
Two-Step Azidoalkenylation of Terminal Alkenes Using Iodomethyl Sulfones


Abstract

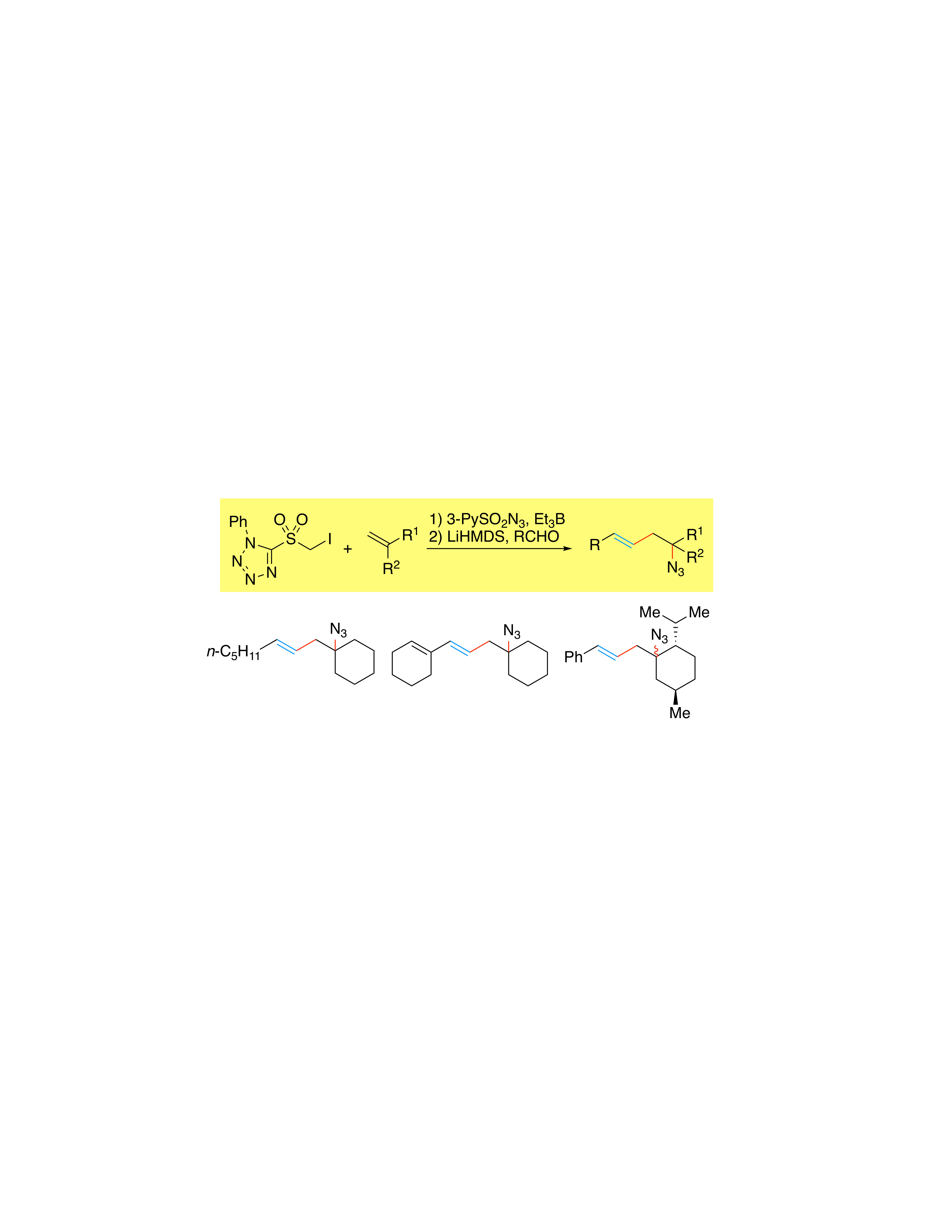{kind=link}
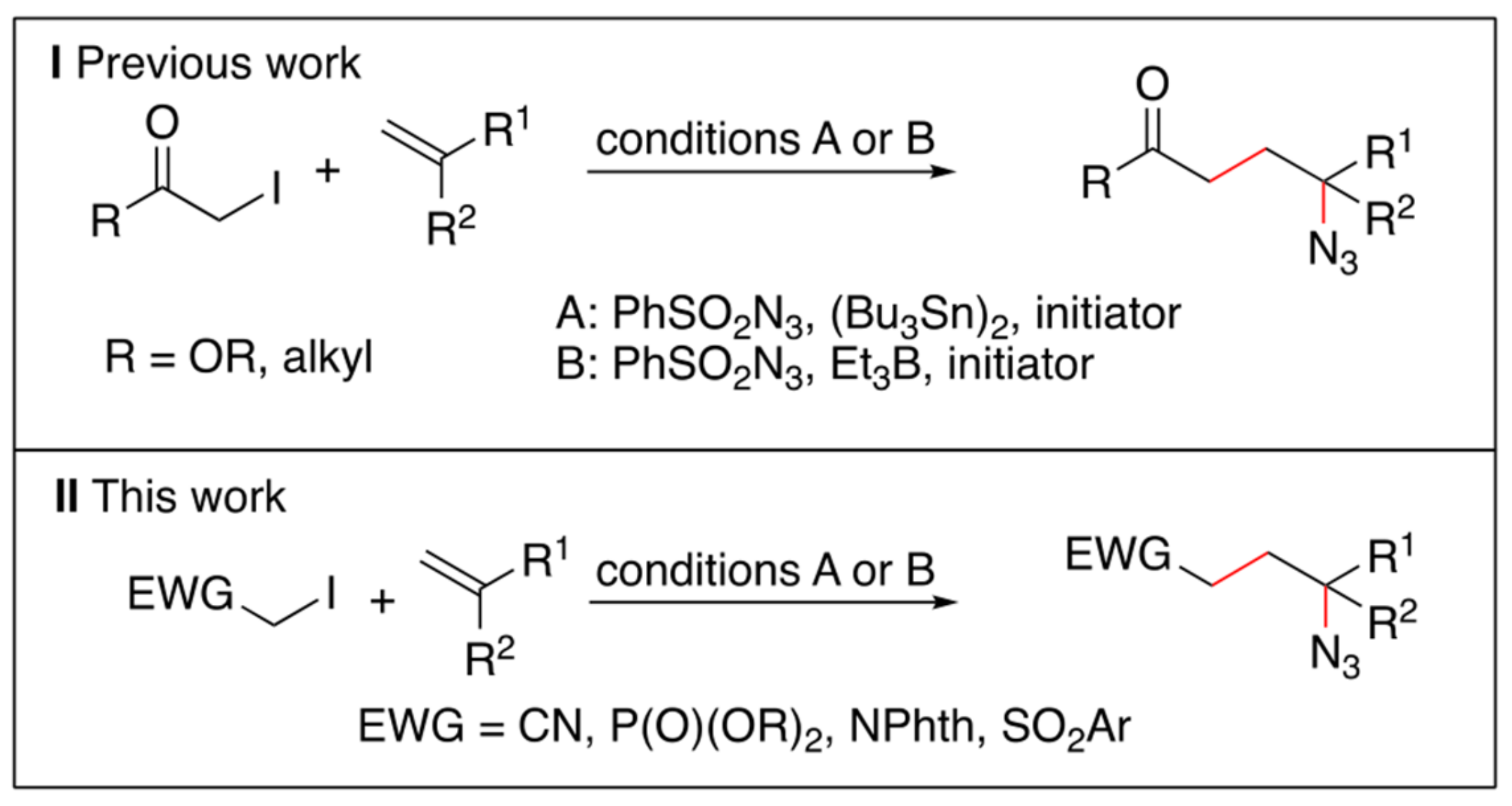{kind=link}
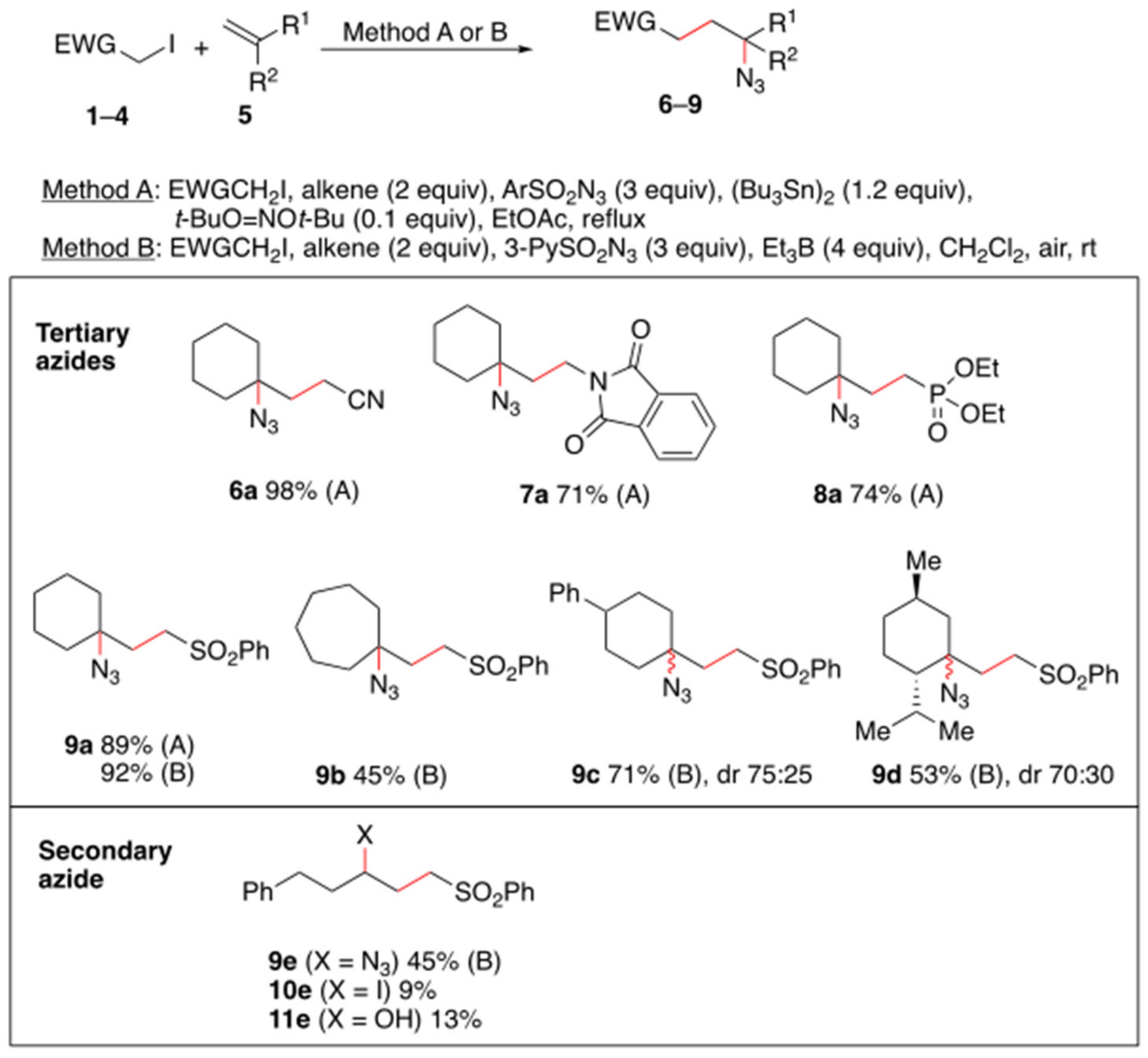{kind=link}
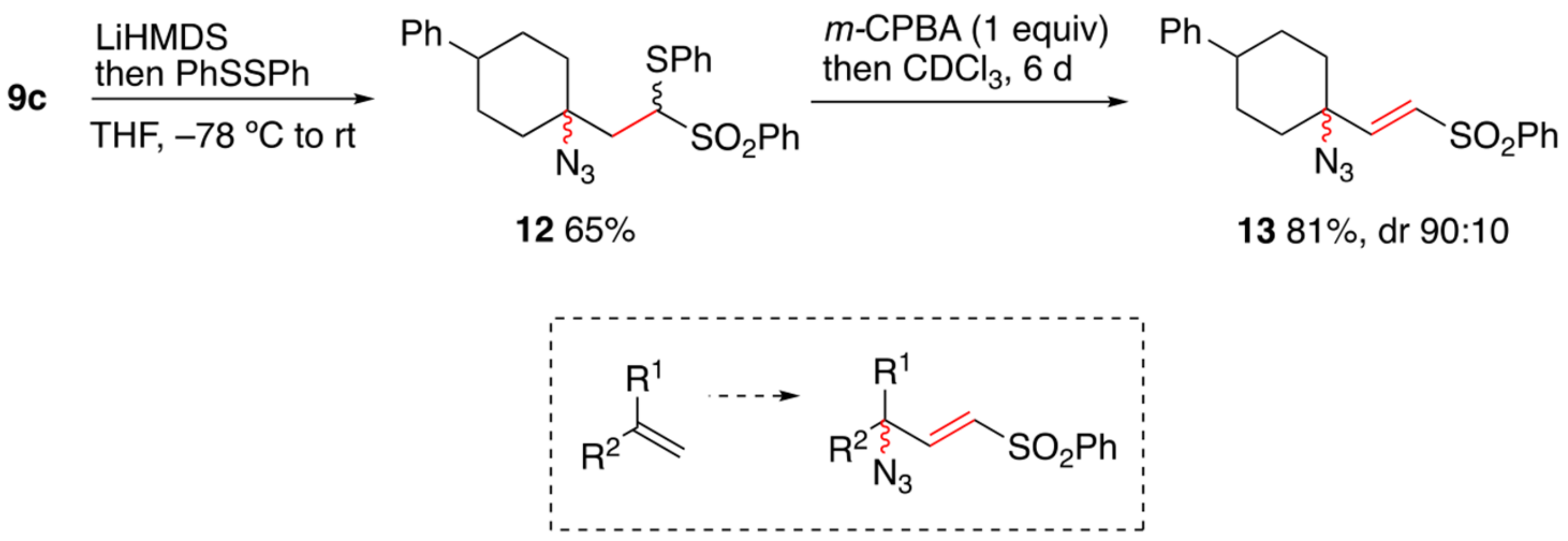{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
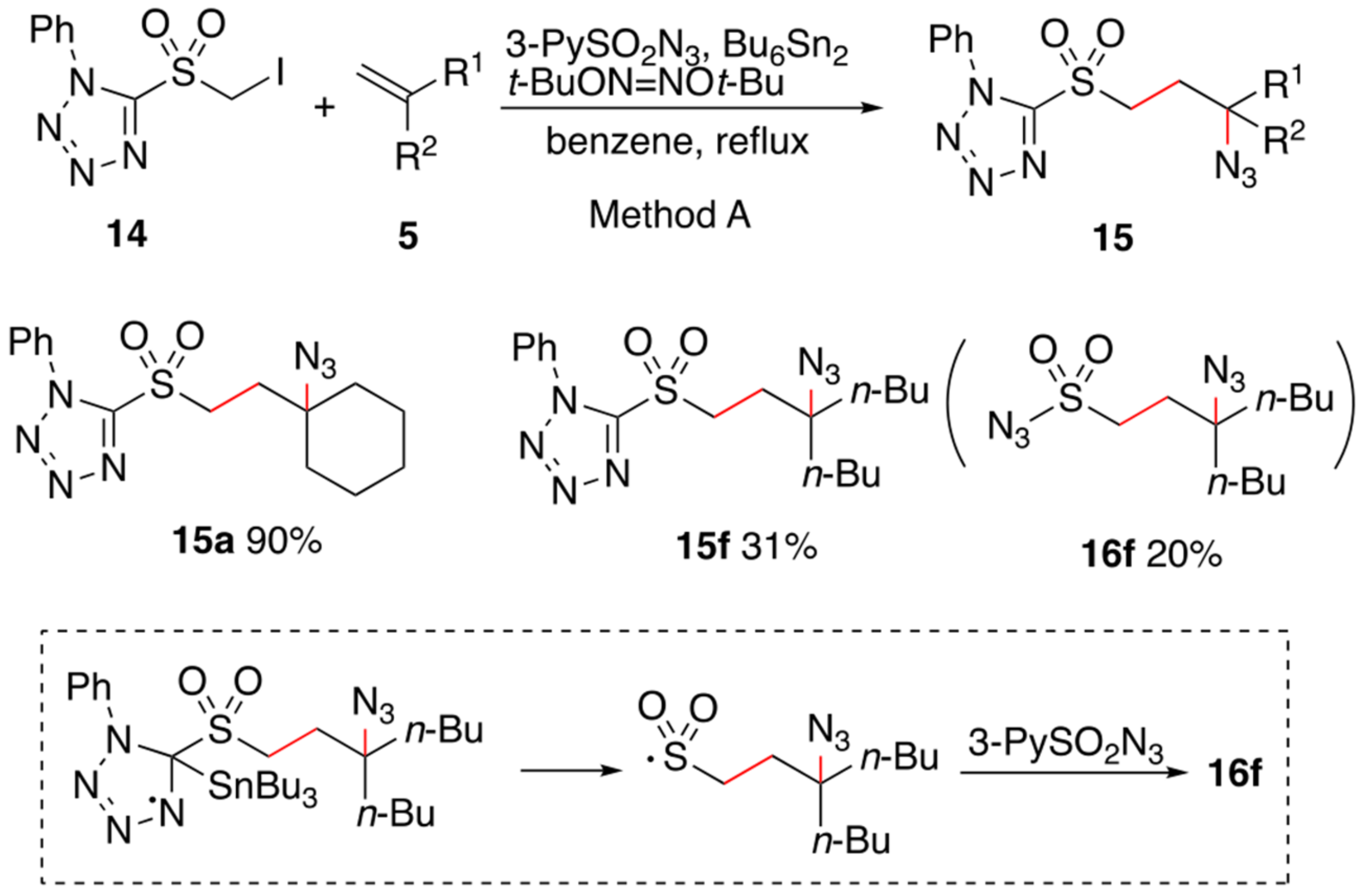
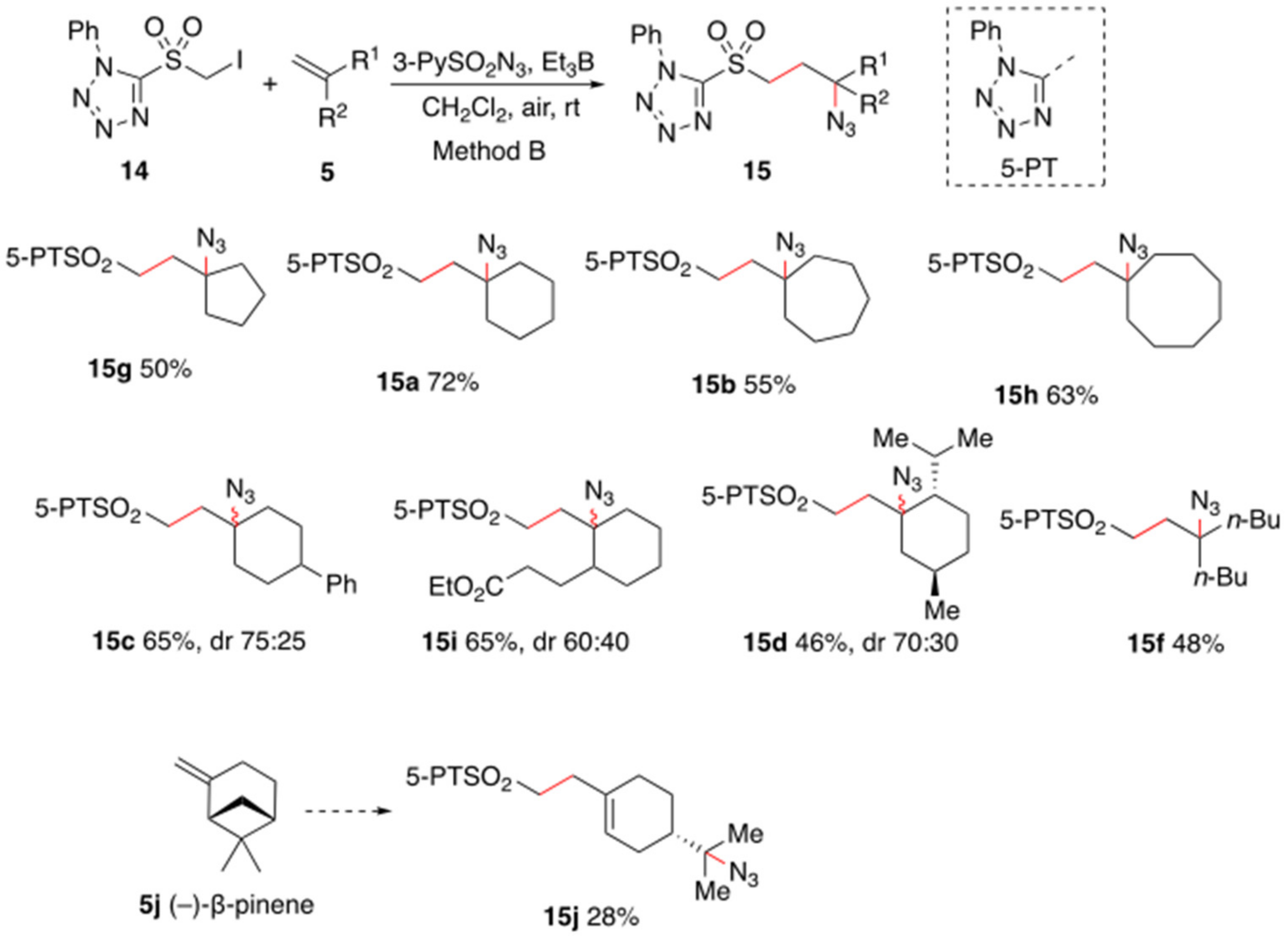
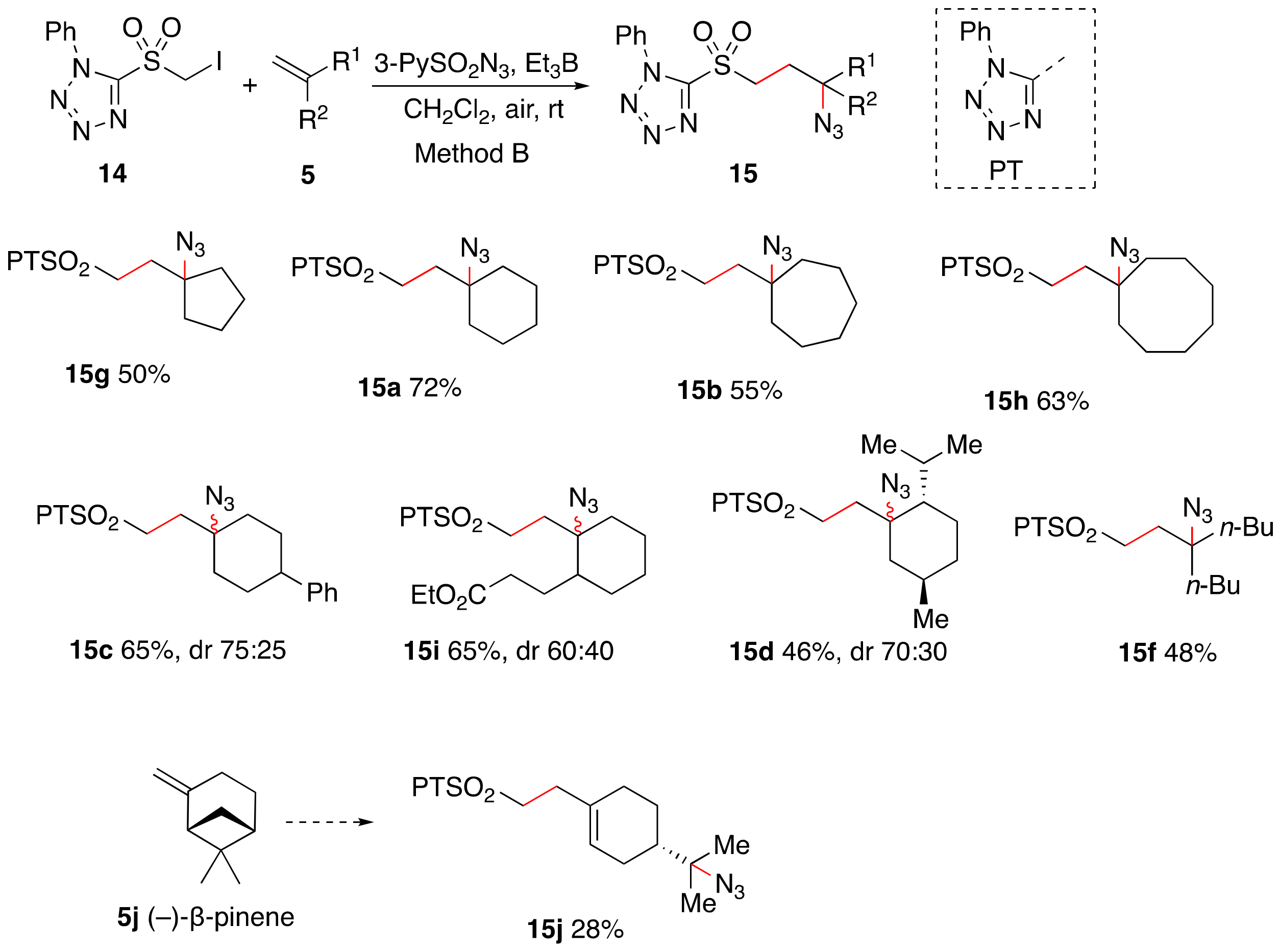
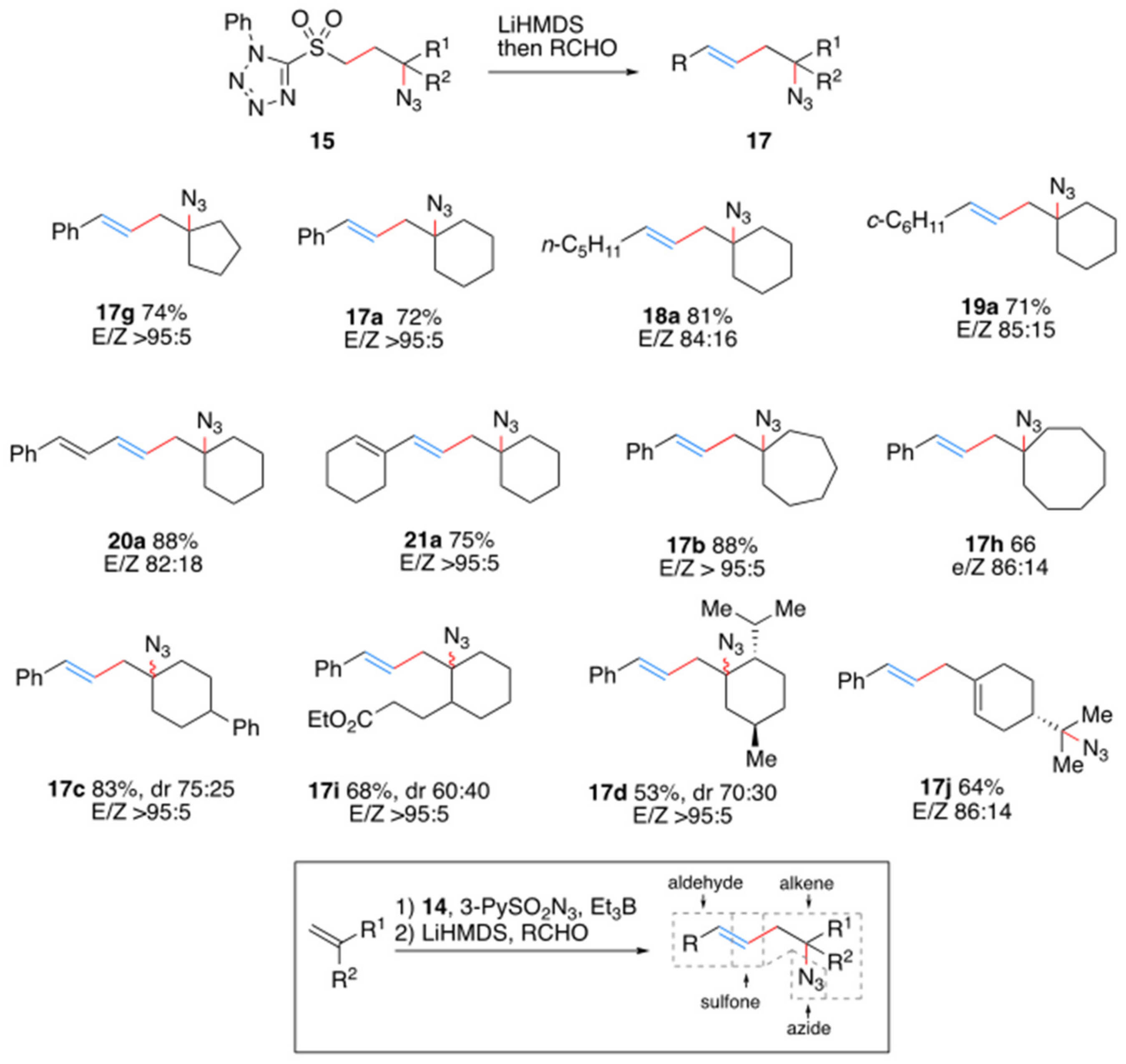
2. Results
3. Experimental Procedures
3.1. General Methods
3.2. General Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bräse, S.; Banert, K. Organic Azides: Syntheses and Applications; John Wiley & Sons: Chichester, UK, 2010; ISBN 978-0-470-51998-1. [Google Scholar]
- Tanimoto, H.; Kakiuchi, K. Recent applications and developments of organic azides in total synthesis of natural products. Nat. Prod. Commun. 2013, 8, 1021–1034. [Google Scholar] [CrossRef]
- Chiba, S. Application of organic azides for the synthesis of nitrogen-containing molecules. Synlett 2012, 23, 21–44. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides. An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Gritsan, N.; Platz, M. Photochemistry of azides: The azide/nitrene interface. In Organic Azides—Syntheses and Applications; Bräse, S., Banert, K., Eds.; John Wiley & Sons: Chichester, UK, 2010; pp. 311–372. ISBN 978-0-470-68251-7. [Google Scholar]
