
Photophysical Properties of Spirobifluorene-Based o-Carboranyl Compounds Altered by Structurally Rotating the Carborane Cages

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. General Considerations
2.2. Synthesis of (9,9-Spirobi[Fluoren]-2-Ylethynyl)Trimethylsilane (S1)
2.3. Synthesis of (9,9′-Spirobi[Fluoren]-4-Ylethynyl)Trimethylsilane (S2)
2.4. Synthesis of 2-Ethynyl-9,9′-Spirobi[Fluorene] (E1)
2.5. Synthesis of 4-Ethynyl-9,9′-Spirobi[Fluorene] (E2)
2.6. Synthesis of C1
2.7. Synthesis of C2
2.8. UV/Vis Absorption and Photoluminescence (PL) Experiments
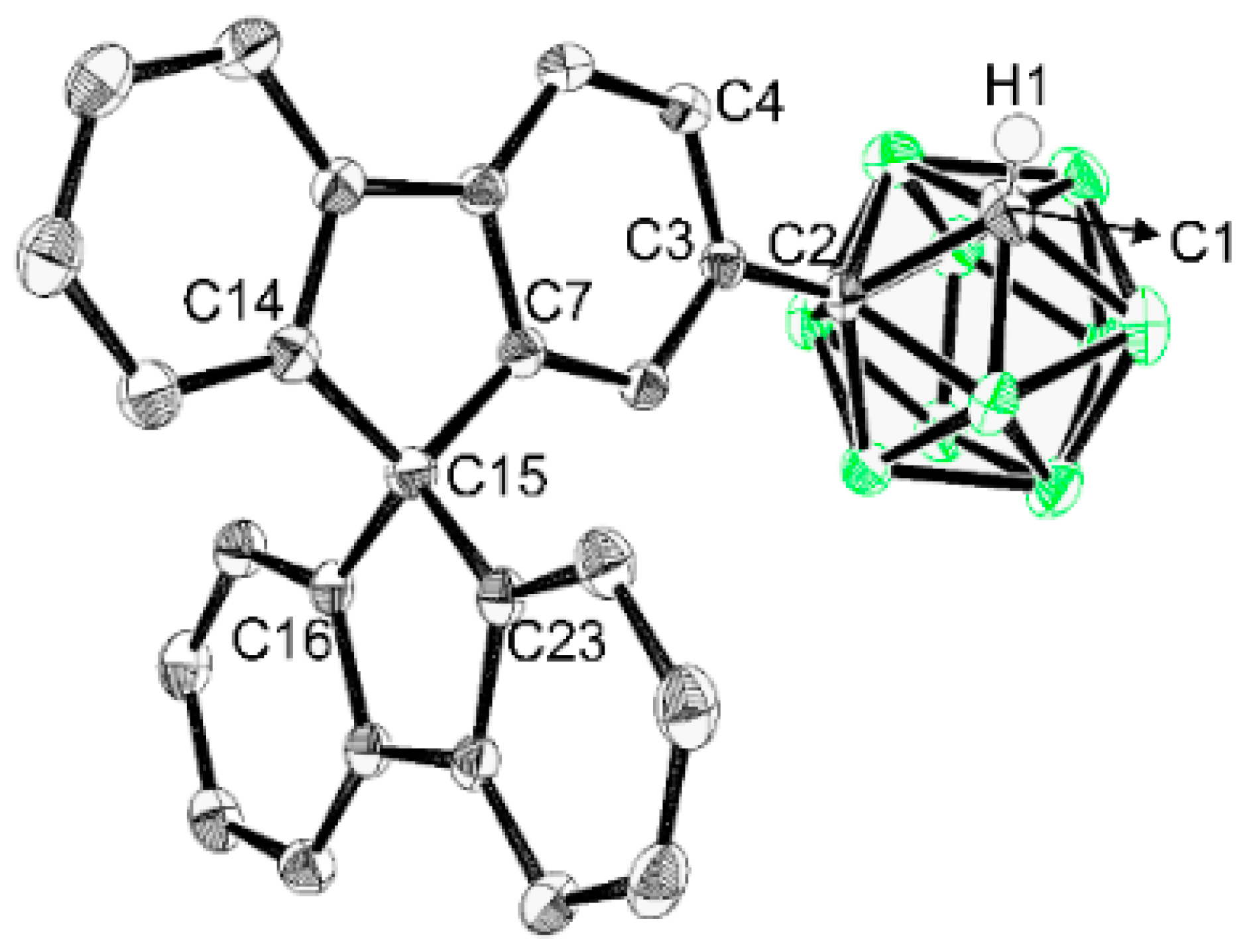
2.9. X-Ray Crystallography
2.10. Computational Studies
3. Results and Discussion
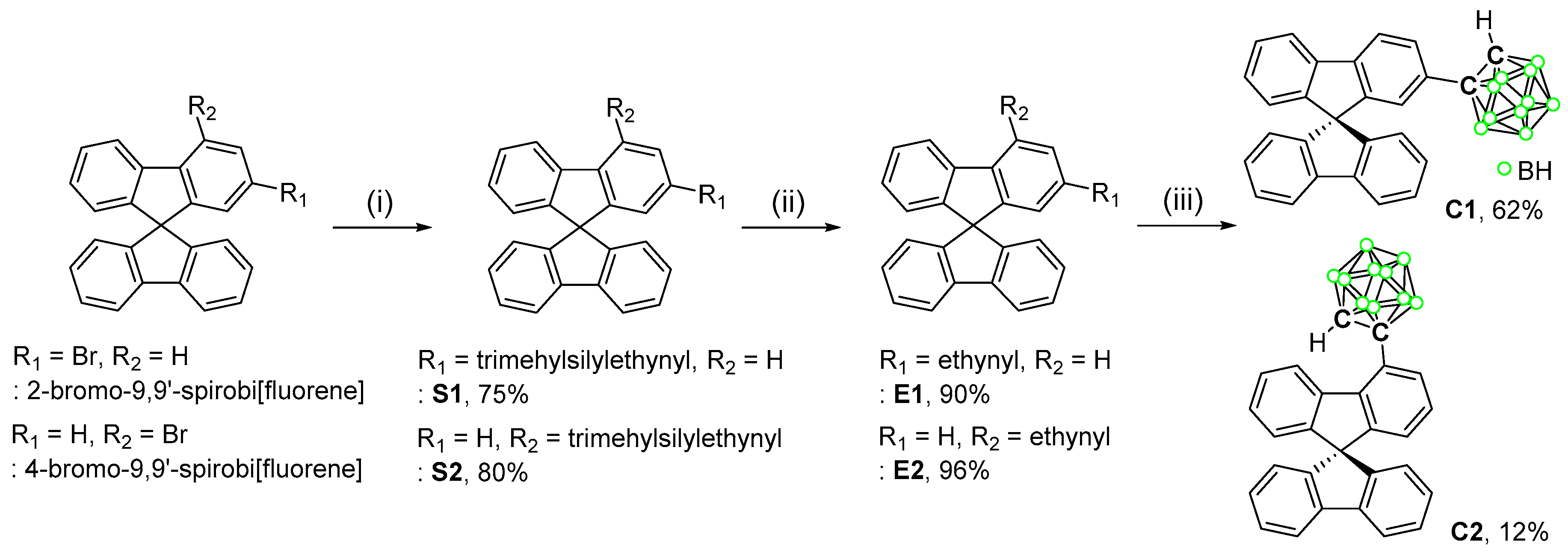
3.1. Synthesis and Characterization
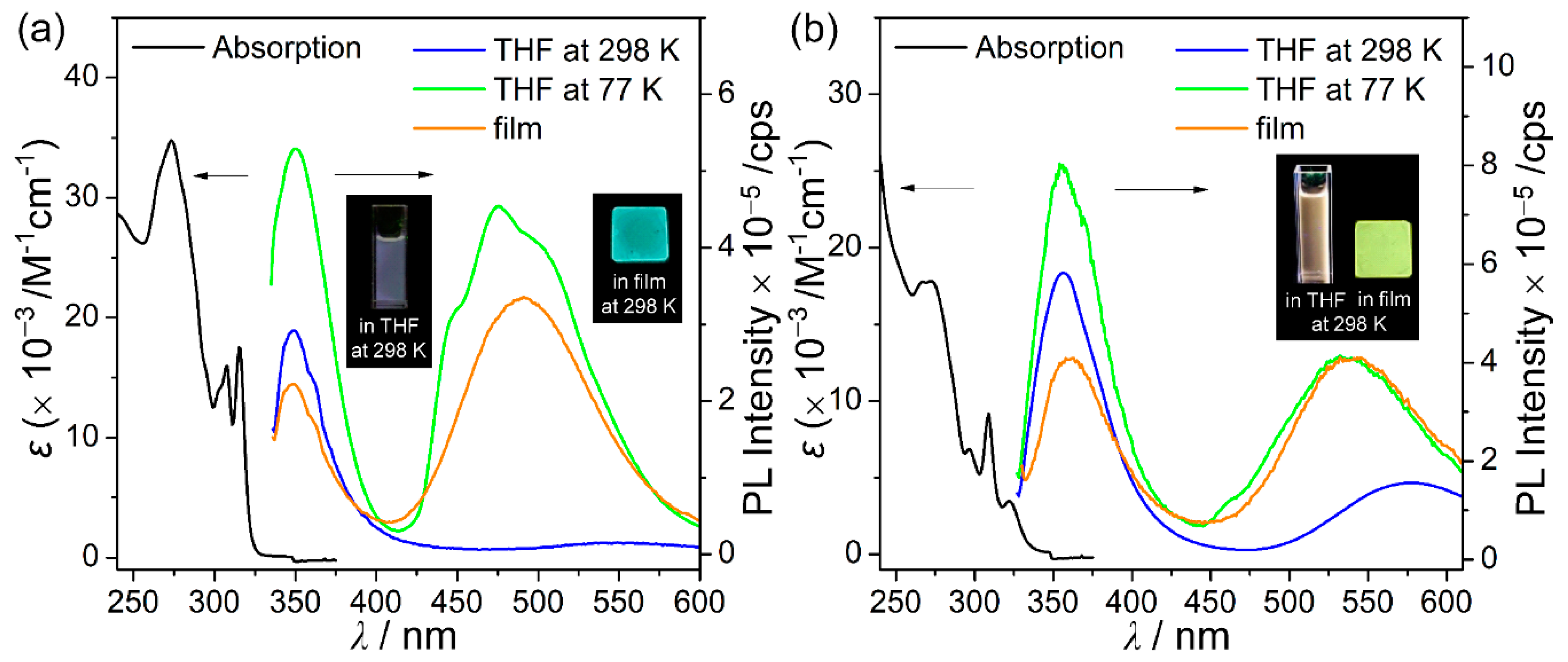
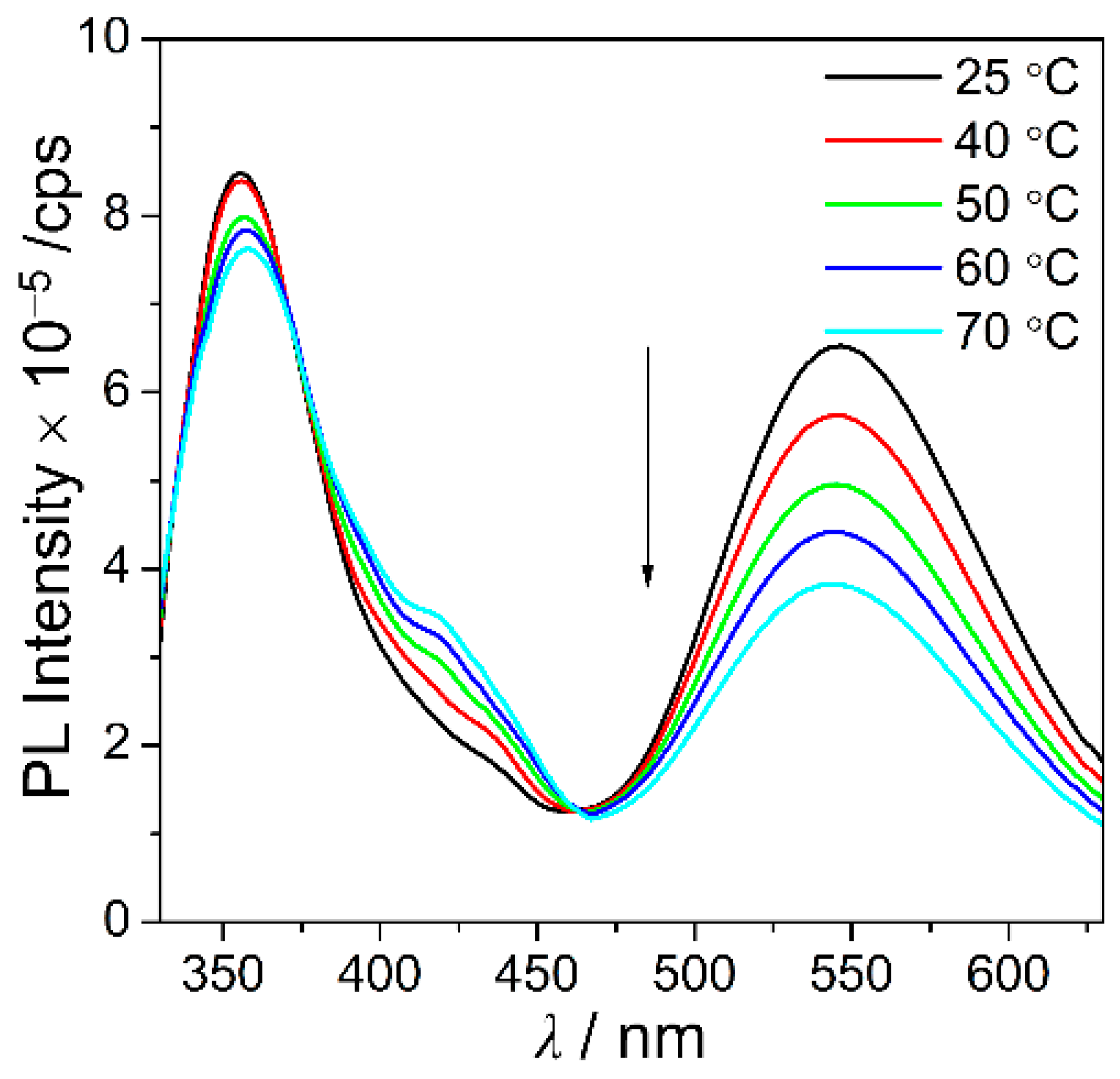
3.2. Photophysical Properties
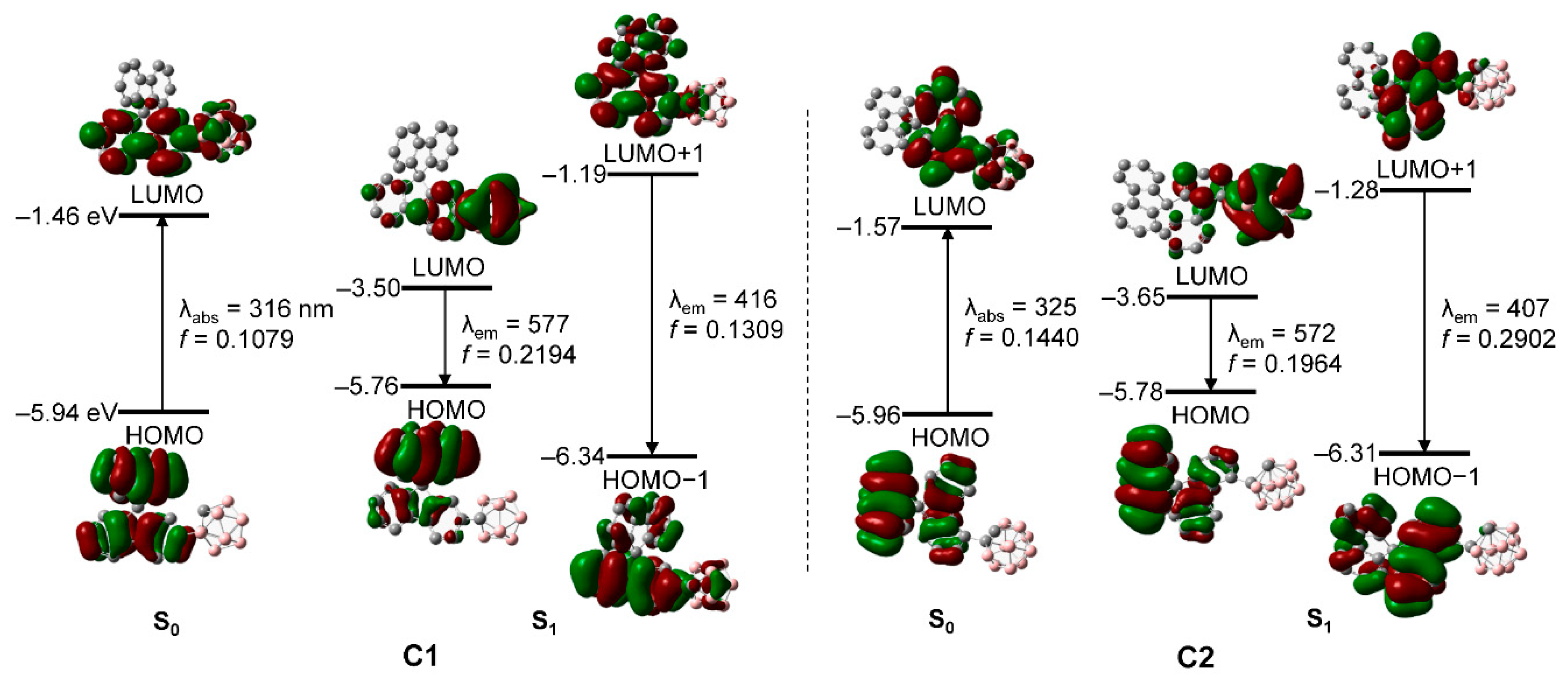
3.3. Computational Chemistry and Orbital Analyses
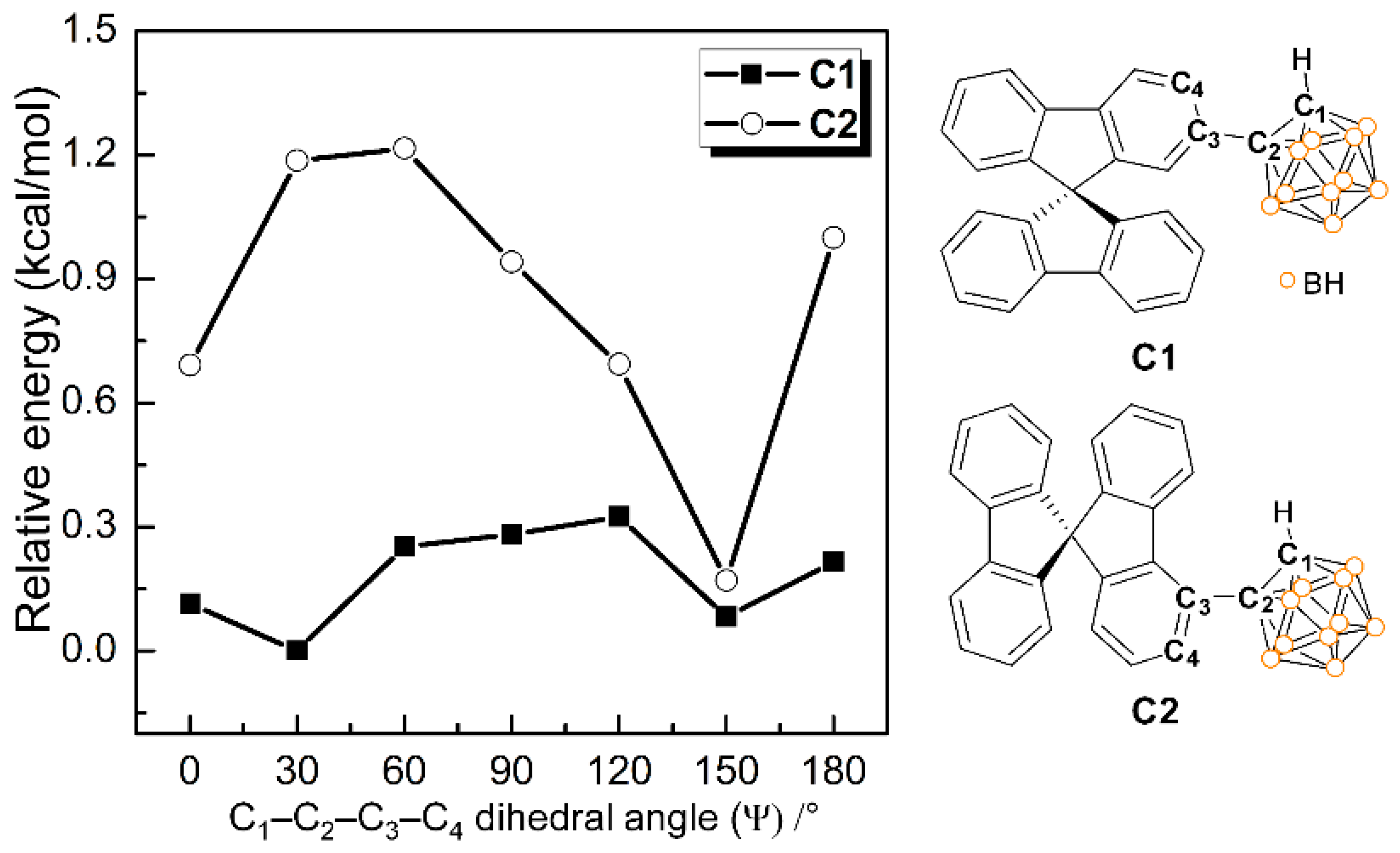
3.4. DFT Energy-Barrier Calculations for Rotational Motion of the o-Carboranyl Cage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- S Bregadze, V.I. Dicarba-closo-dodecaboranes C2B10H12 and their derivatives. Chem. Rev. 1992, 92, 209–223. [Google Scholar] [CrossRef]
- González-Campo, A.; Juárez-Pérez, E.J.; Viñas, C.; Boury, B.; Sillanpää, R.; Kivekäs, R.; Núñez, R. Carboranyl Substituted Siloxanes and Octasilsesquioxanes: Synthesis, Characterization, and Reactivity. Macromolecules 2008, 41, 8458–8466. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in Drug Discovery: Carboranes as Unique Pharmacophores in Biologically Active Compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Wee, K.-R.; Cho, Y.-J.; Jeong, S.; Kwon, S.; Lee, J.-D.; Suh, I.-H.; Kang, S.O. Carborane-Based Optoelectronically Active Organic Molecules: Wide Band Gap Host Materials for Blue Phosphorescence. J. Am. Chem. Soc. 2012, 134, 17982–17990. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Ugalde, A.; Juárez-Pérez, E.J.; Teixidor, F.; Viñas, C.; Núñez, R. Synthesis, Characterization, and Thermal Behavior of Carboranyl–Styrene Decorated Octasilsesquioxanes: Influence of the Carborane Clusters on Photoluminescence. Chem. Eur. J. 2013, 19, 17021–17030. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Kim, H.; Lee, K.M.; Lee, Y.S.; Lee, M.H. Phosphorescence Color Tuning of Cyclometalated Iridium Complexes by o-Carborane Substitution. Inorg. Chem. 2013, 52, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.J.; Chung, J.; Kim, H.; Park, J.; Lee, K.M.; Koh, T.-W.; Lee, Y.S.; Yoo, S.; Do, Y.; Lee, M.H. Deep Red Phosphorescence of Cyclometalated Iridium Complexes by o-Carborane Substitution. Inorg. Chem. 2014, 53, 128–138. [Google Scholar] [CrossRef]
