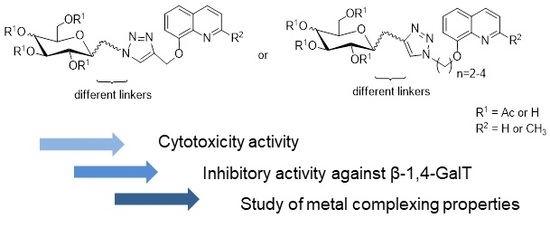
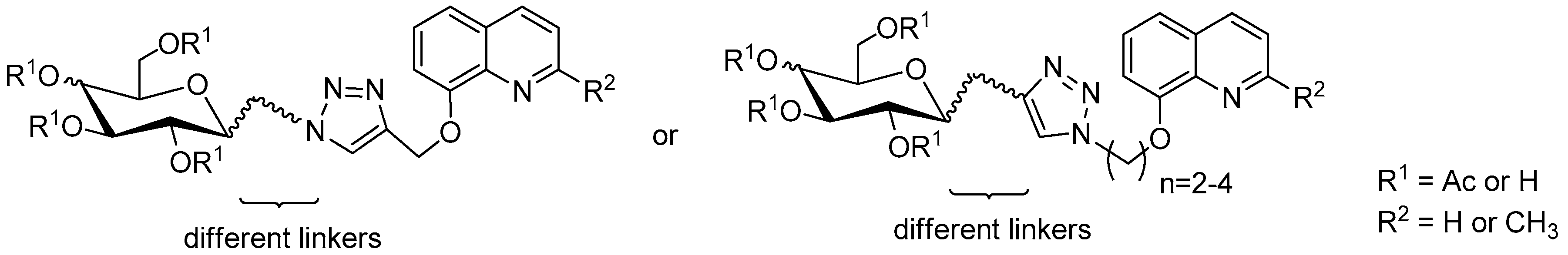
3.2.4. Synthesis of Glycoconjugates 45–108
The appropriate derivatives of 8-hydoxyquinoline 3–10 (0.5 mmol) and sugar derivatives 21–24, 27–30, 33–44 (0.5 mmol) were dissolved in dry THF (5 mL) and i-PrOH (5 mL). To the obtained solution, CuSO4·5H2O (0.1 mmol, 25.0 mg) dissolved in H2O (2.5 mL) and sodium ascorbate (0.2 mmol, 39.6 mg) dissolved in H2O (2.5 mL) were added. The reaction mixture was stirred for 24 h at room temperature. The progress of the reaction was monitored on TLC in an CHCl3:CH3OH eluents system (20:1 for protected or 2:1 for unprotected compounds). After completion, the reaction mixture was concentrated in vacuo and purified using column chromatography (dry loading; toluene:AcOEt, 2:1 and CHCl3:MeOH, 100:1 for fully protected glycoconjugates or CHCl3:MeOH, gradient: 50:1 to 2:1 for glycoconjugates with unprotected sugar part) to give products 45–108.
Glycoconjugate 45: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 21 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a yellow solid (219.2 mg, 73%); m.p.: 130–133 °C; [α]24D = −36.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.84, 1.97, 2.01, 2.07 (4s, 12H, CH3CO), 3.66 (ddd, 1H, J = 2.3 Hz, J = 4.6 Hz, J = 9.8 Hz, H-5glu), 4.10 (dd, 1H, J = 2.3 Hz, J = 12.3 Hz, H-6aglu), 4.21 (dd, 1H, J = 4.6 Hz, J = 12.3 Hz, H-6bglu), 4.63 (d, 1H, J = 8.0 Hz, H-1glu), 4.59–4.70 (m, 2H, CH2N), 4.80 and 4.88 (qAB, 2H, J = 12.7 Hz, CH2C), 4.95 (dd, 1H, J = 8.0 Hz, J = 9.4 Hz H-2glu), 5.01 (t, 2H, J = 5.2 Hz, CH2O), 5.04 (dd-t, 1H, J = 9.4 Hz, J = 9.8 Hz, H-4glu), 5.13 (dd-t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-3glu), 7.05 (dd, 1H, J = 2.1 Hz, J = 6.4 Hz, H-7chin), 7.42–7.55 (m, 3H, H-3chin, H-5chin, H-6chin), 8.21 (d, 1H, J = 8.1 Hz, H-4chin), 8.27 (s, 1H, H-5triaz), 8.99 (d, 1H, J = 2.9 Hz, H-2chin); 13C NMR (100 MHz, CDCl3): δ 20.47, 20.48, 20.59, 20.75, 49.75, 61.84, 62.40, 67.89, 68.34, 71.17, 71.82, 72.86, 99.32, 110.31, 121.07, 121.95, 125.10, 126.87, 129.65, 136.68, 139.55, 143.93, 149.26, 153.45, 169.36, 169.40, 170.18, 170.66; HRMS (ESI-TOF): calcd for C28H33N4O11 ([M + H]+): m/z 601.2146; found: m/z 601.2148.
Glycoconjugate 46: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 22 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a yellow solid (222.2 mg, 74%); m.p.: 69–72 °C; [α]24D = −30.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.85, 1.95, 2.05, 2.13 (4s, 12H, CH3CO), 3.83-3.92 (m, 1H, H-5gal), 4.10-4.16 (m, 2H, H-6agal, H-6bgal), 4.60 (d, 1H, J = 8.0 Hz, H-1gal), 4.62–4.69 (m, 2H, CH2N), 4.79 and 4.90 (qAB, 2H, J = 12.6 Hz, CH2C), 4.94 (dd, 1H, J = 3.4 Hz, J = 10.4 Hz H-3gal), 4.97-5.05 (m, 2H, CH2O), 5.17 (dd, 1H, J = 8.0 Hz, J = 10.4 Hz, H-2gal), 5.35 (dd, 1H, J = 0.7 Hz, J = 3.4 Hz, H-4gal), 6.99–7.12 (m, 1H, H-7chin), 7.38–7.58 (m, 3H, H-3chin, H-5chin, H-6chin), 8.12–8.34 (m, 2H, H-4chin, H-5triaz), 8.99 (bs, 1H, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 20.26, 20.28, 20.35, 20.47, 49.18, 61.17, 61.76, 67.25, 67.43, 68.53, 69.91, 70.19, 98.99, 110.74, 120.57, 121.98, 124.93, 126.84, 129.15, 136.26, 139.30, 143.07, 149.00, 153.50, 169.03, 169.41, 169.85, 169.88; HRMS (ESI-TOF): calcd for C28H33N4O11 ([M + H]+): m/z 601.2146; found: m/z 601.2145.
Glycoconjugate 47: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 21 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a yellow solid (307.3 mg, 100%); m.p.: 119-123 °C; [α]24D = −32.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.83, 1.91, 1.98, 2.02 (4s, 12H, CH3CO), 2.69 (s, 3H, CH3), 3.93 (ddd, 1H, J = 2.4 Hz, J = 4.9 Hz, J = 10.0 Hz, H-5glu), 4.01 (dd, 1H, J = 2.4 Hz, J = 12.3 Hz, H-6aglu), 4.15 (dd, 1H, J = 4.9 Hz, J = 12.3 Hz, H-6bglu), 4.59 (t, 2H, J = 4.9 Hz, CH2N), 4.65 and 4.81 (qAB, 2H, J = 12.3 Hz, CH2C), 4.74 (dd, 1H, J = 8.0 Hz, J = 9.6 Hz H-2glu), 4.87 (d, 1H, J = 8.0 Hz, H-1glu), 4.86–4.95 (m, 3H, CH2O, H-4glu), 5.20 (dd-t, 1H, J = 9.6 Hz, J = 9.6 Hz, H-3glu), 7.20 (dd, 1H, J = 1.2 Hz, J = 7.7 Hz, H-7chin), 7.39-7.46 (m, 2H, H-3chin, H-6chin), 7.50 (dd, 1H, J = 1.2 Hz, J = 8.2 Hz, H-5chin), 8.20 (d, 1H, J = 8.4 Hz, H-4chin), 8.59 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 20.15, 20.22, 20.34, 20.46, 25.01, 49.18, 61.60, 61.90, 67.58, 68.04, 70.58, 70.80, 72.05, 98.51, 111.21, 120.48, 122.52, 125.45, 125.65, 127.35, 136.04, 139.29, 142.89, 153.20, 157.55, 168.88, 169.20, 169.45, 169.99; HRMS (ESI-TOF): calcd for C29H35N4O11 ([M + H]+): m/z 615.2302; found: m/z 615.2304.
Glycoconjugate 48: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 22 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a yellow solid (218.2 mg, 71%); m.p.: 52–56 °C; [α]23D = −20.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.83, 1.89, 2.01, 2.11 (4s, 12H, CH3CO), 2.69 (s, 3H, CH3), 3.99–4.10 (m, 2H, H-5gal, H-6agal), 4.13–4.21 (m, 1H, H-6bgal), 4.59 (t, 2H, J = 5.0 Hz, CH2N), 4.64 and 4.81 (qAB, 2H, J = 12.3 Hz, CH2C), 4.79 (d, 1H, J = 8.0 Hz, H-1gal), 4.86–4.97 (m, 3H, CH2O, H-2gal), 5.11 (dd, 1H, J = 0.9 Hz, J = 10.3 Hz, H-4gal), 5.24 (dd, 1H, J = 3.6 Hz, J = 10.3 Hz, H-3gal), 7.20 (dd, 1H, J = 1.2 Hz, J = 7.7 Hz, H-7chin), 7.38-7.47 (m, 2H, H-3chin, H-6chin), 7.50 (dd, 1H, J = 1.2 Hz, J = 8.2 Hz, H-5chin), 8.21 (d, 1H, J = 8.4 Hz, H-4chin), 8.60 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 20.25, 20.28, 20.35, 20.47, 25.01, 49.19, 61.19, 61.76, 67.26, 67.59, 68.53, 69.89, 70.20, 98.94, 111.22, 120.48, 122.52, 125.40, 125.66, 127.35, 136.04, 139.31, 142.97, 153.21, 157.56, 168.98, 169.40, 169.85, 169.89; HRMS (ESI-TOF): calcd for C29H35N4O11 ([M + H]+): m/z 615.2302; found: m/z 615.2303.
Glycoconjugate 49: Starting from propargyl β-D-glucopyranoside 23 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a yellow solid (205.4 mg, 95%); m.p.: 42-45 °C; [α]25D = −17.2 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.92-3.09 (m, 2H, H-2glu, H-5glu), 3.10-3.17 (m, 2H, H-3glu, H-4glu), 3.40–3.50 (m, 1H, H-6aglu), 3.66–3.75 (m, 1H, H-6bglu), 4.08 (bs, 1H, OH), 4.27 (d, 1H, J = 7.8 Hz, H-1glu), 4.57 (bs, 1H, OH), 4.60–4.66 (m, 3H, CH2N, CH2C), 4.83–4.95 (m, 4H, CH2O, CH2C, OH), 5.02 (bs, 1H, OH), 7.25 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz, H-7chin), 7.47–7.58 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.40 (s, 1H, H-5triaz), 8.89 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 49.12, 61.15, 61.47, 67.36, 70.10, 73.37, 76.70, 76.94, 102.14, 110.57, 120.51, 121.91, 124.96, 126.71, 129.08, 135.85, 139.69, 143.78, 149.27, 153.68; HRMS (ESI-TOF): calcd for C20H25N4O7 ([M + H]+): m/z 433.1723; found: m/z 433.1723.
Glycoconjugate 50: Starting from propargyl β-D-galactopyranoside 24 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a yellow solid (144.9 mg, 67%); m.p.: 44–45 °C; [α]23D = −12.9 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 3.21–3.39 (m, 3H, H-2gal, H-3gal, H-4gal), 3.40–3.48 (m, 1H, H-5gal), 3.50–3.57 (m, 1H, H-6agal), 3.59–3.66 (m, 1H, H-6bgal), 4.21 (d, 1H, J = 7.4 Hz, H-1gal), 4.61 and 4.83 (qAB, 2H, J = 12.2 Hz, CH2C), 4.63 (t, 2H, J = 5.2 Hz, CH2N), 4.90 (t, 2H, J = 5.2 Hz, CH2O), 7.25 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz, H-7chin), 7.47–7.58 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.40 (s, 1H, H-5triaz), 8.89 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 49.11, 60.52, 61.31, 67.37, 68.18, 70.44, 73.40, 75.32, 102.69, 110.58, 120.51, 121.92, 124.91, 126.70, 129.07, 135.83, 139.70, 143.85, 149.30, 153.69; HRMS (ESI-TOF): calcd for C20H25N4O7 ([M + H]+): m/z 433.1723; found: m/z 433.1725.
Glycoconjugate 51: Starting from propargyl β-D-glucopyranoside 23 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a yellow solid (196.4 mg, 88%); m.p.: 120–121 °C; [α]24D = −20.0 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.69 (s, 3H, CH3), 2.92–3.08 (m, 2H, H-2glu, H-5glu), 3.09-3.19 (m, 2H, H-3glu, H-4glu), 3.40–3.49 (m, 1H, H-6aglu), 3.66–3.73 (m, 1H, H-6bglu), 4.09 (bs, 1H, OH), 4.27 (d, 1H, J = 7.8 Hz, H-1glu), 4.33 (bs, 1H, OH), 4.53 (t, 1H, J = 5.6 Hz, OH), 4.60 (t, 2H, J = 5.2 Hz, CH2N), 4.63 and 4.87 (qAB, 2H, J = 12.1 Hz, CH2C), 4.89 (t, 2H, J = 5.2 Hz, CH2O), 4.99 (d, 1H, J = 4.9 Hz, OH), 7.21 (dd, 1H, J = 1.3 Hz, J = 7.7 Hz, H-7chin), 7.39–7.45 (m, 2H, H-3chin, H-6chin), 7.50 (dd, 1H, J = 1.2 Hz, J = 8.2 Hz, H-5chin), 8.20 (d, 1H, J = 8.4 Hz, H-4chin), 8.54 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.11, 49.11, 61.14, 61.48, 67.56, 70.08, 73.35, 76.70, 76.93, 102.19, 111.16, 120.45, 122.54, 125.21, 125.66, 127.35, 136.01, 139.28 143.77, 153.21, 157.61; HRMS (ESI-TOF): calcd for C21H27N4O7 ([M + H]+): m/z 447.1880; found: m/z 447.1882.