- Kim, S. Radical cyclizations involving the evolution of nitrogen. Pure Appl. Chem. 1996, 68, 623–626. [Google Scholar] [CrossRef]
- Kim, S.; Joe, G.H.; Do, J.Y. Novel radical cyclizations of alkyl azides. A new route to N-Heterocycles. J. Am. Chem. Soc. 1994, 116, 5521–5522. [Google Scholar] [CrossRef]
- Montevecchi, P.C.; Navacchia, M.L.; Spagnolo, P. A study of vinyl radical cyclization onto the azido group by addition of sulfanyl, stannyl, and silyl radicals to alkynyl azides. Eur. J. Org. Chem. 1998, 1219–1226. [Google Scholar] [CrossRef]
- Wyler, B.; Brucelle, F.; Renaud, P. Preparation of the core structure of aspidosperma and strychnos alkaloids from aryl azides by a cascade radical cyclization. Org. Lett. 2016, 18, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Brucelle, F.; Renaud, P. Synthesis of a leucomitosane via a diastereoselective radical cascade. J. Org. Chem. 2013, 78, 6245–6252. [Google Scholar] [CrossRef] [PubMed]
- Minozzi, M.; Nanni, D.; Spagnolo, P. From azides to nitrogen-centered radicals: Applications of azide radical chemistry to organic synthesis. Chem. Eur. J. 2009, 15, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
- Wrobleski, A.; Coombs, T.C.; Huh, C.W.; Li, S.-W.; Aubé, J. The Schmidt reaction. Org. React. 2012, 78, 1–320. [Google Scholar]
- Aubé, J.; Fehl, C.; Liu, R.; McLeod, M.C.; Motiwala, H.F. Hofmann, Curtius, Schmidt, Lossen, and related reactions. Compr. Org. Synth. 2014, 6, 598–635. [Google Scholar]