- Asay, M.J.; Fisher, S.P.; Lee, S.E.; Tham, F.S.; Borchardt, D.; Lavallo, V. Synthesis of unsymmetrical N-carboranyl NHCs: Directing effect of the carborane anion. Chem. Commun. 2015, 51, 5359–5362. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.; Jo, S.-J.; Kim, M.; Lee, J.; Lee, S.U.; Lee, M.H. Manipulation of Phosphorescence Efficiency of Cyclometalated Iridium Complexes by Substituted o-Carboranes. Chem. Eur. J. 2015, 21, 2052–2061. [Google Scholar] [CrossRef]
- Núñez, R.; Tarrés, M.; Ferrer-Ugalde, A.; Fabrizi de Biani, F.; Teixidor, F. Electrochemistry and Photoluminescence of Icosahedral Carboranes, Boranes, Metallacarboranes, and Their Derivatives. Chem. Rev. 2016, 116, 14307–14378. [Google Scholar] [CrossRef]
- Mukherjee, S.; Thilagar, P. Boron clusters in luminescent materials. Chem. Commun. 2016, 52, 1070–1093. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, R.M.; Saleh, L.M.A.; Axtell, J.C.; Martin, J.L.; Stevens, S.L.; Royappa, A.T.; Rheingold, A.L.; Spokoyny, A.M. B–N, B–O, and B–CN Bond Formation via Palladium-Catalyzed Cross-Coupling of B-Bromo-Carboranes. J. Am. Chem. Soc. 2016, 138, 9081–9084. [Google Scholar] [CrossRef] [PubMed]
- Kirlikovali, K.O.; Axtell, J.C.; Gonzalez, A.; Phung, A.C.; Khan, S.I.; Spokoyny, A.M. Luminescent metal complexes featuring photophysically innocent boron cluster ligands. Chem. Sci. 2016, 7, 5132–5138. [Google Scholar] [CrossRef]
- Saleh, L.M.A.; Dziedzic, R.M.; Khan, S.I.; Spokoyny, A.M. Forging Unsupported Metal–Boryl Bonds with Icosahedral Carboranes. Chem. Eur. J. 2016, 22, 8466–8470. [Google Scholar] [CrossRef] [PubMed]
- Eleazer, B.J.; Smith, M.D.; Popov, A.A.; Peryshkov, D.V. (BB)-Carboryne Complex of Ruthenium: Synthesis by Double B–H Activation at a Single Metal Center. J. Am. Chem. Soc. 2016, 138, 10531–10538. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.O.; Smith, M.D.; Peryshkov, D.V. Synthesis of the First Example of the 12-Vertex-closo/12-Vertex-nido Biscarborane Cluster by a Metal-Free B−H Activation at a Phosphorus(III) Center. Chem. Eur. J. 2016, 22, 6764–6767. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.L.; Estrada, J.; Kefalidis, C.E.; Lavallo, V. Changing the Charge: Electrostatic Effects in Pd-Catalyzed Cross-Coupling. Organometallics. 2016, 35, 3257–3260. [Google Scholar] [CrossRef]
- Fisher, S.P.; El-Hellani, A.; Tham, F.S.; Lavallo, V. Anionic and zwitterionic carboranyl N-heterocyclic carbene Au(I) complexes. Dalton Trans. 2016, 45, 9762–9765. [Google Scholar] [CrossRef]