Glycoconjugate 52: Starting from propargyl β-D-galactopyranoside 24 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a yellow solid (174.1 mg, 78%); m.p.: 52–54 °C; [α]24D = −15.0 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.69 (s, 3H, CH3), 3.21–3.38 (m, 4H, H-2gal, H-3gal, H-4gal, H-5gal), 3.49–3.57 (m, 1H, H-6agal), 3.60–3.66 (m, 1H, H-6bgal), 4.22 (d, 1H, J = 7.3 Hz, H-1gal), 4.32 (d, 1H, J = 4.5 Hz, OH), 4.58 (t, 1H, J = 5.6 Hz, OH), 4.60 (t, 2H, J = 5.2 Hz, CH2N), 4.60 and 4.87 (qAB, 2H, J = 12.0 Hz, CH2C), 4.66 (d, 1H, J = 5.2 Hz, OH), 4.83 (t, 1H, J = 2.2 Hz, OH), 4.89 (t, 2H, J = 5.2 Hz, CH2O), 7.21 (dd, 1H, J = 1.3 Hz, J = 7.7 Hz, H-7chin), 7.38–7.46 (m, 2H, H-3chin, H-6chin), 7.50 (dd, 1H, J = 1.2 Hz, J = 8.2 Hz, H-5chin), 8.20 (d, 1H, J = 8.4 Hz, H-4chin), 8.54 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.10, 49.11, 60.45, 61.28, 67.57, 68.13, 70.41, 73.42, 75.28, 102.72, 111.18, 120.45, 122.53, 125.19, 125.66, 127.35, 136.01, 139.28, 143.79, 153.21, 157.62; HRMS (ESI-TOF): calcd for C21H27N4O7 ([M + H]+): m/z 447.1880; found: m/z 447.1879.
Glycoconjugate 53: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 21 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a yellow oil (307.3 mg, 100%); [α]25D = −21.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.88, 1.92, 1.98, 2.03 (4s, 12H, CH3CO), 2.43 (p, 2H, J = 6.5 Hz, CH2), 3.99 (ddd, 1H, J = 2.4 Hz, J = 4.9 Hz, J = 10.0 Hz, H-5glu), 4.10 (dd, 1H, J = 2.4 Hz, J = 12.3 Hz, H-6aglu), 4.15–4.24 (m, 3H, CH2N, H-6bglu), 4.66 (t, 2H, J = 6.9 Hz, CH2O), 4.65 and 4.80 (qAB, 2H, J = 12.1 Hz, CH2C), 4.76 (dd, 1H, J = 8.0 Hz, J = 9.6 Hz H-2glu), 4.88 (d, 1H, J = 8.0 Hz, H-1glu), 4.91 (dd-t, 1H, J = 9.6 Hz, J = 9.7 Hz, H-4glu), 5.25 (dd-t, 1H, J = 9.6 Hz, J = 9.6 Hz, H-3glu), 7.19 (dd, 1H, J = 1.9 Hz, J = 7.1 Hz, H-7chin), 7.47–7.53 (m, 2H, H-3chin, H-6chin), 7.56 (dd, 1H, J = 4.0 Hz, J = 8.2 Hz, H-5chin), 8.24 (s, 1H, H-5triaz), 8.33 (dd, 1H, J = 1.6 Hz, J = 8.2 Hz, H-4chin), 8.89 (bs, 1H, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 20.23, 20.25, 20.35, 20.47, 29.57, 46.66, 61.64, 61.85, 65.39, 68.11, 70.61, 70.83, 72.03, 98.47, 109.82, 119.93, 121.85, 124.50, 126.77, 129.03, 135.78, 139.79, 142.94, 149.01, 154.20, 168.92, 169.22, 169.48, 170.01; HRMS (ESI-TOF): calcd for C29H35N4O11 ([M + H]+): m/z 615.2302; found: m/z 615.2303.
Glycoconjugate 54: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 22 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a yellow oil (307.3 mg, 100%); [α]25D = −16.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.89, 1.90, 2.01, 2.11 (4s, 12H, CH3CO), 2.43 (p, 2H, J = 6.4 Hz, CH2), 4.01–4.11 (m, 2H, H-5gal, H-6agal), 4.15–4.24 (m, 3H, H-6bgal, CH2N), 4.64 and 4.79 (qAB, 2H, J = 12.4 Hz, CH2C), 4.66 (t, 2H, J = 6.8 Hz, CH2O), 4.79 (d, 1H, J = 8.0 Hz, H-1gal), 4.92 (dd, 1H, J = 8.0 Hz, J = 10.3 Hz, H-2gal), 5.15 (dd, 1H, J = 3.6 Hz, J = 10.3 Hz, H-3gal), 5.25 (dd, 1H, J = 0.9 Hz, J = 3.6 Hz, H-4gal), 7.20 (dd, 1H, J = 1.5 Hz, J = 7.4 Hz, H-7chin), 7.46–7.61 (m, 3H, H-3chin, H-5chin, H-6chin), 8.24 (s, 1H, H-5triaz), 8.31–8.35 (m, 1H, H-4chin), 8.90 (bs, 1H, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 20.28, 20.29, 20.35, 20.47, 29.58, 46.66, 61.22, 61.72, 65.39, 67.28, 68.57, 69.91, 70.19, 98.89, 109.81, 119.94, 124.47, 125.27, 126.76, 128.16, 128.86, 135.77, 143.00, 148.99, 154.22, 169.03, 169.42, 169.86, 169.89; HRMS (ESI-TOF): calcd for C29H35N4O11 ([M + H]+): m/z 615.2302; found: m/z 615.2302.
Glycoconjugate 55: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 21 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a yellow oil (204.3 mg, 65%); [α]23D = −20.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.88, 1.92, 1.98, 2.03 (4s, 12H, CH3CO), 2.42 (p, 2H, J = 6.5 Hz, CH2), 2.67 (s, 3H, CH3), 3.98 (ddd, 1H, J = 2.4 Hz, J = 4.9 Hz, J = 10.0 Hz, H-5glu), 4.04 (dd, 1H, J = 2.4 Hz, J = 12.4 Hz, H-6aglu), 4.15–4.24 (m, 3H, CH2N, H-6bglu), 4.64 and 4.80 (qAB, 2H, J = 12.3 Hz, CH2C), 4.65 (t, 2H, J = 6.9 Hz, CH2O), 4.75 (dd, 1H, J = 8.0 Hz, J = 9.6 Hz H-2glu), 4.87 (d, 1H, J = 8.0 Hz, H-1glu), 4.91 (dd-t, 1H, J = 9.6 Hz, J = 9.8 Hz, H-4glu), 5.24 (dd-t, 1H, J = 9.6 Hz, J = 9.6 Hz, H-3glu), 7.16 (dd, 1H, J = 1.2 Hz, J = 7.6 Hz, H-7chin), 7.38–7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.25 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 20.23, 20.24, 20.35, 20.48, 25.05, 29.52, 46.64, 61.64, 61.83, 65.57, 68.11, 70.60, 70.83, 72.03, 98.46, 110.37, 119.86, 122.44, 124.47, 125.70, 127.32, 135.98, 139.32, 142.93, 153.67, 157.30, 168.92, 169.23, 169.48, 170.01; HRMS (ESI-TOF): calcd for C30H37N4O11 ([M + H]+): m/z 629.2459; found: m/z 629.2458.
Glycoconjugate 56: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 22 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a yellow oil (242.0 mg, 77%); [α]24D = −8.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.89, 1.90, 2.01, 2.11 (4s, 12H, CH3CO), 2.42 (p, 2H, J = 6.3 Hz, CH2), 2.67 (s, 3H, CH3), 4.01–4.11 (m, 2H, H-5gal, H-6agal), 4.15–4.24 (m, 3H, H-6agal, CH2N), 4.64 and 4.79 (qAB, 2H, J = 12.6 Hz, CH2C), 4.66 (t, 2H, J = 6.9 Hz, CH2O), 4.78 (d, 1H, J = 8.0 Hz, H-1gal), 4.92 (dd, 1H, J = 8.0 Hz, J = 10.4 Hz, H-2gal), 5.14 (dd, 1H, J = 3.6 Hz, J = 10.4 Hz, H-3gal), 5.25 (dd, 1H, J = 0.9 Hz, J = 3.6 Hz, H-4gal), 7.16 (dd, 1H, J = 0.8 Hz, J = 7.5 Hz, H-7chin), 7.37–7.51 (m, 3H, H-3chin, H-5chin, H-6chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.25 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ18.53, 20.28, 20.29, 20.35, 20.48, 29.53, 46.64, 61.23, 61.70, 65.55, 68.57, 69.91, 70.19, 72.20, 98.88, 110.33, 119.87, 122.45, 124.46, 125.71, 127.31, 135.96, 139.32, 143.02, 153.26, 158.61, 169.03, 169.43, 169.87, 169.90; HRMS (ESI-TOF): calcd for C30H37N4O11 ([M + H]+): m/z 629.2459; found: m/z 629.2455.
Glycoconjugate 57: Starting from propargyl β-D-glucopyranoside 23 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (223.2 mg, 100%); m.p.: 77–79 °C; [α]24D = −19.2 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.42 (p, 2H, J = 6.4 Hz, CH2), 2.94-3.09 (m, 2H, H-2glu, H-5glu), 3.10-3.17 (m, 2H, H-3glu, H-4glu), 3.40–3.50 (m, 1H, H-6aglu), 3.67–3.75 (m, 1H, H-6bglu), 4.05–4.11 (m, 1H, OH), 4.21 (t, 2H, J = 6.1 Hz, CH2N), 4.27 (d, 1H, J = 7.8 Hz, H-1glu), 4.34 (t, 1H, J = 5.1 Hz, OH), 4.57 (t, 1H, J = 5.9 Hz, OH), 4.64 (t, 1H, J = 7.1 Hz, CH2O), 4.64 and 4.85 (qAB, 2H, J = 12.2 Hz, CH2C), 5.01 (d, 1H, J = 4.9 Hz, OH), 7.20 (dd, 1H, J = 2.2 Hz, J = 6.8 Hz, H-7chin), 7.47–7.58 (m, 3H, H-3chin, H-5chin, H-6chin), 8.29 (s, 1H, H-5triaz), 8.32 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.90 (dd, 1H, J = 1.6 Hz, J = 4.1 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 29.61, 46.61, 61.16, 61.57, 65.43, 70.11, 73.39, 76.69, 76.94, 102.14, 109.84, 119.92, 121.86, 124.42, 126.79, 129.04, 135.81, 139.76, 143.89, 149.07, 154.19; HRMS (ESI-TOF): calcd for C21H27N4O7 ([M + H]+): m/z 447.1880; found: m/z 447.1880.
Glycoconjugate 58: Starting from propargyl β-D-galactopyranoside 24 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a yellow solid (169.7 mg, 76%); m.p.: 50–54 °C; [α]24D = −11.2 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.42 (p, 2H, J = 6.5 Hz, CH2), 3.22-3.40 (m, 4H, H-2gal, H-3gal, H-4gal, H-5gal), 3.50-3.58 (m, 1H, H-6agal), 3.60–3.66 (m, 1H, H-6bgal), 4.20 (t, 2H, J = 6.0 Hz, CH2N), 4.21 (d, 1H, J = 6.3 Hz, H-1gal), 4.34 (d, 1H, J = 4.4 Hz, OH), 4.57–4.71 (m, 2H, OH), 4.62 and 4.83 (qAB, 2H, J = 12.4 Hz, CH2C), 4.64 (t, 2H, J = 7.0 Hz, CH2O), 4.86 (d, 1H, J = 5.7 Hz, OH), 7.20 (dd, 1H, J = 2.1 Hz, J = 6.8 Hz, H-7chin), 7.46–7.59 (m, 3H, H-3chin, H-5chin, H-6chin), 8.28 (s, 1H, H-5triaz), 8.32 (dd, 1H, J = 1.6 Hz, J = 8.4 Hz, H-4chin), 8.90 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 29.62, 46.61, 60.53, 61.42, 65.44, 68.18, 70.47, 73.39, 75.32, 102.70, 109.85, 119.91, 121.85, 124.36, 126.79, 129.04, 135.79, 139.76, 143.96, 149.08, 154.19; HRMS (ESI-TOF): calcd for C21H27N4O7 ([M + H]+): m/z 447.1880; found: m/z 447.1879.
Glycoconjugate 59: Starting from propargyl β-D-glucopyranoside 23 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a yellow solid (200.3 mg, 87%); m.p.: 47-50 °C; [α]23D = −16.6 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.42 (p, 2H, J = 6.5 Hz, CH2), 2.68 (s, 3H, CH3), 2.93–3.09 (m, 2H, H-2glu, H-5glu), 3.10–3.20 (m, 2H, H-3glu, H-4glu), 3.40–3.50 (m, 1H, H-6aglu), 3.66–3.75 (m, 1H, H-6bglu), 4.20 (t, 2H, J = 6.2 Hz, CH2N), 4.26 (d, 1H, J = 7.8 Hz, H-1glu), 4.34 (t, 2H, J = 5.0 Hz, OH), 4.56 (t, 1H, J = 5.8 Hz, OH), 4.63 and 4.84 (qAB, 2H, J = 12.2 Hz, CH2C), 4.64 (t, 2H, J = 6.9 Hz, CH2O), 5.00 (d, 1H, J = 4.9 Hz, OH), 7.16 (dd, 1H, J = 1.3 Hz, J = 7.6 Hz, H-7chin), 7.38–7.49 (m, 3H, H-3chin, H-5chin, H-6chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.26 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.09, 29.57, 46.60, 61.16, 61.56, 65.57, 70.12, 73.40, 76.70, 76.95, 102.15, 110.35, 119.86, 122.48, 124.40, 125.75, 131.21, 136.01, 139.31, 143.15, 150.40, 153.68; HRMS (ESI-TOF): calcd for C22H29N4O7 ([M + H]+): m/z 461.2036; found: m/z 461.2039.