- Nyfeler, E.; Renaud, P. Intramolecular Schmidt reaction: Applications in natural product synthesis. CHIMIA Int. J. Chem. 2006, 60, 276–284. [Google Scholar] [CrossRef]
- Palacios, F.; Alonso, C.; Aparicio, D.; Rubiales, G.; de los Santos, J.M. Aza-Wittig reaction in natural product syntheses. In Organic Azides; Wiley: Chichester, UK, 2010; pp. 437–467. ISBN 978-0-470-68251-7. [Google Scholar]
- Binder, W.H.; Kluger, C. Azide/alkyne-”click” reactions: Applications in material science and organic synthesis. Curr. Org. Chem. 2006, 10, 1791–1815. [Google Scholar] [CrossRef]
- Schilling, C.; Jung, N.; Bräse, S. Cycloaddition Reactions with azides: An overview. In Organic Azides—Syntheses and Applications; Bräse, S., Banert, K., Eds.; John Wiley & Sons: Chichester, UK, 2010; pp. 269–284. ISBN 978-0-470-68251-7. [Google Scholar]
- Pinho e Melo, T.M.V.D. Synthesis of azides. In Organic Azides—Syntheses and Applications; Bräse, S., Banert, K., Eds.; John Wiley & Sons: Chichester, UK, 2010; pp. 53–94. ISBN 978-0-470-68251-7. [Google Scholar]
- Jimeno, C.; Renaud, P. Radical chemistry with azides. In Organic Azides—Syntheses and Applications; Bräse, S., Banert, K., Eds.; John Wiley & Sons: Chichester, UK, 2010; pp. 239–267. ISBN 978-0-470-68251-7. [Google Scholar]
- Lapointe, G.; Kapat, A.; Weidner, K.; Renaud, P. Radical azidation reactions and their application in the synthesis of alkaloids. Pure Appl. Chem. 2012, 84, 1633–1641. [Google Scholar] [CrossRef]