- Kim, Y.; Park, S.; Lee, Y.H.; Jung, J.; Yoo, S.; Lee, M.H. Homoleptic Tris-Cyclometalated Iridium Complexes with Substituted o-Carboranes: Green Phosphorescent Emitters for Highly Efficient Solution-Processed Organic Light-Emitting Diodes. Inorg. Chem. 2016, 55, 909–917. [Google Scholar] [CrossRef]
- Tu, D.; Leong, P.; Guo, S.; Yan, H.; Lu, C.; Zhao, Q. Highly Emissive Organic Single-Molecule White Emitters by Engineering o-Carborane-Based Luminophores. Angew. Chem. Int. Ed. 2017, 56, 11370–11374. [Google Scholar] [CrossRef]
- Kirlikovali, K.O.; Axtell, J.C.; Anderson, K.; Djurovich, P.I.; Rheingold, A.L.; Spokoyny, A.M. Fine-Tuning Electronic Properties of Luminescent Pt(II) Complexes via Vertex-Differentiated Coordination of Sterically Invariant Carborane-Based Ligands. Organometallics 2018, 37, 3122–3131. [Google Scholar] [CrossRef]
- Nar, I.; Atsay, A.; Altındal, A.; Hamuryudan, E. o-Carborane, Ferrocene, and Phthalocyanine Triad for High-Mobility Organic Field-Effect Transistors. Inorg. Chem. 2018, 57, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.N. Carboranes, 2nd ed.; Academic Press: London, UK, 2011. [Google Scholar]
- Spokoyny, A.M. New ligand platforms featuring boron-rich clusters as organomimetic substituents. Pure Appl. Chem. 2013, 85, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. π Aromaticity and Three-Dimensional Aromaticity: Two sides of the Same Coin? Angew. Chem. Int. Ed. 2014, 53, 12191–12195. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. Hückel’s Rule of Aromaticity Categorizes Aromatic closo Boron Hydride Clusters. Chem. Eur. J. 2016, 22, 7437–7443. [Google Scholar] [CrossRef]
- Núñez, R.; Romero, I.; Teixidor, F.; Viñas, C. Icosahedral boron clusters: A perfect tool for the enhancement of polymer features. Chem. Soc. Rev. 2016, 45, 5147–5173. [Google Scholar] [CrossRef]
- Cabrera-González, J.; Sánchez-Arderiu, V.; Viñas, C.; Parella, T.; Teixidor, F.; Náñez, R. Redox-Active Metallacarborane-Decorated Octasilsesquioxanes. Electrochemical and Thermal Properties. Inorg. Chem. 2016, 55, 11630–11634. [Google Scholar] [CrossRef]
- Kokado, K.; Chujo, Y. Multicolor Tuning of Aggregation-Induced Emission through Substituent Variation of Diphenyl-o-carborane. J. Org. Chem. 2011, 76, 316–319. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Gaillard, E.R.; Norton, K.M.; Maguire, J.A.; Chug, N.; Hosmane, N.S. Enhanced π-Conjugation and Emission via Icosahedral Carboranes: Synthetic and Spectroscopic Investigation. Inorg. Chem. 2011, 50, 5485–5493. [Google Scholar] [CrossRef]