Glycoconjugate 60: Starting from propargyl β-D-galactopyranoside 24 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a brown solid (149.7 mg, 65%); m.p.: 50–54 °C; [α]24D = −4.6 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.42 (p, 2H, J = 6.5 Hz, CH2), 2.67 (s, 3H, CH3), 3.22–3.42 (m, 4H, H-2gal, H-3gal, H-4gal, H-5gal), 3.49–3.57 (m, 1H, H-6agal), 3.60–3.66 (m, 1H, H-6bgal), 4.20 (t, 2H, J = 5.9 Hz, CH2N), 4.21 (d, 1H, J = 7.0 Hz, H-1gal), 4.34 (bs, 1H, OH), 4.54-4.72 (m, 4H, CH2O, CH2C, OH), 4.77-4.89 (m, 2H, CH2C, OH), 7.16 (dd, 1H, J = 1.2 Hz, J = 7.6 Hz, H-7chin), 7.37-7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.26 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.00, 29.56, 46.58, 60.52, 61.40, 65.59, 68.18, 70.48, 73.40, 75.31, 102.71, 110.44, 119.85, 122.51, 124.32, 125.81, 127.34, 136.18, 139.12, 143.96, 153.58, 157.34; HRMS (ESI-TOF): calcd for C22H29N4O7 ([M + H]+): m/z 461.2036; found: m/z 461.2038.
Glycoconjugate 61: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 21 and 8-(4-azidobutoxy)quinoline 7, product was obtained as a yellow oil (301.7 mg, 96%); [α]23D = −17.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.61 (bs, 2H, CH2), 1.92, 1.98, 2.01, 2.08 (4s, 12H, CH3CO), 2.26 (p, 2H, J = 7.1 Hz, CH2), 3.69 (ddd, 1H, J = 2.4 Hz, J = 4.7 Hz, J = 9.9 Hz, H-5glu), 4.12 (dd, 1H, J = 2.4 Hz, J = 12.3 Hz, H-6aglu), 4.24 (dd, 1H, J = 4.7 Hz, J = 12.3 Hz, H-6aglu), 4.28 (t, 2H, J = 6.1 Hz, CH2N), 4.58 (t, 2H, J = 6.9 Hz, CH2O), 4.68 (d, 1H, J = 8.0 Hz, H-1glu), 4.81 and 4.93 (qAB, 2H, J = 12.6 Hz, CH2C), 5.00 (dd, 1H, J = 8.0 Hz, J = 9.5 Hz H-2glu), 5.08 (dd-t, 1H, J = 9.4 Hz, J = 9.8 Hz, H-4glu), 5.17 (dd-t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-3glu), 7.05 (dd, 1H, J = 1.4 Hz, J = 7.5 Hz, H-7chin), 7.38–7.49 (m, 3H, H-3chin, H-5chin, H-6chin), 7.95 (s, 1H, H-5triaz), 8.14 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.93 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz, H-2chin); 13C NMR (100 MHz, CDCl3): δ 20.60, 20.67, 20.75, 20.79, 25.61, 27.83, 49.93, 61.85, 62.86, 68.36, 71.24, 71.89, 72.86, 77.22, 99.66, 108.81, 119.91, 121.71, 123.74, 126.69, 129.54, 135.96, 140.35, 143.77, 149.32, 154.59, 169.35, 169.42, 170.19, 170.65; HRMS (ESI-TOF): calcd for C29H35N4O11 ([M + H]+): m/z 629.2459; found: m/z 629.2458.
Glycoconjugate 62: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 22 and 8-(4-azidobutoxy)quinoline 7, product was obtained as a yellow oil (292.3 mg, 93%); [α]25D = −12.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.78–1.87 (m, 2H, CH2), 1.90, 1.90, 2.01, 2.10 (4s, 12H, CH3CO), 2.05–2.10 (m, 2H, CH2), 4.01-4.11 (m, 2H, H-5gal, H-6agal), 4.16-4.25 (m, 3H, H-6bgal, CH2N), 4.54 (t, 2H, J = 7.0 Hz, CH2O), 4.64 and 4.80 (qAB, 2H, J = 12.4 Hz, CH2C), 4.81 (d, 1H, J = 8.0 Hz, H-1gal), 4.92 (dd, 1H, J = 8.0 Hz, J = 10.3 Hz, H-2gal), 5.15 (dd, 1H, J = 3.6 Hz, J = 10.3 Hz, H-3gal), 5.25 (dd, 1H, J = 0.9 Hz, J = 3.6 Hz, H-4gal), 7.17–7.21 (m, 1H, H-7chin), 7.47–7.57 (m, 3H, H-3chin, H-5chin, H-6chin), 8.25 (s, 1H, H-5triaz), 8.31 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.85 (bs, 1H, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 20.29, 20.32, 20.35, 20.48, 25.53, 27.04, 49.08, 61.21, 61.81, 67.28, 67.85, 68.58, 69.90, 70.19, 98.97, 109.37, 119.55, 121.79, 124.44, 126.80, 129.00, 135.76, 139.72, 142.87, 148.92, 154.38, 169.05, 169.42, 169.86, 169.89; HRMS (ESI-TOF): calcd for C30H37N4O11 ([M + H]+): m/z 629.2459; found: m/z 629.2459.
Glycoconjugate 63: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 21 and 2-methyl-8-(4-azidobutoxy)quinoline 10, product was obtained as a yellow oil (295.6 mg, 92%); [α]23D = −18.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.80 (p, 2H, J = 6.6 Hz, CH2), 1.88, 1.91, 1.97, 2.02 (4s, 12H, CH3CO), 2.08 (p, 2H, J = 7.4 Hz, CH2), 2.63 (s, 3H, CH3), 3.97 (ddd, 1H, J = 2.4 Hz, J = 4.9 Hz, J = 10.0 Hz, H-5glu), 4.03 (dd, 1H, J = 2.4 Hz, J = 12.4 Hz, H-6aglu), 4.14–4.24 (m, 3H, CH2N, H-6bglu), 4.59 (t, 2H, J = 6.9 Hz, CH2O), 4.64 and 4.80 (qAB, 2H, J = 12.4 Hz, CH2C), 4.75 (dd, 1H, J = 8.0 Hz, J = 9.7 Hz H-2glu), 4.90 (d, 1H, J = 8.0 Hz, H-1glu), 4.91 (dd-t, 1H, J = 9.7 Hz, J = 9.7 Hz, H-4glu), 5.24 (dd-t, 1H, J = 9.6 Hz, J = 9.6 Hz, H-3glu), 7.15 (dd, 1H, J = 1.8 Hz, J = 7.2 Hz, H-7chin), 7.38–7.47 (m, 3H, H-3chin, H-5chin, H-6chin), 8.18 (d, 1H, J = 8.4 Hz, H-4chin), 8.34 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 20.23, 20.30, 20.34, 20.47, 24.93, 25.25, 27.23, 49.08, 61.64, 61.92, 68.09, 68.14, 70.60, 70.82, 72.03, 98.53, 109.64, 119.42, 122.41, 124.62, 125.74, 127.26, 135.96, 139.19, 142.70, 153.89, 157.21, 168.92, 169.21, 169.46, 170.00; HRMS (ESI-TOF): calcd for C31H39N4O11 ([M + H]+): m/z 643.2615; found: m/z 643.2618.
Glycoconjugate 64: Starting from propargyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 22 and 2-methyl-8-(4-azidobutoxy)quinoline 10, product was obtained as a yellow oil (321.3 mg, 100%); [α]24D = −13.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.81 (p, 2H, J = 6.6 Hz, CH2), 1.89, 1.89, 2.00, 2.10 (4s, 12H, CH3CO), 2.03–2.10 (m, 2H, CH2), 2.63 (s, 3H, CH3), 4.01–4.11 (m, 2H, H-5gal, H-6agal), 4.15–4.25 (m, 3H, H-6agal, CH2N), 4.59 (t, 2H, J = 7.0 Hz, CH2O), 4.64 and 4.80 (qAB, 2H, J = 12.9 Hz, CH2C), 4.81 (d, 1H, J = 8.0 Hz, H-1gal), 4.92 (dd, 1H, J = 8.0 Hz, J = 10.3 Hz, H-2gal), 5.14 (dd, 1H, J = 3.6 Hz, J = 10.3 Hz, H-3gal), 5.24 (dd, 1H, J = 0.9 Hz, J = 3.6 Hz, H-4gal), 7.15 (dd, 1H, J = 1.8 Hz, J = 7.2 Hz, H-7chin), 7.38–7.47 (m, 3H, H-3chin, H-5chin, H-6chin), 8.18 (d, 1H, J = 8.4 Hz, H-4chin), 8.33 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 20.22, 20.28, 20.34, 20.47, 24.93, 25.26, 27.25, 49.08, 61.20, 61.78, 67.28, 68.14, 68.57, 69.89, 70.19, 98.95, 109.64, 119.41, 122.40, 124.57, 125.74, 127.26, 135.96, 139.19, 142.78, 153.89, 157.21, 168.02, 169.41, 169.84, 169.88; HRMS (ESI-TOF): calcd for C31H39N4O11 ([M + H]+): m/z 643.2615; found: m/z 643.2612.
Glycoconjugate 65: Starting from propargyl β-D-glucopyranoside 23 and 8-(4-azidobutoxy)quinoline 7, product was obtained as a yellow solid (165.8 mg, 72%); m.p.: 49–51 °C; [α]24D = −20.2 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 1.77-1.89 (m, 2H, CH2), 2.03-2.15 (m, 2H, CH2), 2.94–3.09 (m, 2H, H-2glu, H-5glu), 3.10–3.17 (m, 2H, H-3glu, H-4glu), 3.40–3.50 (m, 1H, H-6aglu), 3.67–3.75 (m, 1H, H-6bglu), 4.05–4.11 (m, 1H, OH), 4.21 (m, 2H, CH2N), 4.27 (d, 1H, J = 7.8 Hz, H-1glu), 4.34 (t, 1H, J = 5.1 Hz, OH), 4.52 (t, 1H, J = 6.8 Hz, CH2O), 4.57 (t, 1H, J = 5.9 Hz, OH), 4.64 and 4.85 (qAB, 2H, J = 12.2 Hz, CH2C), 5.01 (d, 1H, J = 4.9 Hz, OH), 7.17–7.22 (m, 1H, H-7chin), 7.45–7.61 (m, 3H, H-3chin, H-5chin, H-6chin), 8.28 (s, 1H, H-5triaz), 8.31–8.35 (m, 1H, H-4chin), 8.87 (bs, 1H, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 25.61, 26.96, 49.06, 61.16, 61.59, 67.84, 70.11, 73.38, 76.70, 76.95, 102.15, 109.35, 119.64, 121.85, 124.42, 126.84, 128.86, 135.68, 139.79, 143.73, 148.98, 154.35; HRMS (ESI-TOF): calcd for C22H29N4O7 ([M + H]+): m/z 461.2036; found: m/z 461.2036.
Glycoconjugate 66: Starting from propargyl β-D-galactopyranoside 24 and 8-(4-azidobutoxy)quinoline 7, product was obtained as a yellow solid (207.2 mg, 90%); m.p.: 62–64 °C; [α]24D = −13.0 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 1.83 (p, 2H, J = 6.3 Hz, CH2), 2.08 (p, 2H, J = 7.1 Hz, CH2), 3.24–3.30 (m, 1H, H-2gal), 3.33–3.48 (m, 3H, H-3gal, H-4gal, H-5gal), 3.50–3.57 (m, 1H, H-6agal), 3.61–3.66 (m, 1H, H-6bgal), 4.21 (t, 2H, J = 6.3 Hz, CH2N), 4.22 (d, 1H, J = 7.5 Hz, H-1gal), 4.31–4.36 (m, 2H, OH), 4.52 (t, 2H, J = 7.0 Hz, CH2O), 4.62 and 4.83 (qAB, 2H, J = 12.1 Hz, CH2C), 4.67 (d, 1H, J = 5.1 Hz, OH), 4.87 (d, 1H, J = 4.6 Hz, OH), 7.17–7.21 (m, 1H, H-7chin), 7.46–7.57 (m, 3H, H-3chin, H-5chin, H-6chin), 8.27 (s, 1H, H-5triaz), 8.31 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.87 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 25.59, 26.98, 49.03, 60.52, 61.45, 67.82, 68.18, 70.47, 73.40, 75.32, 102.73, 109.39, 119.55, 121.80, 124.33, 126.80, 129.00, 135.75, 139.71, 143.77, 148.99, 154.38; HRMS (ESI-TOF): calcd for C22H29N4O7 ([M + H]+): m/z 461.2036; found: m/z 461.2035.
Glycoconjugate 67: Starting from propargyl β-D-glucopyranoside 23 and 2-methyl-8-(4-azidobutoxy)quinoline 10, product was obtained as an orange solid (180.3 mg, 76%); m.p.: 75–78 °C; [α]24D = −17.6 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 1.82 (p, 2H, J = 6.5 Hz, CH2), 2.08 (p, 2H, J = 7.0 Hz, CH2), 2.64 (s, 3H, CH3), 2.94–3.09 (m, 2H, H-2glu, H-5glu), 3.10–3.19 (m, 2H, H-3glu, H-4glu), 3.41–3.49 (m, 1H, H-6aglu), 3.66–3.74 (m, 1H, H-6bglu), 4.20 (t, 2H, J = 6.3 Hz, CH2N), 4.27 (d, 1H, J = 7.8 Hz, H-1glu), 4.55 (d, 1H, J = 5.9 Hz, OH), 4.56 (t, 2H, J = 6.9 Hz, CH2O), 4.64 and 4.86 (qAB, 2H, J = 12.2 Hz, CH2C), 4.89 (t, 2H, J = 5.2 Hz, OH), 4.91 (t, 1H, J = 4.7 Hz, OH), 5.01 (d, 1H, J = 4.9 Hz, OH), 7.15 (dd, 1H, J = 1.8 Hz, J = 7.1 Hz, H-7chin), 7.38–7.47 (m, 3H, H-3chin, H-5chin, H-6chin), 8.18 (d, 1H, J = 8.4 Hz, H-4chin), 8.31 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.40, 25.44, 27.09, 49.07, 61.13, 61.55, 68.07, 70.09, 73.37, 76.69, 76.93, 102.13, 109.62, 119.44, 122.43, 124.51, 125.78, 127.28, 135.98, 139.16, 145.72, 148.08, 153.85; HRMS (ESI-TOF): calcd for C23H31N4O7 ([M + H]+): m/z 475.2193; found: m/z 475.2194.