- Panchaud, P.; Chabaud, L.; Landais, Y.; Ollivier, C.; Renaud, P.; Zigmantas, S. Radical amination with sulfonyl azides: A powerful method for the formation of CN bonds. Chem. Eur. J. 2004, 10, 3606–3614. [Google Scholar] [CrossRef]
- Renaud, P.; Ollivier, C.; Panchaud, P. Radical carboazidation of alkenes: An efficient tool for the preparation of pyrrolidinone derivatives. Angew. Chem. Int. Edit. 2002, 41, 3460–3462. [Google Scholar] [CrossRef]
- Panchaud, P.; Ollivier, C.; Renaud, P.; Zigmantas, S. Radical carboazidation: Expedient assembly of the core structure of various alkaloid families. J. Org. Chem. 2004, 69, 2755–2759. [Google Scholar] [CrossRef]
- Chabaud, L.; Landais, Y.; Renaud, P. Total synthesis of hyacinthacine A(1) and 3-epi-hyacinthacine A(1). Org. Lett. 2005, 7, 2587–2590. [Google Scholar] [CrossRef]
- Schär, P.; Renaud, P. Total synthesis of the marine alkaloid (+/−)-lepadiformine via a radical carboazidation. Org. Lett. 2006, 8, 1569–1571. [Google Scholar] [CrossRef]
- Weidner, K.; Giroult, A.; Panchaud, P.; Renaud, P. Efficient carboazidation of alkenes using a radical desulfonylative azide transfer process. J. Am. Chem. Soc. 2010, 132, 17511–17515. [Google Scholar] [CrossRef]
- Lapointe, G.; Schenk, K.; Renaud, P. Concise synthesis of pyrrolidine and indolizidine alkaloids by a highly convergent three-component reaction. Chem. Eur. J. 2011, 17, 3207–3212. [Google Scholar] [CrossRef]