- Wee, K.-R.; Han, W.-S.; Cho, D.W.; Kwon, S.; Pac, C.; Kang, S.O. Carborane photochemistry triggered by aryl substitution: Carborane-based dyads with phenyl carbazoles. Angew. Chem. Int. Ed. 2012, 51, 2677–2680. [Google Scholar] [CrossRef]
- Weber, L.; Kahlert, J.; Brockhinke, R.; Böhling, L.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Harder, R.A.; Fox, M.A. Luminescence Properties of C-Diazaborolyl-ortho-Carboranes as Donor–Acceptor Systems. Chem. Eur. J. 2012, 18, 8347–8357. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.J.; Kim, H.; Lee, K.M.; Kim, T.; Eo, M.; Lee, Y.S.; Do, Y.; Lee, M.H. Heteroleptic tris-cyclometalated iridium(III) complexes supported by an o-carboranyl-pyridine ligand. Dalton Trans. 2013, 42, 8549–8552. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.; Kahlert, J.; Brockhinke, R.; Böhling, L.; Halama, J.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Nervi, C.; Harder, R.A.; et al. C,C′-Bis(benzodiazaborolyl)dicarba-closo-dodecaboranes: Synthesis, structures, photophysics and electrochemistry. Dalton Trans. 2013, 42, 10982–10996. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.; Kahlert, J.; Böhling, L.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Harder, R.A.; Low, P.J.; Fox, M.A. Electrochemical and spectroelectrochemical studies of C-benzodiazaborolyl-ortho-carboranes. Dalton Trans. 2013, 42, 2266–2281. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Wee, K.-R.; Cho, Y.-J.; Kang, S.O. Carborane Dyads for Photoinduced Electron Transfer: Photophysical Studies on Carbazole and Phenyl-o-carborane Molecular Assemblies. Chem. Eur. J. 2014, 20, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Ugalde, A.; González-Campo, A.; Viñas, C.; Rodríguez-Romero, J.; Santillan, R.; Farfán, N.; Sillanpää, R.; Sousa-Pedrares, A.; Núñez, R.; Teixidor, F. Fluorescence of New o-Carborane Compounds with Different Fluorophores: Can it be Tuned? Chem. Eur. J. 2014, 20, 9940–9951. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.J.; Kim, H.; Lee, K.M.; Kim, T.; Lee, Y.S.; Do, Y.; Lee, M.H. Through-space charge transfer and emission color tuning of di-o-carborane substituted benzene. Dalton Trans. 2014, 43, 4978–4985. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.; Lee, J.; Lee, S.U.; Lee, M.H. Iridium Cyclometalates with Tethered o-Carboranes: Impact of Restricted Rotation of o-Carborane on Phosphorescence Efficiency. J. Am. Chem. Soc. 2015, 137, 8018–8021. [Google Scholar] [CrossRef]
- Naito, H.; Morisaki, Y.; Chujo, Y. o-Carborane-Based Anthracene: A Variety of Emission Behaviors. Angew. Chem. Int. Ed. 2015, 54, 5084–5087. [Google Scholar] [CrossRef]