Glycoconjugate 68: Starting from propargyl β-D-galactopyranoside 24 and 2-methyl-8-(4-azidobutoxy)quinoline 10, product was obtained as an orange solid (156.6 mg, 66%); m.p.: 38–41 °C; [α]23D = −6.0 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 1.82 (m, 2H, CH2), 2.08 (m, 2H, CH2), 2.64 (s, 3H, CH3), 3.23–3.43 (m, 4H, H-2gal, H-3gal, H-4gal, H-5gal), 3.49–3.58 (m, 1H, H-6agal), 3.61–3.66 (m, 1H, H-6bgal), 4.15–4.25 (m 3H, H-1gal, CH2N), 4.56 (t, 2H, J = 6.8 Hz, CH2O), 4.62 and 4.83 (qAB, 2H, J = 12.1 Hz, CH2C), 7.16 (m, 1H, H-7chin), 7.36–7.52 (m, 3H, H-3chin, H-5chin, H-6chin), 8.19 (d, 1H, J = 8.2 Hz, H-4chin), 8.29 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 24.94, 25.40, 27.10, 49.06, 60.50, 61.43, 68.08, 68.17, 70.47, 73.41, 75.31, 102.73, 109.74, 119.43, 122.45, 124.40, 125.81, 127.28, 136.09, 139.06, 143.74, 153.83, 157.26; HRMS (ESI-TOF): calcd for C23H31N4O7 ([M + H]+): m/z 475.2193; found: m/z 475.2199.
Glycoconjugate 69: Starting from 2-azidoethyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 27 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a yellow solid (234.2 mg, 78%); m.p.: 52–55 °C; [α]22D = −13.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.92, 1.98, 2.02, 2.06 (4s, 12H, CH3CO), 3.67 (ddd, 1H, J = 2.4 Hz, J = 4.7 Hz, J = 10.0 Hz, H-5glu), 3.93–3.99 (m, 1H, CH2O), 4.11 (dd, 1H, J = 2.4 Hz, J = 12.4 Hz, H-6aglu), 4.19–4.24 (m, 2H, H-6bglu, CH2O.), 4.48 (d, 1H, J = 7.9 Hz, H-1glu), 4.49–4.62 (m, 2H, CH2N), 4.95 (dd, 1H, J = 7.9 Hz, J = 9.5 Hz H-2glu), 5.04 (dd, 1H, J = 9.4 Hz, J = 10.0 Hz, H-4glu), 5.16 (dd-t, 1H, J = 9.4 Hz, J = 9.5 Hz, H-3glu), 5.54 (s, 2H, CH2Ochin), 7.32–7.50 (m, 4H, H-3chin, H-5chin, H-6chin, H-7chin), 7.84 (s, 1H, H-5triaz), 8.14 (d, 1H, J = 8.0 Hz, H-4chin), 8.94 (bs, 1H, H-2chin); 13C NMR (100 MHz, CDCl3): δ 20.53, 20.55, 20.64, 20.70, 50.06, 61.74, 62.79, 67.58, 68.25, 70.90, 71.99, 72.52, 100.54, 110.09, 120.29, 121.63, 124.62, 126.77, 129.50, 136.05, 140.21, 143.85, 149.26, 153.90, 169.29, 169.35, 170.08, 170.54; HRMS (ESI-TOF): calcd for C28H33N4O11 ([M + H]+): m/z 601.2146; found: m/z 601.2149.
Glycoconjugate 70: Starting from 2-azidoethyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 28 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a yellow solid (186.2 mg, 62%); m.p.: 55–58 °C; [α]23D = −18.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.90, 2.00, 2.09, 2.10 (4s, 12H, CH3CO), 3.91–3.99 (m, 1H, CH2O), 4.00-4.22 (m, 4H, H-6agal, H-6bgal, H-5gal, CH2O), 4.53–4.66 (m, 2H, CH2N), 4.74 (d, 1H, J = 8.0 Hz, H-1gal), 4.91 (dd, 1H, J = 8.0 Hz, J = 10.4, H-2gal), 5.12 (dd, 1H, J = 3.6 Hz, J = 10.4 Hz, H-3gal), 5.25 (dd, 1H, J = 0.8 Hz, J = 3.6 Hz, H-4gal), 5.35 (s, 2H, CH2Ochin), 7.41 (dd, 1H, J = 3.4 Hz, J = 5.6 Hz, H-7chin), 7.50–7.57 (m, 3H, H-3chin, H-5chin, H-6chin), 8.15 (s, 1H, H-5triaz), 8.31 (dd, 1H, J = 1.6 Hz, J = 8.3 Hz, H-4chin), 8.83 (bs, 1H, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 20.28, 20.32, 20.34, 20.47, 49.34, 61.21, 61.88, 67.26, 68.31, 69.98, 70.08, 79.14, 99.61, 110.10, 120.05, 121.82, 124.97, 126.72, 129.03, 135.75, 139.75, 142.46, 148.92, 153.85, 169.13, 169.43, 169.86, 169.88; HRMS (ESI-TOF): calcd for C28H33N4O11 ([M + H]+): m/z 601.2146; found: m/z 601.2140.
Glycoconjugate 71: Starting from 2-azidoethyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 27 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a pink solid (298.1 mg, 97%); m.p.: 50–53 °C; [α]24D = −13.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.89, 1.92, 1.98, 2.01 (4s, 12H, CH3CO), 2.63 (s, 3H, CH3), 3.91–4.00 (m, 2H, H-5glu, CH2O), 4.04 (dd, 1H, J = 2.5 Hz, J = 12.4 Hz, H-6aglu), 4.09–4.13 (m, 1H, CH2O), 4.17 (dd, 1H, J = 5.0 Hz, J = 12.4 Hz, H-6bglu), 4.52–4.66 (m, 2H, CH2N), 4.75 (dd, 1H, J = 8.1 Hz, J = 9.7 Hz, H-2glu), 4.85 (d, 1H, J = 8.1 Hz, H-1glu), 4.90 (dd-t, 1H, J = 9.4 Hz, J = 9.8 Hz, H-4glu), 5.23 (dd-t, 1H, J = 9.4 Hz, J = 9.7 Hz, H-3glu), 5.34 (s, 2H, CH2Ochin), 7.37 (dd, 1H, J = 1.7 Hz, J = 7.4 Hz, H-7chin), 7.41 (d, 1H, J = 8.4 Hz, H-3chin), 7.43 (dd, 1H, J = 7.4 Hz, J = 8.1 Hz, H-6chin), 7.47 (dd, 1H, J = 1.7 Hz, J = 8.1 Hz, H-5chin), 8.16 (s, 1H, H-5triaz), 8.18 (d, 1H, J = 8.4 Hz, H-4chin); 13C NMR (100 MHz, DMSO-d6): δ 20.19, 20.20, 20.31, 20.43, 24.85, 49.27, 61.60, 61.71, 67.39, 68.07, 70.51, 71.87, 79.12, 99.12, 110.26, 119.83, 122.40, 125.05, 125.60, 127.29, 135.90, 139.17, 142.50, 153.34, 157.19, 168.98, 169.19, 169.44, 169.97; HRMS (ESI-TOF): calcd for C29H35N4O11 ([M + H]+): m/z 615.2302; found: m/z 615.2303.
Glycoconjugate 72: Starting from 2-azidoethyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 28 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a pink solid (236.6 mg, 77%); m.p.: 60–63 °C; [α]24D = −13.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.89, 1.90, 2.00, 2.10 (4s, 12H, CH3CO), 2.62 (s, 3H, CH3), 3.95 (m, 1H, CH2O), 4.00–4.08 (m, 2H, H-6agal, H-6bgal), 4.08–4.21 (m, 2H, CH2O, H-5gal), 4.52–4.66 (m, 2H, CH2N), 4.74 (d, 1H, J = 8.0 Hz, H-1gal), 4.92 (dd, 1H, J = 8.0 Hz, J = 10.4, H-2gal), 5.12 (dd, 1H, J = 3.6 Hz, J = 10.4 Hz H-3gal), 5.25 (dd, 1H, J = 0.8 Hz, J = 3.6 Hz, H-4gal), 5.34 (s, 2H, CH2Ochin), 7.37 (dd, 1H, J = 1.6 Hz, J = 7.4 Hz, H-7chin), 7.41 (d, 1H, J = 8.4 Hz, H-3chin), 7.43 (dd, 1H, J = 7.4 Hz, J = 8.2 Hz, H-6chin), 7.47 (dd, 1H, J = 1.6 Hz, J = 8.2 Hz, H-5chin), 8.15 (s, 1H, H-5triaz), 8.18 (d, 1H, J = 8.4 Hz, H-4chin); 13C NMR (100 MHz, DMSO-d6): δ 20.30, 20.37, 20.50, 21.05, 24.89, 49.36, 61.23, 61.79, 67.29, 68.31, 70.01, 70.11, 79.17, 99.65, 110.33, 119.90, 122.46, 125.05, 125.67, 127.35, 135.96, 139.23, 142.58, 153.40, 157.25, 169.18, 169.46, 169.89, 169.9k2; HRMS (ESI-TOF): calcd for C29H35N4O11 ([M + H]+): m/z 615.2302; found: m/z 615.2305.
Glycoconjugate 73: Starting from 2-azidoethyl β-D-glucopyranoside 29 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a beige solid (134.1 mg, 62%); m.p.: 60–63 °C; [α]23D = −9.6 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.95-3.08 (m, 1H, H-5glu), 3.10–3.19 (m, 3H, H-2glu, H-3glu, H-4glu), 3.40-3.48 (m, 1H, H-6aglu), 3.65–3.72 (m, 1H, H-6bglu), 3.91–3.98 (m, 1H, CH2O), 4.07-4.14 (m, 1H, CH2O), 4.26 (d, 1H, J = 7.8 Hz, H-1glu), 4.52–4.56 (m, 1H, OH), 4.60–4.66 (m, 2H, CH2N), 4.88–4.96 (m, 2H, OH), 5.10–5.14 (d, 1H, J = 4.7 Hz, OH), 5.34 (s, 2H, CH2Ochin), 7.39–7.45 (m, 1H, H-7chin), 7.50–7.57 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.8 Hz, J = 8.1 Hz, H-4chin), 8.38 (s, 1H, H-5triaz), 8.83 (dd, 1H, J = 1.8 Hz, J = 4.1 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 49.74, 61.06, 61.82 67.36, 70.01, 73.27, 76.59, 76.96, 102.95, 109.96, 119.95, 121.81, 125.69, 126.73, 129.03, 135.78, 139.67, 142.32, 148.96, 153.84; HRMS (ESI-TOF): calcd for C20H25N4O7 ([M + H]+): m/z 433.1723; found: m/z 433.1719.
Glycoconjugate 74: Starting from 2-azidoethyl β-D-galactopyranoside 30 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a brown oil (118.9 mg, 55%); [α]25D = 10.8 (c = 1.0, MeOH); 1H NMR (400 MHz, D2O): δ 3.44-3.54 (m, 1H, H-2gal), 3.57–3.74 (m, 4H, H-3gal, H-5gal, H-6agal, H-6bgal), 3.91 (d, 1H, J = 3.2 Hz, H-4gal), 4.11-4.21 (m, 1H, CH2O), 4.30–4.40 (m, 2H, CH2O, H-1gal), 4.72-4.78 (m, 2H, CH2Ntriaz), 5.66 (s, 2H, CH2Ochin), 7.78 (dd, 1H, J = 2.3 Hz, J = 6.4 Hz, H-7chin), 7.85-7.97 (m, 2H, H-3chin, H-5chin), 8.06–8.16 (m, 1H, H-6chin), 8.35 (s, 1H, H-5triaz), 9.05 (d, 1H, J = 4.7 Hz, H-2chin), 9.14 (d, 1H, J = 8.1 Hz, H-4chin); 13C NMR (100 MHz, D2O): δ 50.49, 60.83, 62.27, 67.96, 68.45, 70.52, 72.54, 75.05, 102.97, 114.63, 120.83, 122.17, 126.24, 129.37, 129.92, 130.37, 142.07, 143.05, 147.30, 147.5; HRMS (ESI-TOF): calcd for C20H25N4O7 ([M + H]+): m/z 433.1723; found: m/z 433.1720.
Glycoconjugate 75: Starting from 2-azidoethyl β-D-glucopyranoside 29 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a brown oil (129.5 mg, 58%); [α]24D = 1.6 (c = 1.0, MeOH); 1H NMR (400 MHz, D2O): δ 3.01 (s, 3H, CH3), 3.18 (dd-t, 1H, J = 8.2 Hz, J = 8.3 Hz, H-4glu), 3.29 (dd-t, 1H, J = 8.6 Hz, J = 9.0 Hz, H-2glu), 3.38–3.47 (m, 2H, H-3glu, H-5glu), 3.63 (dd, 1H, J = 4.7 Hz, J = 12.3 Hz, H-6aglu), 3.81–3.90 (m, 1H, H-6bglu), 4.10–4.22 (m, 1H, CH2O), 4.27–4.37 (m, 1H, CH2O), 4.41 (d, 1H, J = 7.8 Hz, H-1glu), 4.68–4.77 (m, 2H, CH2N), 5.64 (s, 2H, CH2Ochin), 7.64–7.96 (m, 4H, H-3chin, H-5chin, H-6chin, H-7chin), 8.30 (s, 1H, H-5triaz), 8.91 (d, 1H, J = 8.3 Hz, H-4chin); 13C NMR (100 MHz, D2O): δ 19.99, 50.45, 60.60, 62.10, 67.96, 69.49, 72.88, 75.53, 75.82, 102.32, 114.65, 120.85, 124.20, 126.25, 128.08, 129.08, 129.44, 146.30, 146.98, 155.63, 157.49; HRMS (ESI-TOF): calcd for C21H27N4O7 ([M + H]+): m/z 447.1880; found: m/z 447.1880.