- Lapointe, G.; Schenk, K.; Renaud, P. Total synthesis of (±)-cylindricine C. Org. Lett. 2011, 13, 4774–4777. [Google Scholar] [CrossRef]
- Gonçalves-Martin, M.G.; Zigmantas, S.; Renaud, P. Formal synthesis of (−)-cephalotaxine. Helv. Chim. Acta 2012, 95, 2502–2514. [Google Scholar] [CrossRef]
- Huang, W.-Y.; Lü, L. The reaction of perfluoroalkanesulfinates VII. Fenton reagent-initiated addition of sodium perfluoroalkanesulfinates to alkenes. Chin. J. Chem. 1992, 10, 365–372. [Google Scholar] [CrossRef]
- Wei, X.-H.; Li, Y.-M.; Zhou, A.-X.; Yang, T.-T.; Yang, S.-D. Silver-catalyzed carboazidation of arylacrylamides. Org. Lett. 2013, 15, 4158–4161. [Google Scholar] [CrossRef]
- Bunescu, A.; Ha, T.M.; Wang, Q.; Zhu, J. Copper-catalyzed three-component carboazidation of clkenes with acetonitrile and sodium azide. Angew. Chem. Int. Ed. 2017, 56, 10555–10558. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Y.; Wu, C.-S.; Wang, Z.; Luo, Y. Fe-Catalyzed three-component carboazidation of alkenes with alkanes and trimethylsilyl azide. Chem. Commun. 2018, 54, 11013–11016. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Ramkumar, N.; Chiou, M.-F.; Jian, W.; Li, Y.; Su, J.-H.; Zhang, X.; Bao, H. Iron-catalyzed carboazidation of alkenes and alkynes. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, C.; Renaud, P. Formation of carbon-nitrogen bonds via a novel radical azidation process. J. Am. Chem. Soc. 2000, 122, 6496–6497. [Google Scholar] [CrossRef]