- Kim, T.; Lee, J.; Lee, S.U.; Lee, M.H. o-Carboranyl–Phosphine as a New Class of Strong-Field Ancillary Ligand in Cyclometalated Iridium (III) Complexes: Toward Blue Phosphorescence. Organometallics 2015, 34, 3455–3458. [Google Scholar] [CrossRef]
- Choi, B.H.; Lee, J.H.; Hwang, H.; Lee, K.M.; Park, M.H. Novel Dimeric o-Carboranyl Triarylborane: Intriguing Ratiometric Color-Tunable Sensor via Aggregation-Induced Emission by Fluoride Anions. Organometallics 2016, 35, 1771–1777. [Google Scholar] [CrossRef]
- Wee, K.-R.; Cho, Y.-J.; Song, J.K.; Kang, S.O. Multiple photoluminescence from 1,2-dinaphthyl-ortho-carborane. Angew. Chem. Int. Ed. 2013, 52, 9682–9685. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Solid-State Emission of the Anthracene-o-Carborane Dyad from the Twisted-Intramolecular Charge Transfer in the Crystalline State. Angew. Chem. Int. Ed. 2017, 56, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Guo, J.; Cao, Y.; Zhao, J.; Jia, W.; Chen, Y.; Jia, D. Mechanically triggered reversible stepwise tricolor switching and thermochromism of anthracene-o-carborane dyad. Chem. Sci. 2018, 9, 5270–5277. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, C.; Peng, X.; Chen, Y.; Qi, Q.; Luo, X.; Lai, W.-Y.; Huang, W. Stimuli-responsive solid-state emission from o-carborane–tetraphenylethene dyads induced by twisted intramolecular charge transfer in the crystalline state. J. Mater. Chem. C 2018, 6, 19–28. [Google Scholar] [CrossRef]
- Nishino, K.; Yamamoto, H.; Tanaka, K.; Chujo, Y. Development of Solid-State Emissive Materials Based on Multifunctional o-Carborane–Pyrene Dyads. Org. Lett. 2016, 18, 4064–4067. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Zhao, J.; Che, Y.; Jia, D.; Chen, Y. Multifunctional luminescent molecules of o-carborane-pyrene dyad/triad: Flexible synthesis and study of the photophysical properties. Dyes Pigm. 2018, 154, 44–51. [Google Scholar] [CrossRef]
- Marsh, A.V.; Cheetham, N.J.; Little, M.; Dyson, M.; White, A.J.P.; Beavis, P.; Warriner, C.N.; Swain, A.C.; Stavrinou, P.N.; Heeney, M. Carborane-Induced Excimer Emission of Severely Twisted Bis-o-Carboranyl Chrysene. Angew. Chem. Int. Ed. 2018, 57, 10640–10645. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Cho, Y.-J.; Jin, G.F.; Han, W.-S.; Son, H.-J.; Cho, D.W.; Kang, S.O. Intriguing emission properties of triphenylamine–carborane systems. Phys. Chem. Chem. Phys. 2015, 17, 15679–15682. [Google Scholar] [CrossRef]
- Wan, Y.; Li, J.; Peng, X.; Huang, C.; Qi, Q.; Lai, W.-Y.; Huang, W. Intramolecular charge transfer induced emission from triphenylamine-o-carborane dyads. RSC Adv. 2017, 7, 35543–35548. [Google Scholar] [CrossRef]