Glycoconjugate 76: Starting from 2-azidoethyl β-D-galactopyranoside 30 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a pink solid (131.7 mg, 59%); m.p.: 102-105 °C; [α]23D = 4.8 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.63 (s, 3H, CH3), 3.33–3.39 (m, 2H, H-2gal, H-3gal), 3.40–3.48 (m, 1H, H-5gal), 3.49–3.55 (m, 2H, H-6agal, H-6bgal), 3.63 (dd-t, 1H, J = 0.8 Hz, J = 3.6 Hz, H-4gal), 3.88–3.96 (m, 1H, CH2O), 4.06–4.14 (m, 1H, CH2O), 4.19 (d, 1H, J = 7.3 Hz, H-1gal), 4.35 (d, 1H, J = 4.6 Hz, OH), 4.53–4.64 (m, 3H, CH2N, OH), 4.69 (d, 1H, J = 5.0 Hz, OH), 4.95 (d, 1H, J = 4.4 Hz, OH), 5.33 (s, 2H, CH2Ochin), 7.38 (dd, 1H, J = 2.0 Hz, J = 7.2 Hz, H-7chin), 7.41 (d, 1H, J = 8.4 Hz, H-3chin), 7.44 (dd, 1H, J = 7.2 Hz, J = 7.8 Hz, H-6chin), 7.47 (dd, 1H, J = 1.8 Hz, J = 7.8 Hz, H-5chin), 8.18 (d, 1H, J = 8.4 Hz, H-4chin), 8.37 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 24.89, 49.75, 60.46, 61.66, 67.16, 68.14, 70.35, 73.29, 75.39, 103.53, 110.20, 119.77, 122.42, 125.65, 125.69, 127.30, 135.93, 139.16, 142.39, 153.37, 157.23; HRMS (ESI-TOF): calcd for C21H27N4O7 ([M + H]+): m/z 447.1880; found: m/z 447.1881.
Glycoconjugate 77: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)azidoacetamide 33 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a light yellow solid (205.5 mg, 67%); m.p.: 97–100 °C; [α]25D = 4.4 (c = 0.6, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.94, 1.97, 1.99, 2.00 (4s, 12H, CH3CO), 3.99 (m, 1H, H-6aglu), 4.10–4.18 (m, 2H, H-5glu, H-6bglu), 4.87 (dd-t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-4glu), 4.93 (dd-t, 1H, J = 9.7 Hz, J = 9.7 Hz, H-2glu), 5.16 and 5.22 (qAB, 2H, J = 16.5 Hz, CH2CO), 5.34–5.41 (m, 3H, H-1glu, CH2Ochin), 5.45 (dd-t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-3glu), 7.41 (dd, 1H, J = 3.6 Hz, J = 5.4 Hz, H-7chin), 7.49–7.57 (m, 3H, H-3chin, H-5chin, H-6chin), 8.24 (s, 1H, H-5triaz), 8.32 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.83 (d, 1H, J = 2.7 Hz, H-2chin), 9.25 (d, 1H, J = 9.3 Hz, CONH); 13C NMR (100 MHz, DMSO-d6): δ 20.27, 20.30, 20.33, 20.48, 51.40, 61.73, 67.72, 70.58, 72.13, 72.64, 76.85, 79.12, 109.94, 119.95, 121.81, 126.38, 126.69, 129.02, 135.73, 139.70, 142.37, 148.94, 153.82, 166.17, 169.13, 169.25, 169.44, 169.94; HRMS (ESI-TOF): calcd for C28H32N5O11 ([M + H]+): m/z 614.2098; found: m/z 614.2095.
Glycoconjugate 78: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)azidoacetamide 34 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a yellow solid (227.0 mg, 74%); m.p.: 100–103 °C; [α]22D = 14.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.92, 1.98, 1.99, 2.12 (4s, 12H, CH3CO), 3.98 (dd, 1H, J = 6.7 Hz, J = 11.3 Hz, H-6agal), 4.05 (dd, 1H, J = 5.9 Hz, J = 11.3 Hz, H-6bgal), 4.35 (m, 1H, H-5gal), 5.08 (dd, 1H, J = 9.0 Hz, J = 9.4 Hz, H-2gal), 5.13 and 5.21 (qAB, 2H, J = 16.5 Hz, CH2CO), 5.27-5.44 (m, 5H, H-1gal, H-3gal, H-4gal, CH2Ochin), 7.42 (dd, 1H, J = 3.2 Hz, J = 5.8 Hz, H-7chin), 7.50–7.58 (m, 3H, H-3chin, H-5chin, H-6chin), 8.24 (s, 1H, H-5triaz), 8.28–8.35 (m, 2H, H-4chin, H-2chin), 9.34 (d, 1H, J = 9.5 Hz, CONH); 13C NMR (100 MHz, DMSO-d6): δ 20.33, 20.38, 20.48, 20.72, 51.37, 61.42, 61.75, 67.54, 68.24, 70.66, 71.43, 77.22, 109.95, 119.98, 121.88, 124.11, 126.39, 126.70, 135.73, 139.72, 142.38, 148.91, 153.85, 166.09, 169.31, 169.37, 169.78, 169.85; HRMS (ESI-TOF): calcd for C28H32N5O11 ([M + H]+): m/z 614.2098; found: m/z 614.2091.
Glycoconjugate 79: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)azidoacetamide 33 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a pink solid (254.2 mg, 81%); m.p.: 93–96 °C; [α]27D = 5.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.94, 1.97, 1.99, 2.00 (4s, 12H, CH3CO), 2.63 (s, 3H, CH3), 3.94–4.03 (m, 1H, H-6aglu), 4.06–4.18 (m, 2H, H-5glu, H-6bglu), 4.87 (dd-t, J = 9.4 Hz, J = 10.4 Hz, H-4glu), 4.92 (dd-t, J = 9.7 Hz, J = 9.8 Hz, H-2glu), 5.15 and 5.21 (qAB, 2H, J = 16.5 Hz, CH2CO), 5.33–5.48 (m, 4H, H-1glu, H-3glu, CH2Ochin), 7.38 (dd, 1H, J = 1.6 Hz, J = 7.5 Hz, H-7chin), 7.41 (d, 1H, J = 8.4 Hz, H-3chin), 7.43 (dd, 1H, J = 7.5 Hz, J = 8.0 Hz, H-6chin), 7.47 (dd, 1H, J = 1.6 Hz, J = 8.0 Hz, H-5chin), 8.18 (d, 1H, J = 8.4 Hz, H-4chin), 8.24 (s, 1H, H-5triaz), 9.25 (d, 1H, J = 9.3 Hz, CONH); 13C NMR (100 MHz, DMSO-d6): δ 20.21, 20.24, 20.27, 20.41, 24.85, 51.34, 61.53, 67.65, 70.51, 72.06, 72.57, 76.78, 79.06, 110.09, 119.72, 122.38, 125.54, 126.38, 127.27, 135.85, 139.11, 142.37, 153.27, 157.15, 166.10, 169.06, 169.19, 169.37, 169.87; HRMS (ESI-TOF): calcd for C29H34N5O11 ([M + H]+): m/z 628.2255; found: m/z 628.2256.
Glycoconjugate 80: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)azidoacetamide 34 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a pink solid (244.8 mg, 78%); m.p.: 98–101 °C; [α]27D = 11.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.92, 1.98, 1.99, 2.12 (4s, 12H, CH3CO), 2.63 (s, 3H, CH3), 3.98 (dd, 1H, J = 6.6 Hz, J = 11.3 Hz, H-6agal), 4.06 (dd, 1H, J = 5.9 Hz, J = 11.3 Hz, H-6bgal), 4.35 (m, 1H, H-5gal,), 5.07 (dd-t, 1H, J = 9.4 Hz, J = 9.4 Hz, H-2gal), 5.12 and 5.20 (qAB, 2H, J = 16.5 Hz, CH2CO), 5.26–5.44 (m, 5H, H-1gal, H-3gal, H-4gal, CH2Ochin), 7.32–7.54 (m, 4H, H-3chin, H-5chin, H-6chin, H-7chin)8.21 (d, 1H, J = 7.9 Hz, H-4chin), 8.24 (s, 1H, H-5triaz), 9.33 (d, 1H, J = 9.5 Hz, CONH); 13C NMR (100 MHz, DMSO-d6): δ 20.23, 20.26, 20.31, 20.40, 24.84, 51.30, 61.54, 67.47, 68.16, 70.59, 71.35, 77.14, 79.06, 110.10, 119.72, 122.36, 125.54, 126.38, 127.24, 135.85, 139.11, 142.36, 153.27, 157.15, 166.02, 169.23, 169.29, 169.71, 169.77; HRMS (ESI-TOF): calcd for C29H34N5O11 ([M + H]+): m/z 628.2255; found: m/z 628.2253.
Glycoconjugate 81: Starting from N-(β-D-glucopyranosyl)azidoacetamide 35 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a brown solid (133.6 mg, 60%); m.p.: 160-163 °C; [α]25D = 16.7 (c = 2.0, DMSO); 1H NMR (400 MHz, DMSO-d6): δ 3.04–3.15 (m, 3H, H-2glu, H-4glu, H-5glu), 3.21 (dd-t, 1H, J = 8.4 Hz, J = 8.6 Hz, H-3glu), 3.42 (m, 1H, H-6aglu), 3.63 (m, 1H, H-6bglu), 4.00–4.50 (m, 4H, OH), 4.71 (dd-t, 1H, J = 8.9 Hz, J = 9.0 Hz, H-1glu), 5.17 and 5.22 (qAB, 2H, J = 16.5 Hz, CH2CO), 5.57 (s, 2H, CH2Ochin), 7.60–8.10 (m, 5H, H-3chin, H-4chin, H-5chin, H-6chin, H-7chin), 8.39 (s, 1H, H-5triaz), 8.96 (d, 1H, J = 7.4 Hz, H-2chin), 9.04 (d, 1H, J = 8.8 Hz, CONH); 13C NMR (100 MHz, DMSO-d6): δ 51.55, 60.72, 62.36, 69.74, 72.44, 77.18, 78.65, 79.63, 112.94, 120.36, 122.56, 126.79, 128.94, 132.86, 136.25, 136.88, 141.50, 146.25, 151.30, 165.67; HRMS (ESI-TOF): calcd for C20H24N5O7 ([M + H]+): m/z 446.1676; found: m/z 446.1679.
Glycoconjugate 82: Starting from N-(β-D-galactopyranosyl)azidoacetamide 36 and 8-(2-propyn-1-yloxy)quinoline 3, product was obtained as a brown solid (129.2 mg, 58%); m.p.: 170-173 °C; [α]25D = 14.5 (c = 2.0, DMSO); 1H NMR (400 MHz, DMSO-d6): δ 3.34 (dd, 1H, J = 3.2 Hz, J = 9.4 Hz, H-3gal), 3.36–3.46 (m, 3H, H-2gal, H-5gal, H-6agal), 3.50 (dd, 1H, J = 5.8 Hz, J = 10.5 Hz, H-6bgal), 3.69 (d, 1H, J = 3.0 Hz, H-4gal), 4.09–4.42 (m, 4H, OH), 4.69 (dd-t, 1H J = 8.9 Hz, J = 9.0 Hz, H-1gal), 5.18 (s, 2H, CH2CO), 5.60 (s, 2H, CH2Ochin), 7.56–8.16 (m, 4H, H-3chin, H-5chin, H-6chin, H-7chin), 8.41 (s, 1H, H-5triaz), 8.85–9.20 (m, 3H, H-2chin, H-4chin, CONH); 13C NMR (100 MHz, DMSO-d6): δ 51.62, 60.31, 68.02, 69.66, 73.85, 76.75, 79.08, 80.07, 112.53, 120.38, 126.83, 126.96, 129.01, 130.10, 141.66, 142.90, 146.07, 150.24, 152.06, 165.58; HRMS (ESI-TOF): calcd for C20H24N5O7 ([M + H]+): m/z 446.1676; found: m/z 446.1670.
Glycoconjugate 83: Starting from N-(β-D-glucopyranosyl)azidoacetamide 35 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a brown solid (140.1 mg, 61%); m.p.: 135–138 °C; [α]26D = −2.8 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.92 (s, 3H, CH3), 3.04–3.24 (m, 3H, H-2glu, H-4glu, H-5glu), 3.39–3.47 (m, 2H, H-3glu, H-6aglu,), 3.62 (dd, 1H, J = 1.6 Hz, J = 12.1 Hz, H-6bglu), 4.69 (dd-t, 1H, J = 8.9 Hz, J = 9.0 Hz, H-1glu), 5.15 and 5.20 (qAB, 2H, J = 16.5 Hz, CH2CO), 5.60 (s, 2H, CH2Ochin), 7.73–7.93 (m, 4H, H-3chin, H-5chin, H-6chin, H-7chin), 8.34 (s, 1H, H-5triaz), 8.89 (d, 1H, J = 8.3 Hz, H-4chin), 9.04 (d, 1H, J = 8.8 Hz, CONH); 13C NMR (100 MHz, DMSO-d6): δ 18.45, 51.52, 60.71, 62.31, 69.74, 72.42, 77.18, 78.65, 79.62, 113.85, 120.30, 124.31, 126.87, 127.70, 128.48, 141.31, 143.79, 148.61, 158.29, 163.67, 165.65; HRMS (ESI-TOF): calcd for C21H26N5O7 ([M + H]+): m/z 460.1832; found: m/z 460.1830.
Glycoconjugate 84: Starting from N-(β-D-galactopyranosyl)azidoacetamide 36 and 2-methyl-8-(2-propyn-1-yloxy)quinoline 4, product was obtained as a brown solid (179.2 mg, 78%); m.p.: 145–148 °C; [α]24D = 5.0 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.88 (s, 3H, CH3), 3.34 (dd, 1H, J = 3.2 Hz, J = 9.4 Hz, H-3gal), 3.37-3.54 (m, 4H, H-2gal, H-5gal, H-6agal, H-6bgal), 3.69 (d, 1H, J = 3.0 Hz, H-4gal), 3.95–4.31 (m, 4H, OH), 4.67 (dd-t, 1H, J = 8.9 Hz, J = 9.0 Hz, H-1gal), 5.17 (s, 2H, CH2CO), 5.59 (s, 2H, CH2Ochin), 7.62–7.98 (m, 4H, H-3chin, H-5chin, H-6chin, H-7chin), 8.34 (s, 1H, H-5triaz), 8.84 (bs, 1H, H-4chin), 9.01 (d, 1H, J = 9.0 Hz, CONH); 13C NMR (100 MHz, DMSO-d6): δ 18.45, 51.55, 60.30, 62.27, 68.01, 69.65, 73.85, 76.74, 80.06, 113.46, 120.27, 124.12, 126.87, 128.23, 131.53, 141.40, 142.91, 149.12, 158.22, 162.36, 165.59; HRMS (ESI-TOF): calcd for C21H26N5O7 ([M + H]+): m/z 460.1832; found: m/z 460.1831.