- Ollivier, C.; Renaud, P. A novel approach for the formation of carbon - nitrogen bonds: Azidation of alkyl radicals with sulfonyl azides. J. Am. Chem. Soc. 2001, 123, 4717–4727. [Google Scholar] [CrossRef]
- Panchaud, P.; Renaud, P. A convenient tin-free procedure for radical carboazidation and azidation. J. Org. Chem. 2004, 69, 3205–3207. [Google Scholar] [CrossRef] [PubMed]
- Panchaud, P.; Renaud, P. 3-Pyridinesulfonyl azide. In Encyclopedia of Reagents for Organic Synthesis; John Wiley Sons, Ltd.: Chichester, UK, 2006; ISBN 978-0-470-84289-8. [Google Scholar]
- Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley Sons, Ltd.: Chichester, UK, 2000; ISBN 978-0-471-89149-9. [Google Scholar]
- Cren, S.; Schär, P.; Renaud, P.; Schenk, K. Diastereoselectivity control of the radical carboazidation of substituted methylenecyclohexanes. J. Org. Chem. 2009, 74, 2942–2946. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.R.; Chern, C.Y.; Jaszberenyi, J.C. The Invention of radical reactions. XXXIII. Homologation reactions of carboxylic acids by radical chain chemistry. Aust. J. Chem. 1995, 48, 407–425. [Google Scholar] [CrossRef]
- Vita, M.V.; Caramenti, P.; Waser, J. Enantioselective synthesis of homoallylic azides and nitriles via palladium-catalyzed decarboxylative allylation. Org. Lett. 2015, 17, 5832–5835. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, P.R.; Cole, W.J.; Kocieński, P.J.; Morley, A. A stereoselective synthesis of trans-1,2-sisubstituted alkenes based on the condensation of aldehydes with metallated 1-phenyl-1H-tetrazol-5-yl sulfones. Synlett 1998, 1998, 26–28. [Google Scholar] [CrossRef]