- Nishino, K.; Uemura, K.; Gon, M.; Tanaka, K.; Chujo, Y. Enhancement of Aggregation-Induced Emission by Introducing Multiple o-Carborane Substitutions into Triphenylamine. Molecules 2017, 22, 2009. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Uemura, K.; Tanaka, K.; Morisaki, Y.; Chujo, Y. Modulation of the cis- and trans-Conformations in Bis-ocarborane Substituted Benzodithiophenes and Emission Enhancement Effect on Luminescent Efficiency by Solidification. Eur. J. Org. Chem. 2018, 12, 1507–1512. [Google Scholar] [CrossRef]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Luminescence Color Tuning from Blue to Near Infrared of Stable Luminescent Solid Materials Based on Bis-o-Carborane-Substituted Oligoacenes. Chem. J. 2017, 12, 2134–2138. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Highly-efficient solid-state emissions of anthracene–o-carborane dyads with various substituents and their thermochromic luminescence properties. J. Mater. Chem. C 2017, 5, 10047–10054. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Quan, Y.; Jia, W.; Jia, D.; Chen, Y.; Xie, Z. Cage carbon-substitute does matter for aggregation-induced emission features of o-carborane-functionalized anthracene triads. J. Mater. Chem. C 2018, 6, 4140–4149. [Google Scholar] [CrossRef]
- Mori, H.; Nishino, K.; Wada, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Modulation of luminescence chromic behaviors and environment-responsive intensity changes by substituents in bis-o-carborane-substituted conjugated molecules. Mater. Chem. Front. 2018, 2, 573–579. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, J.; Wu, X.; Jia, D.; Tong, F. Color-tuning aggregation-induced emission of o-Carborane-bis(1,3,5-triaryl-2-pyrazoline) triads: Preparation and investigation of the photophysics. Dyes Pigm. 2018, 148, 180–188. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Lee, J.-D.; Cho, Y.-J.; Son, M.R.; Son, H.-J.; Cho, D.W.; Kang, S.O. Excitation spectroscopic and synchronous fluorescence spectroscopic analysis of the origin of aggregation-induced emission in N,N-diphenyl-1-naphthylamine-o-carborane derivatives. Phys. Chem. Chem. Phys. 2018, 20, 17458–17463. [Google Scholar] [CrossRef]
- Shin, N.; Yu, S.; Lee, J.H.; Hwang, H.; Lee, K.M. Biphenyl- and Fluorene-Based o-Carboranyl Compounds: Alteration of Photophysical Properties by Distortion of Biphenyl Rings. Organometallics 2017, 36, 1522–1529. [Google Scholar] [CrossRef]