Glycoconjugate 85: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)propiolamide 37 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a yellow solid (220.9 mg, 72%); m.p.: 89–94 °C; [α]23D = −18.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.86, 1.94, 1.98, 2.00 (4s, 12H, CH3CO), 3.98 (dd, 1H, J = 3.9 Hz, J = 14.0 Hz, H-6aglu), 4.07–4.18 (m, 2H, H-5glu, H-6bglu), 4.66 (t, 2H, J = 5.1 Hz, CH2N), 4.90 (dd-t, 1H, J = 9.6 Hz, J = 9.6 Hz, H-4glu), 4.98 (t, 2H, J = 5.1 Hz, CH2O), 5.20 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-2glu), 5.38 (dd-t, 1H, J = 9.5 Hz, J = 9.5 Hz, H-1glu), 5.59 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-3glu), 7.25 (dd, 1H, J = 1.4 Hz, J = 7.5 Hz, H-7chin), 7.47–7.59 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.7 Hz, J = 8.4 Hz, H-4chin), 8.87 (dd, 1H, J = 1.5 Hz, J = 4.1 Hz, H-2chin), 8.98 (s, 1H, H-5triaz), 9.20 (d, 1H, J = 9.5 Hz, NHCO); 13C NMR (100 MHz, DMSO-d6): δ 20.30, 20.31, 20.37, 20.50, 49.56, 61.85, 67.07, 67.85, 70.49, 72.04, 73.03, 76.79, 110.69, 120.60, 121.93, 126.69, 128.20, 129.08, 135.86, 139.71, 141.83, 149.25, 153.60, 160.03, 169.02, 169.32, 169.54, 169.97; HRMS (ESI-TOF): calcd for C28H32N5O11 ([M + H]+): m/z 614.2098; found: m/z 614.2097.
Glycoconjugate 86: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)propiolamide 38 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a white solid (285.3 mg, 93%); m.p.: 187–189 °C; [α]24D = −6.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.96, 2.00, 2.01, 2.17 (4s, 12H, CH3CO), 4.03–4.17 (m, 3H, H-5gal, H-6agal, H-6bgal), 4.63 (t, 2H, J = 4.9 Hz, CH2N), 5.02 (t, 2H, J = 4.9 Hz, CH2O), 5.15 (dd, 1H, J = 3.3 Hz, J = 10.1 Hz, H-3gal), 5.30 (dd-t, 1H, J = 9.3 Hz, J = 10.1 Hz, H-2gal), 5.41 (dd-t, 1H, J = 9.3 Hz, J = 9.5 Hz, H-1gal), 5.45 (dd, 1H, J = 0.6 Hz, J = 3.3 Hz, H-4gal), 7.04 (dd, 1H, J = 1.7 Hz, J = 7.2 Hz, H-7chin), 7.40–7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 7.83 (d, 1H, J = 9.5 Hz, NHCO); 8.14 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.95 (s, 1H, H-5triaz), 9.00 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz. H-2chin); 13C NMR (100 MHz, CDCl3): δ 20.57, 20.63, 20.64, 20.67, 50.14, 61.27, 67.24, 67.53, 68.08, 71.17, 72.32, 78.15, 110.28, 121.34, 122.02, 126.42, 128.22, 129.64, 135.98, 140.36, 142.35, 149.81, 153.67, 160.32, 169.90, 170.15, 170.35, 170.46; HRMS (ESI-TOF): calcd for C28H32N5O11 ([M + H]+): m/z 614.2098; found: m/z 614.2095.
Glycoconjugate 87: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)propiolamide 37 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a yellow solid (207.1 mg, 66%); m.p.: 167–172 °C; [α]24D = −14.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.85, 1.93, 1.98, 2.00 (4s, 12H, CH3CO), 2.72 (s, 3H, CH3), 3.98 (dd, 1H, J = 4.5 Hz, J = 14.3 Hz, H-6aglu), 4.07–4.18 (m, 2H, H-5glu, H-6bglu), 4.54–4.67 (m, 2H, CH2N), 4.90 (dd-t, 1H, J = 9.4 Hz, J = 9.5 Hz, H-4glu), 4.98 (t, 2H, J = 5.0 Hz, CH2O), 5.21 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-2glu),5.38 (dd-t, 1H, J = 9.5 Hz, J = 9.5 Hz, H-1glu), 5.60 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-3glu), 7.21 (dd, 1H, J = 1.2 Hz, J = 7.7 Hz, H-7chin), 7.38–7.46 (m, 2H, H-3chin, H-6chin), 7.50 (dd, 1H, J = 1.1 Hz, J = 8.2 Hz, H-5chin), 8.20 (d, 1H, J = 8.5, H-4chin), 9.17 (d, 1H, J = 9.5 Hz, NHCO), 9.20 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 20.29, 20.31, 20.37, 20.50, 25.00, 49.59, 61.86, 67.17, 67.85, 70.52, 72.01, 73.01, 76.75, 111.06, 120.46, 122.58, 125.63, 127.30, 128.62, 136.02, 139.28, 141.93, 153.06, 157.80, 160.08, 169.01, 169.32, 169.53, 169.97; HRMS (ESI-TOF): calcd for C29H33N5O11 ([M + H]+): m/z 628.2255; found: m/z 628.2252.
Glycoconjugate 88: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)propiolamide 38 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a white solid (257.3 mg, 82%); m.p.: 107–109 °C; [α]25D = −7.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.95, 2.00, 2.02, 2.17 (4s, 12H, CH3CO), 2.88 (s, 3H, CH3), 4.03–4.17 (m, 3H, H-5gal, H-6agal, H-6bgal), 4.59 (t, 2H, J = 4.9 Hz, CH2N), 5.00 (t, 2H, J = 4.8 Hz, CH2O), 5.15 (dd, 1H, J = 3.4 Hz, J = 10.2 Hz, H-3gal), 5.30 (dd-t, 1H, J = 9.4 Hz, J = 10.2 Hz, H-2gal), 5.42 (dd-t, 1H, J = 9.4 Hz, J = 9.5 Hz, H-1gal), 5.45 (dd, 1H, J = 0.7 Hz, J = 3.4 Hz, H-4gal), 7.02 (dd, 1H, J = 1.3 Hz, J = 7.5 Hz, H-7chin), 7.32–7.39 (m, 2H, H-3chin, H-6chin), 7.42 (dd, 1H, J = 1.3 Hz, J = 8.2 Hz, H-5chin), 7.84 (d, 1H, J = 9.6 Hz, NHCO), 8.02 (d, 1H, J = 8.4 Hz, H-4chin), 9.26 (s, 1H, H-5triaz); 13C NMR (100 MHz, CDCl3): δ 20.57, 20.61, 20.64, 20.66, 25.62, 50.18, 61.23, 67.23, 67.45, 68.08, 71.18, 72.25, 78.11, 110.54, 121.14, 122.90, 125.38, 127.79, 128.65, 136.04, 139.94, 142.40, 153.15, 159.03, 160.36, 169.90, 170.15, 170.34, 170.41; HRMS (ESI-TOF): calcd for C29H34N5O11 ([M + H]+): m/z 628.2255; found: m/z 628.2254.
Glycoconjugate 89: Starting from N-(β-D-glucopyranosyl)propiolamide 39 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a white solid (133.6 mg, 60%); m.p.: 169–171 °C; [α]28D = 5.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO-d6): δ 3.05-3.11 (m, 1H, H-5glu), 3.13–3.18 (m, 2H, H-2glu, H-4glu), 3.33–3.37 (m, 1H, H-3glu), 3.38–3.44 (m, 1H, H-6aglu), 3.60–3.67 (m, 1H, H-6bglu), 4.48 (t, 1H, J = 5.9 Hz, 6-OH), 4.66 (t, 2H, J = 4.6 Hz, CH2N), 4.83–4.93 (m, 3H, H-1glu, OH), 4.95–5.01 (m, 3H, CH2O, OH), 7.25 (d, 1H, J = 7.5 Hz, H-7chin), 7.47–7.62 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (d, 1H, J = 10.4 Hz, H-4chin), 8.68 (d, 1H, J = 9.1 Hz, NHCO), 8.88 (bs, 1H, H-2chin), 8.94 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 49.52, 60.97, 67.11, 69.97, 71.83, 77.44, 78.70, 79.51, 110.64, 120.60, 121.96, 126.69, 127.68, 129.08, 135.84, 139.71, 142.53, 149.29, 153.63, 160.08; HRMS (ESI-TOF): calcd for C20H24N5O7 ([M + H]+): m/z 446.1676; found: m/z 446.1678.
Glycoconjugate 90: Starting from N-(β-D-galactopyranosyl)propiolamide 40 and 8-(2-azidoethoxy)quinoline 5, product was obtained as a white solid (189.3 mg, 85%); m.p.: 162–164 °C; [α]25D = 24.8 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 3.34-3.54 (m, 4H, H-2gal, H-3gal, H-4gal H-5gal), 3.57–3.65 (m, 1H, H-6agal), 3.66–3.72 (m, 1H, H-6bgal), 4.29 (d, 1H, J = 4.9 Hz, OH), 4.56 (t, 1H, J = 5.4, 6-OH), 4.66 (t, 2H, J = 5.1 Hz, CH2N), 4.74 (d, 1H, J = 5.5 Hz, OH), 4.79 (d, 1H, J = 5.5 Hz, OH), 4.87 (dd-t, 1H, J = 9.0 Hz, J = 9.1 Hz, H-1gal), 4.98 (t, 2H, J = 5.0 Hz, CH2O), 7.26 (dd, 1H, J = 1.4 Hz, J = 7.5 Hz, H-7chin), 7.47–7.59 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.5 Hz, J = 8.2 Hz, H-4chin), 8.48 (d, 1H, J = 9.1 Hz, NHCO), 8.88 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2chin), 8.95 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 49.52, 60.42, 67.13, 68.37, 69.33, 74.03, 76.82, 79.93, 110.69, 120.60, 121.93, 126.68, 127.61, 129.07, 135.84, 139.72, 142.47, 149.29, 153.62, 159.97; HRMS (ESI-TOF): calcd for C20H24N5O7 ([M + H]+): m/z 446.1676; found: m/z 446.1675.
Glycoconjugate 91: Starting from N-(β-D-glucopyranosyl)propiolamide 39 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a white solid (144.7 mg, 63%); m.p.: 159–161 °C; [α]25D = −3.6 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.72 (s, 3H, CH3), 3.03–3.12 (m, 1H, H-5glu), 3.13–3.27 (m, 2H, H-2glu, H-4glu), 3.34–3.46 (m, 2H, H-3glu, H-6aglu), 3.60–3.68 (m, 1H, H-6bglu), 4.47 (t, 1H, J = 5.9 Hz, 6-OH), 4.55–4.67 (m, 2H, CH2N), 4.86–4.93 (m, 3H, H-1glu, OH), 4.94–5.02 (m, 3H, CH2O, OH), 7.21 (dd, 1H, J = 1.2 Hz, J = 7.7 Hz, H-7chin), 7.39–7.47 (m, 2H, H-3chin, H-6chin), 7.50 (dd, 1H, J = 1.1 Hz, J = 8.2 Hz, H-5chin), 8.20 (d, 1H, J = 8.4 Hz, H-4chin), 8.63 (d, 1H, J = 9.1 Hz, NHCO), 9.15 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.07, 49.57, 60.99, 67.27, 69.97, 71.85, 77.42, 78.68, 79.47, 111.02, 120.47, 122.62, 125.64, 127.31, 128.11, 136.02, 139.28, 142.63, 153.11, 157.85, 160.12; HRMS (ESI-TOF): calcd for C20H24N5O7 ([M + H]+): m/z 460.1832; found: m/z 460.1830.
Glycoconjugate 92: Starting from N-(β-D-galactopyranosyl)propiolamide 40 and 2-methyl-8-(2-azidoethoxy)quinoline 8, product was obtained as a white solid (186.1 mg, 81%); m.p.: 148–151 °C; [α]28D = 23.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO-d6): δ 2.72 (s, 3H, CH3), 3.34–3.55 (m, 4H, H-2gal, H-3gal, H-4gal H-5gal), 3.57–3.66 (m, 1H, H-6agal), 3.66–3.72 (m, 1H, H-6bgal), 4.28 (d, 1H, J = 5.0 Hz, OH), 4.52–4.67 (m, 3H, CH2N, 6-OH), 4.73 (d, 1H, J = 5.5 Hz, OH), 4.79 (d, 1H, J = 5.5 Hz, OH), 4.88 (dd-t, 1H, J = 9.0 Hz, J = 9.1 Hz, H-1gal), 4.98 (t, 2H, J = 5.0 Hz, CH2O), 7.21 (dd, 1H, J = 1.2 Hz, J = 7.7 Hz, H-7chin), 7.39–7.46 (m, 2H, H-3chin, H-6chin), 7.50 (dd, 1H, J = 1.1 Hz, J = 8.2 Hz, H-5chin), 8.20 (d, 1H, J = 8.4 Hz, H-4chin), 8.43 (d, 1H, J = 9.1 Hz, NHCO), 9.16 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.04, 49.56, 60.40, 67.28, 68.37, 69.36, 74.02, 76.78, 79.89, 111.08, 120.47, 122.59, 125.62, 127.30, 128.04, 136.00, 139.29, 142.57, 153.11, 157.83, 160.00; HRMS (ESI-TOF): calcd for C21H26N5O7 ([M + H]+): m/z 460.1832; found: m/z 460.1836.