- Marko, I.; Pospisil, J. Julia, Julia–Kocienski, and related sulfur-based alkenations. In Category 6, Compounds with All-Carbon Functions, Science of Synthesis; Neier, R., Bellus, D., Eds.; G. Thieme Verlag: Stuttgart, Germany, 2010; Volume 47a, ISBN 978-3-13-119011-6. [Google Scholar]
- Lebrun, M.-E.; Le Marquand, P.; Berthelette, C. Stereoselective synthesis of Z alkenyl halides via Julia olefination. J. Org. Chem. 2006, 71, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bowman, E.J.; Bowman, B.J.; Porco, J.A. Total synthesis of the salicylate enamide macrolide oximidine III: Application of relay ring-closing metathesis. Angew. Chem. Int. Ed. 2004, 43, 3601–3605. [Google Scholar] [CrossRef]
- Kamijo, S.; Kamijo, K.; Murafuji, T. Aryl ketone mediated photoinduced radical coupling for the alkylation of benzazoles employing saturated heterocyclic compounds. Synthesis 2019, 51, 3859–3864. [Google Scholar]
- David Mendenhall, G. The Lewis acid catalyzed reaction of trans-hyponitrite ion with alkyl halides. Tetrahedron Lett. 1983, 24, 451–452. [Google Scholar] [CrossRef]
- Panchaud, P.; Renaud, P. 3-Pyridinesulfonyl azide: A useful reagent for radical azidation. Adv. Synth. Catal. 2004, 346, 925–928. [Google Scholar] [CrossRef]