- Jin, H.; Bae, H.J.; Kim, S.; Lee, J.H.; Hwang, H.; Park, M.H.; Lee, K.M. 2-Phenylpyridine- and 2-(benzo[b]thiophen-2-yl)pyridine-based o-carboranyl compounds: Impact of the structural formation of aromatic rings on photophysical properties. Dalton Trans. 2019, 48, 1467–1476. [Google Scholar] [CrossRef]
- Jin, H.; Kim, S.; Bae, H.J.; Lee, J.H.; Hwang, H.; Park, M.H.; Lee, K.M. Effect of Planarity of Aromatic Rings Appended to o-Carborane on Photophysical Properties: A Series of o-Carboranyl Compounds Based on 2-Phenylpyridine- and 2-(Benzo[b]thiophen-2-yl)pyridine. Molecules 2019, 24, 201. [Google Scholar] [CrossRef] [PubMed]
- Saragi, T.P.I.; Spehr, T.; Siebert, A.; Fuhrmann-Lieker, T.; Salbeck, J. Spiro Compounds for Organic Optoelectronics. Chem. Rev. 2007, 107, 1011–1065. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lin, Y.T.; Chiang, H.H.; Cho, T.Y.; Chen, C.W.; Wong, K.T.; Liao, Y.L.; Lee, G.H.; Peng, S.M. Highly bright blue organic light-emitting devices using spirobifluorene-cored conjugated compounds. Appl. Phys. Lett. 2002, 81, 577. [Google Scholar] [CrossRef]
- Jang, S.E.; Joo, C.W.; Yook, K.S.; Kim, J.-W.; Lee, C.-W.; Lee, J.Y. Thermally stable fluorescent blue organic light-emitting diodes using spirobifluorene based anthracene host materials with different substitution position. Synth. Met. 2010, 160, 1184–1188. [Google Scholar] [CrossRef]
- Li, Z.; Jiao, B.; Wu, Z.; Liu, P.; Ma, L.; Lei, X.; Wang, D.; Zhou, G.; Hu, H.; Hou, X. Fluorinated 9,9′-spirobifluorene derivatives as host materials for highly efficient blue organic light-emitting devices. J. Mater. Chem. C 2013, 1, 2183–2192. [Google Scholar] [CrossRef]
- Usluer, Ö. New spirobifluorene-based hole-transporting semiconductors for electroluminescent devices. J. Mater. Chem. C 2014, 2, 8098–8104. [Google Scholar] [CrossRef]
- Thiery, S.; Tondelier, D.; Geffroy, B.; Jacques, E.; Robin, M.; Métivier, R.; Jeannin, O.; Rault-Berthelot, J.; Poriel, C. Spirobifluorene-2,7-dicarbazole-4′-phosphine Oxide as Host for High-Performance Single-Layer Green Phosphorescent OLED Devices. Org. Lett. 2015, 17, 4682–4685. [Google Scholar] [CrossRef]
- Braveenth, R.; Bae, H.W.; Nguyen, Q.P.B.; Ko, H.M.; Lee, C.H.; Kim, H.J.; Kwon, J.H.; Chai, K.Y. Spirobifluorene Core-Based Novel Hole Transporting Materials for Red Phosphorescence OLEDs. Molecules 2017, 22, 464. [Google Scholar] [CrossRef]
- Heredia, D.; Natera, J.; Gervaldo, M.; Otero, L.; Fungo, F.; Lin, C.-Y.; Wong, K.-T. Spirobifluorene-Bridged Donor/Acceptor Dye for Organic Dye-Sensitized Solar Cells. Org. Lett. 2010, 12, 12–15. [Google Scholar] [CrossRef]
- Wang, M.; Li, C.; Lv, A.; Wang, Z.; Bo, Z. Spirobifluorene-Based Conjugated Polymers for Polymer Solar Cells with High Open-Circuit Voltage. Macromolecules 2012, 45, 3017–3022. [Google Scholar] [CrossRef]