Glycoconjugate 93: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)propiolamide 37 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a yellow solid (210.2 mg, 67%); m.p.: 178–180 °C; [α]24D = −18.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.89, 1.94, 1.99, 2.00 (4s, 12H, CH3CO), 2.40–2.50 (m, 2H, CH2), 4.00 (dd, 1H, J = 4.4 Hz, J = 14.2 Hz, H-6aglu), 4.06–4.18 (m, 2H, H-5glu, H-6bglu), 4.21 (t, 2H, J = 6.0 Hz, CH2N), 4.72 (t, 2H, J = 6.8 Hz, CH2O), 4.91 (dd-t, 1H, J = 9.5 Hz, J = 9.5 Hz, H-4glu), 5.21 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-2glu), 5.39 (dd-t, 1H, J = 9.5 Hz, J = 9.5 Hz, H-1glu), 5.61 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-3glu), 7.20 (dd, 1H, J = 1.8 Hz, J = 7.1 Hz, H-7chin), 7.46–7.59 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.5 Hz, J = 8.1 Hz, H-4chin), 8.90 (dd, 1H, J = 1.7 Hz, J = 4.0 Hz, H-2chin), 8.90 (s, 1H, H-5triaz), 9.17 (d, 1H, J = 9.5 Hz, NHCO); 13C NMR (100 MHz, DMSO-d6): δ 20.32, 20.33, 20.38, 20.52, 29.36, 47.24, 61.85, 65.52, 67.84, 70.52, 72.04, 73.02, 76.80, 109.92, 119.97, 121.84, 126.77, 127.67, 129.03, 135.79, 139.77, 141.83, 149.07, 154.18, 160.10, 169.04, 169.32, 169.54, 169.98; HRMS (ESI-TOF): calcd for C29H34N5O11 ([M + H]+): m/z 628.2255; found: m/z 628.2252.
Glycoconjugate 94: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)propiolamide 38 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (263.6 mg, 84%); m.p.: 98–100 °C; [α]25D = −5.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.99, 2.00, 2.03, 2.17 (4s, 12H, CH3CO), 2.62 (p, 2H, J = 6.2 Hz, CH2), 4.04–4.18 (m, 3H, H-5gal, H-6agal, H-6bgal), 4.24 (t, 2H, J = 5.8 Hz, CH2N), 4.81 (t, 2H, J = 6.6 Hz, CH2O), 5.17 (dd, 1H, J = 3.3 Hz, J = 10.2 Hz, H-3gal), 5.31 (dd-t, 1H, J = 9.4 Hz, J = 10.2 Hz, H-2gal), 5.42 (dd-t, 1H, J = 9.4 Hz, J = 9.5 Hz, H-1gal), 5.46 (dd, 1H, J = 0.6 Hz, J = 3.3 Hz, H-4gal), 7.06 (dd, 1H, J = 1.7 Hz, J = 7.2 Hz, H-7chin), 7.41–7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 7.85 (d, 1H, J = 9.5 Hz, NHCO), 8.16 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.52 (s, 1H, H-5triaz), 9.01 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz. H-2chin); 13C NMR (100 MHz, CDCl3): δ 20.53, 20.57, 20.64, 20.68, 29.67, 47.73, 61.28, 65.36, 67.24, 68.11, 71.16, 72.33, 78.16, 109.94, 120.67, 121.82, 126.62, 127.18, 129.61, 136.09, 140.39, 142.04, 149.55, 154.23, 160.36, 169.90, 170.15, 170.36, 170.45; HRMS (ESI-TOF): calcd for C29H34N5O11 ([M + H]+): m/z 628.2255; found: m/z 628.2253.
Glycoconjugate 95: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)propiolamide 37 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a beige solid (195.7 mg, 61%); m.p.: 197–198 °C; [α]24D = −18.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6): δ 1.88, 1.94, 1.99, 2.00 (4s, 12H, CH3CO), 2.41–2.49 (m, 2H, CH2), 2.67 (s, 3H, CH3), 3.96–4.02 (m, 1H, H-6aglu), 4.05–4.17 (m, 2H, H-5glu, H-6bglu), 4.20 (t, 2H, J = 6.2 Hz, CH2N), 4.71 (t, 2H, J = 6.9 Hz, CH2O), 4.91 (dd-t, 1H, J = 9.5 Hz, J = 9.5 Hz, H-4glu), 5.20 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-2glu), 5.38 (dd-t, 1H, J = 9.5 Hz, J = 9.5 Hz, H-1glu), 5.59 (dd-t, 1H, J = 9.3 Hz, J = 9.4 Hz, H-3glu), 7.17 (dd, 1H, J = 1.3 Hz, J = 7.6 Hz, H-7chin), 7.36–7.44 (m, 2H, H-3chin, H-6chin), 7.47 (dd, 1H, J = 1.2 Hz, J = 8.2 Hz, H-5chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.83 (s, 1H, H-5triaz), 9.17 (d, 1H, J = 9.5 Hz, NHCO); 13C NMR (100 MHz, DMSO-d6): δ 20.31, 20.37, 20.51, 20.74, 25.03, 29.25, 47.14, 61.84, 65.56, 67.83, 70.51, 72.02, 73.01, 76.78, 110.54, 119.91, 122.44, 125.68, 127.32, 127.58, 135.97, 139.34, 141.77, 153.64, 157.35, 160.06, 169.02, 169.31, 169.53, 169.97; HRMS (ESI-TOF): calcd for C30H36N5O11 ([M + H]+): m/z 642.2411; found: m/z 642.2408.
Glycoconjugate 96: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)propiolamide 38 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a white solid (202.1 mg, 63%); m.p.: 121–123 °C; [α]25D = −4.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.98, 2.00, 2.03, 2.17 (4s, 12H, CH3CO), 2.60 (p, 2H, J = 6.4 Hz, CH2), 2.80 (s, 3H, CH3), 4.04–4.17 (m, 3H, H-5gal, H-6agal, H-6bgal), 4.24 (t, 2H, J = 5.9 Hz, CH2N), 4.82 (t, 2H, J = 6.7 Hz, CH2O), 5.16 (dd, 1H, J = 3.4 Hz, J = 10.2 Hz, H-3gal), 5.30 (dd-t, 1H, J = 9.4 Hz, J = 10.1 Hz, H-2gal), 5.40 (dd-t, 1H, J = 9.4 Hz, J = 9.5 Hz, H-1gal), 5.46 (dd, 1H, J = 0.6 Hz, J = 3.3 Hz, H-4gal), 7.04 (dd, 1H, J = 1.8 Hz, J = 7.1 Hz, H-7chin), 7.31–7.42 (m, 3H, H-3chin, H-5chin, H-6chin), 7.84 (d, 1H, J = 9.5 Hz, NHCO), 8.03 (d, 1H, J = 8.4 Hz, H-4chin), 8.38 (s, 1H, H-5triaz); 13C NMR (100 MHz, CDCl3): δ 20.57, 20.63, 20.64, 20.68, 25.65, 29.58, 47.58, 61.27, 65.24, 67.23, 68.10, 71.15, 72.32, 78.14, 110.56, 120.60, 122.68, 125.58, 127.06, 127.85, 136.19, 140.02, 141.93, 153.59, 158.44, 160.30, 169.90, 170.14, 170.36, 170.44; HRMS (ESI-TOF): calcd for C30H36N5O11 ([M + H]+): m/z 642.2411; found: m/z 642.2409.
Glycoconjugate 97: Starting from N-(β-D-glucopyranosyl)propiolamide 39 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (158.5 mg, 69%); m.p.: 185–189 °C; [α]28D = 6.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO-d6): δ 2.46 (p, 2H, J = 6.5 Hz, CH2), 3.06–3.11 (m, 1H, H-5glu), 3.15-3.19 (m, 1H, H-2glu), 3.20–3.25 (m, 1H, H-4glu), 3.33–3.37 (m, 1H, H-3glu), 3.39–3.45 (m, 1H, H-6aglu), 3.63–3.68 (m, 1H, H-6bglu), 4.19 (t, 2H, J = 6.0 Hz, OH), 4.49 (t, 1H, J = 5.9 Hz, 6-OH), 4.72 (t, 2H, J = 6.3 Hz, CH2N), 4.84–4.94 (m, 3H, H-1glu, CH2O), 5.00 (d, 1H, J = 4.6 Hz, OH), 7.20 (dd, 1H, J = 1.5 Hz, J = 7.4 Hz, H-7chin), 7.47-7.62 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.5 Hz, J = 8.0 Hz, H-4chin), 8.66 (d, 1H, J = 9.1 Hz, NHCO), 8.86 (s, 1H, H-5triaz), 8.91 (dd, 1H, J = 1.6 Hz, J = 4.1 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 26.42, 44.13, 58.00, 62.40, 66.98, 68.88, 74.44, 75.69, 76.48, 106.91, 116.97, 118.88, 123.77, 124.17, 132.78, 136.66, 139.53, 142.49, 145.37, 146.13, 157.11; HRMS (ESI-TOF): calcd for C21H26N5O7 ([M + H]+): m/z 460.1832; found: m/z 460.1832.
Glycoconjugate 98: Starting from N-(β-D-galactopyranosyl)propiolamide 40 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (170.0 mg, 74%); m.p.: 129–132 °C; [α]28D = 22.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO-d6): δ 2.41–2.49 (m, 2H, CH2), 3.35–3.56 (m, 4H, H-2gal, H-3gal, H-4gal H-5gal), 3.58–3.66 (m, 1H, H-6agal), 3.68–3.75 (m, 1H, H-6bgal), 4.20 (t, 2H, J = 6.0 Hz, OH), 4.30 (d, 1H, J = 4.9 Hz, OH), 4.58 (t, 1H, J = 5.4 Hz, 6-OH), 4.68–4.77 (m, 3H, CH2N, CH2O), 4.81 (d, 1H, J = 5.5 Hz, CH2O), 4.89 (dd-t, 1H, J = 9.0 Hz, J = 9.1 Hz, H-1gal), 7.20 (dd, 1H, J = 1.8 Hz, J = 7.1 Hz, H-7chin), 7.46–7.60 (m, 3H, H-3chin, H-5chin, H-6chin), 8.32 (dd, 1H, J = 1.6 Hz, J = 8.3 Hz, H-4chin), 8.47 (d, 1H, J = 9.1 Hz, NHCO), 8.87 (s, 1H, H-5triaz), 8.91 (bs, 1H, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 29.41, 47.14, 60.43, 65.43, 68.38, 69.37, 74.04, 76.80, 79.92, 109.90, 119.97, 121.86, 126.77, 127.09, 129.03, 135.78, 139.77, 142.48, 149.12, 154.18, 160.01; HRMS (ESI-TOF): calcd for C21H26N5O7 ([M + H]+): m/z 460.1832; found: m/z 460.1835.
Glycoconjugate 99: Starting from N-(β-D-glucopyranosyl)propiolamide 39 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a brown solid (137.3 mg, 58%); m.p.: 167–169 °C; [α]27D = −7.0 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.46 (p, 2H, J = 6.5 Hz, CH2), 2.68 (s, 3H, CH3), 3.05–3.12 (m, 1H, H-5glu), 3.14–3.19 (m, 1H, H-2glu), 3.20–3.25 (m, 1H, H-4glu), 3.34–3.38 (m, 1H, H-3glu), 3.39–3.46 (m, 1H, H-6aglu), 3.61–3.68 (m, 1H, H-6bglu), 4.18 (t, 2H, J = 6.1 Hz, OH), 4.49 (m, 1H, 6-OH), 4.72 (t, 2H, J = 6.9 Hz, CH2N), 4.87–4.94 (m, 3H, H-1glu, CH2O), 5.00 (bs, 1H, OH), 7.17 (d, 1H, J = 7.5 Hz, H-7chin), 7.38–7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.66 (d, 1H, J = 9.1 Hz, NHCO), 8.80 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.06, 29.32, 47.02, 60.99, 65.43, 69.98, 71.87, 77.44, 78.69, 79.49, 110.51, 119.92, 122.48, 125.71, 127.10, 127.33, 136.01, 142.48, 146.29, 153.63, 157.39, 160.08; HRMS (ESI-TOF): calcd for C22H28N5O7 ([M + H]+): m/z 474.1989; found: m/z 474.1987.
Glycoconjugate 100: Starting from N-(β-D-galactopyranosyl)propiolamide 40 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a brown solid (175.2 mg, 74%); m.p.: 77–80 °C; [α]26D = 8.0 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.41–2.49 (m, 2H, CH2), 2.67 (s, 3H, CH3), 3.35–3.54 (m, 4H, H-2gal, H-3gal, H-4gal H-5gal), 3.57–3.64 (m, 1H, H-6agal), 3.66–3.72 (m, 1H, H-6bgal), 4.15–4.22 (m, 2H, OH), 4.30 (d, 1H, J = 4.9 Hz, OH), 4.37–4.44 (m, 1H, OH), 4.52–4.60 (m, 2H, CH2N), 4.66–4.78 (m, 3H, CH2N, CH2O), 4.81 (d, 1H, J = 5.5 Hz, CH2O), 4.87 (dd-t, 1H, J = 9.0 Hz, J = 9.1 Hz, H-1gal), 7.17 (dd, 1H, J = 1.2 Hz, J = 7.6 Hz, H-7chin), 7.37–7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.46 (d, 1H, J = 9.1 Hz, NHCO), 8.80 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 24.96, 29.23, 46.95, 60.33, 65.36, 68.28, 69.28, 73.94, 76.71, 79.83, 110.43, 119.82, 122.38, 125.62, 126.92, 127.24, 135.92, 139.22, 142.34, 153.54, 157.29, 159.88; HRMS (ESI-TOF): calcd for C22H28N5O7 ([M + H]+): m/z 474.1989; found: m/z 474.1987.
