscuity cliff pathways formed by inhibitors of the human kinome. J. Comput. Aided. Mol. Des. 2019, 33, 559–572. [Google Scholar] [CrossRef]
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The protein data bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef]
- Gilberg, E.; Stumpfe, D.; Bajorath, J. X-ray Structure-based identification of compounds with activity against targets from different families and generation of templates for multitarget ligand design. ACS Omega 2018, 3, 106–111. [Google Scholar] [CrossRef]
- Zenodo Platform. Available online: https://zenodo.org/record/3543341 (accessed on 15 November 2019).
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PD | Number of Compounds |
|---|---|
| All | 216,094 |
| PD = 0 | 129,215 |
| PD = 1 | 46,034 |
| PD ≥ 2 | 40,845 |
| PD ≥ 5 | 7592 |
| PD ≥ 10 | 1067 |
| PD ≥ 15 | 304 |
| Target Class | Number of Proteins |
|---|---|
| Enzymes | 481 |
| G protein-coupled receptors | 74 |
| Transcription factors | 60 |
| Ion channels | 15 |
| Receptors | 13 |
| Transporters | 7 |
| Others | 20 |
| Unclassified | 109 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feldmann, C.; Miljković, F.; Yonchev, D.; Bajorath, J. Identifying Promiscuous Compounds with Activity against Different Target Classes. Molecules 2019, 24, 4185. https://doi.org/10.3390/molecules24224185
Feldmann C, Miljković F, Yonchev D, Bajorath J. Identifying Promiscuous Compounds with Activity against Different Target Classes. Molecules. 2019; 24(22):4185. https://doi.org/10.3390/molecules24224185
Chicago/Turabian StyleFeldmann, Christian, Filip Miljković, Dimitar Yonchev, and Jürgen Bajorath. 2019. "Identifying Promiscuous Compounds with Activity against Different Target Classes" Molecules 24, no. 22: 4185. https://doi.org/10.3390/molecules24224185
APA StyleFeldmann, C., Miljković, F., Yonchev, D., & Bajorath, J. (2019). Identifying Promiscuous Compounds with Activity against Different Target Classes. Molecules, 24(22), 4185. https://doi.org/10.3390/molecules24224185