- Harrowven, D.C.; Guy, I.L. KF–Silica as a stationary phase for the chromatographic removal of tin residues from organic compounds. Chem. Commun. 2004, 1968–1969. [Google Scholar] [CrossRef] [PubMed]
- Merchant, R.R.; Edwards, J.T.; Qin, T.; Kruszyk, M.M.; Bi, C.; Che, G.; Bao, D.-H.; Qiao, W.; Sun, L.; Collins, M.R.; et al. Modular radical cross-coupling with sulfones enables access to sp3-rich (fluoro)alkylated scaffolds. Science 2018, 360, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M.E.; Fier, P.S. Desulfonylative arylation of redox-active alkyl sulfones with aryl bromides. Org. Lett. 2019, 21, 5650–5654. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples are not available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millius, N.; Lapointe, G.; Renaud, P. Two-Step Azidoalkenylation of Terminal Alkenes Using Iodomethyl Sulfones. Molecules 2019, 24, 4184. https://doi.org/10.3390/molecules24224184
Millius N, Lapointe G, Renaud P. Two-Step Azidoalkenylation of Terminal Alkenes Using Iodomethyl Sulfones. Molecules. 2019; 24(22):4184. https://doi.org/10.3390/molecules24224184
Chicago/Turabian StyleMillius, Nicolas, Guillaume Lapointe, and Philippe Renaud. 2019. "Two-Step Azidoalkenylation of Terminal Alkenes Using Iodomethyl Sulfones" Molecules 24, no. 22: 4184. https://doi.org/10.3390/molecules24224184
APA StyleMillius, N., Lapointe, G., & Renaud, P. (2019). Two-Step Azidoalkenylation of Terminal Alkenes Using Iodomethyl Sulfones. Molecules, 24(22), 4184. https://doi.org/10.3390/molecules24224184