Glycoconjugate 101: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)-O-propargyl carbamate 41 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (309.1 mg, 94%); m.p.: 60–65 °C; [α]25D = −2.6 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.00, 2.01, 2.02, 2.07 (4s, 12H, CH3CO), 2.62 (p, 2H, J = 6.4 Hz, CH2), 3.73–3.83 (m, 1H, H-5glu), 4.03–4.13 (m, 1H, H-6aglu), 4.24 (t, 2H, J = 5.9 Hz, CH2N), 4.26–4.34 (m, 1H, H-6bglu), 4.74 (t, 2H, J = 6.7 Hz, CH2O), 4.90 (dd-t, 1H, J = 9.5 Hz, J = 9.5 Hz, H-4glu), 5.00 (dd-t, 1H, J = 9.5 Hz, J = 9.6 Hz, H-3glu), 5.06 (dd-t, 1H, J = 9.7 Hz, J = 9.7 Hz, H-2glu), 5.16 and 5.20 (qAB, 2H, J = 12.7 Hz, CH2OCO), 5.59 (d, 1H, J = 9.5 Hz, H-1glu), 7.04 (dd, 1H, J = 2.6 Hz, J = 6.3 Hz, H-7chin), 7.40–7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 7.78 (s, 1H, H-5triaz), 8.16 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.96 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz, H-2chin); 13C NMR (100 MHz, CDCl3): δ 20.56, 20.57, 20.63, 20.72, 29.66, 47.32, 58.81, 61.58, 65.26, 68.08, 70.11, 72.80, 73.36, 80.82, 109.52, 120.38, 121.74, 124.61, 126.68, 129.58, 136.07, 140.35, 142.38, 149.42, 154.23, 155.23, 169.48, 169.90, 170.55, 170.59; HRMS (ESI-TOF): calcd for C30H36N5O12 ([M + H]+): m/z 658.2360; found: m/z 658.2360.
Glycoconjugate 102: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)-O-propargyl carbamate 42 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (295.9 mg, 90%); m.p.: 66–69 °C; [α]25D = −7.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.98, 2.02, 2.03, 2.13 (4s, 12H, CH3CO), 2.62 (p, 2H, J = 6.4 Hz, CH2), 3.96–4.03 (m, 1H, H-6agal), 4.04–4.16 (m, 2H, H-5gal, H-6agal), 4.25 (t, 2H, J = 5.9 Hz, CH2N), 4.74 (t, 2H, J = 6.7 Hz, CH2O), 4.97 (dd-t, 1H, J = 8.3 Hz, J = 8.8 Hz, H-2gal), 5.03–5.13 (m, 2H, H-3gal, H-4gal), 5.16 and 5.20 (qAB, 2H, J = 12.8 Hz, CH2OCO), 5.58 (d, 1H, J = 9.5 Hz, H-1gal), 7.05 (dd, 1H, J = 2.5 Hz, J = 6.4 Hz, H-7chin), 7.41–7.49 (m, 3H, H-3chin, H-5chin, H-6chin), 7.78 (s, 1H, H-5triaz), 8.16 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.96 (dd, 1H, J = 1.7 Hz, J = 4.2 Hz, H-2chin); 13C NMR (100 MHz, CDCl3): δ 20.52, 20.60, 20.67, 20.71, 29.67, 47.33, 58.79, 61.07, 65.29, 67.10, 67.87, 70.92, 72.08, 81.16, 109.56, 120.39, 121.75, 124.57, 126.68, 129.59, 136.07, 140.37, 142.48, 149.43, 154.25, 155.25, 169.79, 170.04, 170.33, 170.83; HRMS (ESI-TOF): calcd for C30H36N5O12 ([M + H]+): m/z 658.2360; found: m/z 658.2360.
Glycoconjugate 103: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-glucopyranosyl)-O-propargyl carbamate 41 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a white solid (228.4 mg, 68%); m.p.: 56–59 °C; [α]26D = −3.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.01, 2.01, 2.03, 2.07 (4s, 12H, CH3CO), 2.59 (p, 2H, J = 6.4 Hz, CH2), 2.79 (s, 3H, CH3), 3.73–3.83 (m, 1H, H-5glu), 4.01–4.13 (m, 1H, H-6aglul), 4.23 (t, 2H, J = 5.9 Hz, CH2N), 4.26–4.34 (m, 1H, H-6bglu), 4.75 (t, 2H, J = 6.7 Hz, CH2O), 4.89 (dd-t, 1H, J = 9.4 Hz, J = 9.5 Hz, H-4glu), 4.99 (dd-t, 1H, J = 9.5 Hz, J = 9.7 Hz, H-3glu), 5.06 (dd-t, 1H, J = 9.7 Hz, J = 9.8 Hz, H-2glu), 5.13–5.23 (m, 2H, J = 12.7 Hz, CH2OCO), 5.53 (d, 1H, J = 9.5 Hz, H-1glu), 7.03 (dd, 1H, J = 2.3 Hz, J = 6.6 Hz, H-7chin), 7.30–7.43 (m, 3H, H-3chin, H-5chin, H-6chin), 7.83 (s, 1H, H-5triaz), 8.04 (dd, 1H, J = 1.7 Hz, J = 8.4 Hz, H-4chin); 13C NMR (100 MHz, CDCl3): δ 20.58, 20.59, 20.66, 20.74, 25.69, 29.62, 47.25, 58.79, 61.54, 65.32, 68.01, 70.03, 72.75, 73.30, 80.78, 110.04, 120.31, 122.64, 124.70, 125.66, 127.78, 136.21, 139.88, 142.28, 153.62, 155.20, 158.27, 169.50, 169.93, 170.58, 170.63; HRMS (ESI-TOF): calcd for C31H38N5O12 ([M + H]+): m/z 672.2517; found: m/z 672.2516.
Glycoconjugate 104: Starting from 2,3,4,6-tetra-O-acetyl-N-(β-D-galactopyranosyl)-O-propargyl carbamate 42 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a white solid (214.9 mg, 64%); m.p.: 62–65 °C; [α]26D = 6.8 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.98, 2.02, 2.03, 2.14 (4s, 12H, CH3CO), 2.59 (p, 2H, J = 6.4 Hz, CH2), 2.79 (s, 3H, CH3), 3.96–4.03 (m, 1H, H-6agal), 4.04–4.16 (m, 2H, H-5gal, H-6agal), 4.23 (t, 2H, J = 5.9 Hz, CH2N), 4.76 (t, 2H, J = 6.7 Hz, CH2O), 4.96 (dd-t, 1H, J = 8.3 Hz, J = 8.8 Hz, H-2gal), 5.03–5.13 (m, 2H, H-3gal, H-4gal), 5.14–5.23 (m, 2H, CH2OCO), 5.53 (d, 1H, J = 9.3 Hz, H-1gal), 7.03 (dd, 1H, J = 2.5 Hz, J = 6.5 Hz, H-7chin), 7.31–7.43 (m, 3H, H-3chin, H-5chin, H-6chin), 7.83 (s, 1H, H-5triaz), 8.04 (d, 1H, J = 8.4 Hz, H-4chin); 13C NMR (100 MHz, CDCl3): δ 20.53, 20.61, 20.67, 20.72, 25.69, 29.68, 47.28, 58.80, 61.07, 65.45, 67.10, 67.85, 70.93, 72.08, 81.15, 110.24, 120.37, 122.63, 124.62, 125.67, 127.82, 136.19, 139.99, 142.36, 153.71, 155.23, 158.28, 169.80, 170.05, 170.34, 170.83; HRMS (ESI-TOF): calcd for C31H38N5O12 ([M + H]+): m/z 672.2517; found: m/z 672.2518.
Glycoconjugate 105: Starting from N-(β-D-glucopyranosyl)-O-propargyl carbamate 43 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (203.1 mg, 83%); m.p.: 73–75 °C; [α]27D = 0.2 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.42 (p, 2H, J = 6.4 Hz, CH2), 2.96–3.11 (m, 3H, H-2glu, H-4glu, H-5glu), 3.15–3.20 (m, 1H, H-3glu), 3.36–3.44 (m, 1H, H-6aglu), 3.58–3.68 (m, 1H, H-6bglu), 4.09 (d, 1H, J = 5.1 Hz, OH), 4.20 (t, 2H, J = 6.0 Hz, CH2N), 4.45–4.54 (m, 2H, OH), 4.65 (t, 2H, J = 6.9 Hz, CH2O), 4.86 (m, 2H, CH2OCO), 4.95 (m, 1H, H-1glu), 5.07 (s, 1H, OH), 7.20 (dd, 1H, J = 2.2 Hz, J = 6.8 Hz, H-7chin), 7.47–7.59 (m, 3H, H-3chin, H-5chin, H-6chin), 7.89 (d, 1H, J = 9.2 Hz, NHCO), 8.31 (s, 1H, H-5triaz), 8.33 (dd, 1H, J = 1.7 Hz, J = 8.3 Hz, H-4chin), 8.89 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 29.64, 46.70, 57.07, 60.95, 65.43, 69.92, 72.02, 77.56, 78.38, 82.44, 109.85, 119.94, 121.89, 124.98, 126.82, 129.06, 135.84, 139.77, 142.42, 149.09, 154.19, 155.71; HRMS (ESI-TOF): calcd for C22H28N5O8 ([M + H]+): m/z 490.1938; found: m/z 490.1937.
Glycoconjugate 106: Starting from N-(β-D-galactopyranosyl)-O-propargyl carbamate 44 and 8-(3-azidopropoxy)quinoline 6, product was obtained as a white solid (200.7 mg, 82%); m.p.: 119–121 °C; [α]28D = 19.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO-d6): δ 2.42 (p, 2H, J = 6.5 Hz, CH2), 3.33–3.53 (m, 4H, H-2gal, H-3gal, H-4gal H-5gal), 3.63-3.69 (m, 1H, H-6agal), 4.08 (dd, 1H, J = 5.3 Hz, J = 10.5 Hz, H-6bgal), 4.20 (t, 2H, J = 6.1 Hz, CH2N), 4.32 (d, 1H, J = 3.8 Hz, OH), 4.46 (dd-t, 1H, J = 9.0 Hz, J = 9.0 Hz, H-1gal), 4.55 (t, 1H, J = 5.6 Hz, OH), 4.61–4.72 (m, 4H, CH2O, CH2OCO), 5.06 (s, 2H, OH), 7.20 (dd, 1H, J = 2.2 Hz, J = 6.8 Hz, H-7chin), 7.47–7.59 (m, 3H, H-3chin, H-5chin, H-6chin), 7.84 (d, 1H, J = 9.2 Hz, NHCO), 8.29 (s, 1H, H-5triaz), 8.32 (dd, 1H, J = 1.7 Hz, J = 8.2 Hz, H-4chin), 8.89 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz, H-2chin); 13C NMR (100 MHz, DMSO-d6): δ 29.62, 46.68, 57.03, 60.47, 65.44, 68.19, 69.27, 74.21, 76.53, 82.92, 109.86, 119.93, 121.86, 124.92, 126.80, 129.04, 135.80, 139.77, 142.47, 149.06, 154.18, 155.76; HRMS (ESI-TOF): calcd for C22H28N5O8 ([M + H]+): m/z 490.1938; found: m/z 490.1935.
Glycoconjugate 107: Starting from N-(β-D-glucopyranosyl)-O-propargyl carbamate 43 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a white solid (201.4 mg, 80%); m.p.: 57–60 °C; [α]27D = −0.2 (c = 1.0, MeOH); 1H NMR (400 MHz, DMSO-d6): δ 2.41 (p, 2H, J = 6.5 Hz, CH2), 2.67 (s, 3H, CH3), 2.96–3.11 (m, 3H, H-2glu, H-4glu, H-5glu), 3.12–3.20 (m, 1H, H-3glu), 3.36–3.44 (m, 1H, H-6aglu), 3.59–3.68 (m, 1H, H-6bglu), 4.09 (d, 1H, J = 5.3 Hz, OH), 4.19 (t, 2H, J = 6.2 Hz, CH2N), 4.44–4.53 (m, 2H, OH), 4.61–4.69 (m, 2H, CH2O), 4.86 (m, 2H, CH2OCO), 4.94 (d, 1H, J = 4.7 Hz, H-1glu), 5.06 (s, 1H, OH), 7.16 (dd, 1H, J = 1.1 Hz, J = 7.5 Hz, H-7chin), 7.38–7.50 (m, 3H, H-3chin, H-5chin, H-6chin), 7.88 (d, 1H, J = 9.2 Hz, NHCO), 8.02 (d, 1H, J = 9.2 Hz, H-2chin), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.30 (s, 1H, H-5triaz); 13C NMR (100 MHz, DMSO-d6): δ 25.08, 29.57, 46.68, 57.06, 60.94, 65.58, 69.92, 72.02, 77.54, 78.37, 82.43, 110.36, 119.86, 122.48, 124.94, 125.75, 127.34, 136.01, 139.32, 142.39, 153.68, 155.70, 157.35; HRMS (ESI-TOF): calcd for C23H30N5O8 ([M + H]+): m/z 504.2094; found: m/z 504.2090.
Glycoconjugate 108: Starting from N-(β-D-galactopyranosyl)-O-propargyl carbamate 44 and 2-methyl-8-(3-azidopropoxy)quinoline 9, product was obtained as a white solid (196.4 mg, 78%); m.p.: 94–98 °C; [α]26D = 3.0 (c = 1.0, H2O); 1H NMR (400 MHz, DMSO-d6): δ 2.41 (p, 2H, J = 6.6 Hz, CH2), 2.67 (s, 3H, CH3), 3.33–3.53 (m, 4H, H-2gal, H-3gal, H-4gal H-5gal), 3.63–3.69 (m, 1H, H-6agal), 4.08 (dd, 1H, J = 5.3 Hz, J = 10.5 Hz, H-6bgal), 4.19 (t, 2H, J = 6.2 Hz, CH2N), 4.32 (d, 1H, J = 3.8 Hz, OH), 4.46 (m, 1H, H-1gal), 4.55 (t, 1H, J = 5.6 Hz, OH), 4.61–4.73 (m, 4H, CH2O, CH2OCO), 5.06 (s, 2H, OH), 7.16 (dd, 1H, J = 1.3 Hz, J = 7.6 Hz, H-7chin), 7.38–7.49 (m, 3H, H-3chin, H-5chin, H-6chin), 7.83 (d, 1H, J = 9.2 Hz, NHCO), 8.19 (d, 1H, J = 8.4 Hz, H-4chin), 8.29 (s, 1H, H-5triaz), 13C NMR (100 MHz, DMSO-d6): δ 25.07, 29.56, 46.66, 57.03, 60.47, 65.60, 68.18, 69.27, 74.21, 76.52, 82.92, 110.39, 119.86, 122.46, 124.88, 125.73, 127.33, 135.98, 139.33, 142.45, 153.68, 155.76, 157.32; HRMS (ESI-TOF): calcd for C23H30N5O8 ([M + H]+): m/z 504.2094; found: m/z 504.2097.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