- Song, Z.; Liu, J.; Wang, G.; Zuo, W.; Liao, C.; Mei, J. Understanding the Photovoltaic Performance of Perovskite–Spirobifluorene Solar Cells. Chem. Phys. Chem. 2017, 18, 3030–3038. [Google Scholar] [CrossRef] [PubMed]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision, B.01; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner., K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
Sample Availability: Samples of the compounds C1 and C2 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λabs1/nm (ε × 10−3 M−1 cm−1) | λex/nm | λem/nm | |||||
|---|---|---|---|---|---|---|---|---|
| Tol 2 | THF 2 | DCM 2 | 77 K 1 | film 3 | ||||
| C1 | 307 (15.7), 315 (17.5) | 310 | 350 | 349 | 349 | 350, 476 | 349, 490 | |
| C2 | 309 (9.2), 322 (3.5) | 322 | 356, 545 | 356, 577 | 356, 588 | 355, 545 | 359, 539 | |
| Compound | Φem4,5 | τ/ns 5 | kr6/× 108 s−1 | knr7/× 108 s−1 | ||||
| THF 2 | film 2 | THF 2 | film 2 | THF 2 | film 2 | THF 2 | film 2 | |
| C1 | - 8 | 0.02 | - 8 | 1.2 | - | 0.17 | - | 8.3 |
| C2 | 0.07 | 0.41 | 1.4 | 1.5 | 0.50 | 2.7 | 6.6 | 3.9 |
| State | λcalc/nm | fcalc | Assignment | |
|---|---|---|---|---|
| C1 | S0 | 316.26 | 0.1079 | HOMO → LUMO (97.7%) |
| S1 | 576.80 415.74 | 0.2194 0.1309 | HOMO → LUMO (99.8%) HOMO−1 → LUMO+1 (91.2%) | |
| C2 | S0 | 324.89 | 0.1440 | HOMO → LUMO (91.7%) |
| S1 | 571.55 407.40 | 0.1964 0.2902 | HOMO → LUMO (98.6%) HOMO−1 → LUMO+1 (93.1%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; So, H.; Lee, J.H.; Hwang, H.; Kwon, H.; Park, M.H.; Lee, K.M. Photophysical Properties of Spirobifluorene-Based o-Carboranyl Compounds Altered by Structurally Rotating the Carborane Cages. Molecules 2019, 24, 4135. https://doi.org/10.3390/molecules24224135
Kim S, So H, Lee JH, Hwang H, Kwon H, Park MH, Lee KM. Photophysical Properties of Spirobifluorene-Based o-Carboranyl Compounds Altered by Structurally Rotating the Carborane Cages. Molecules. 2019; 24(22):4135. https://doi.org/10.3390/molecules24224135
Chicago/Turabian StyleKim, Seonah, Hyunhee So, Ji Hye Lee, Hyonseok Hwang, Hyoshik Kwon, Myung Hwan Park, and Kang Mun Lee. 2019. "Photophysical Properties of Spirobifluorene-Based o-Carboranyl Compounds Altered by Structurally Rotating the Carborane Cages" Molecules 24, no. 22: 4135. https://doi.org/10.3390/molecules24224135
APA StyleKim, S., So, H., Lee, J. H., Hwang, H., Kwon, H., Park, M. H., & Lee, K. M. (2019). Photophysical Properties of Spirobifluorene-Based o-Carboranyl Compounds Altered by Structurally Rotating the Carborane Cages. Molecules, 24(22), 4135. https://doi.org/10.3390/molecules24224135