
Insertion of Carbenes into Deprotonated nido-Undecaborane, B11H13(2-)


Abstract
:1. Introduction
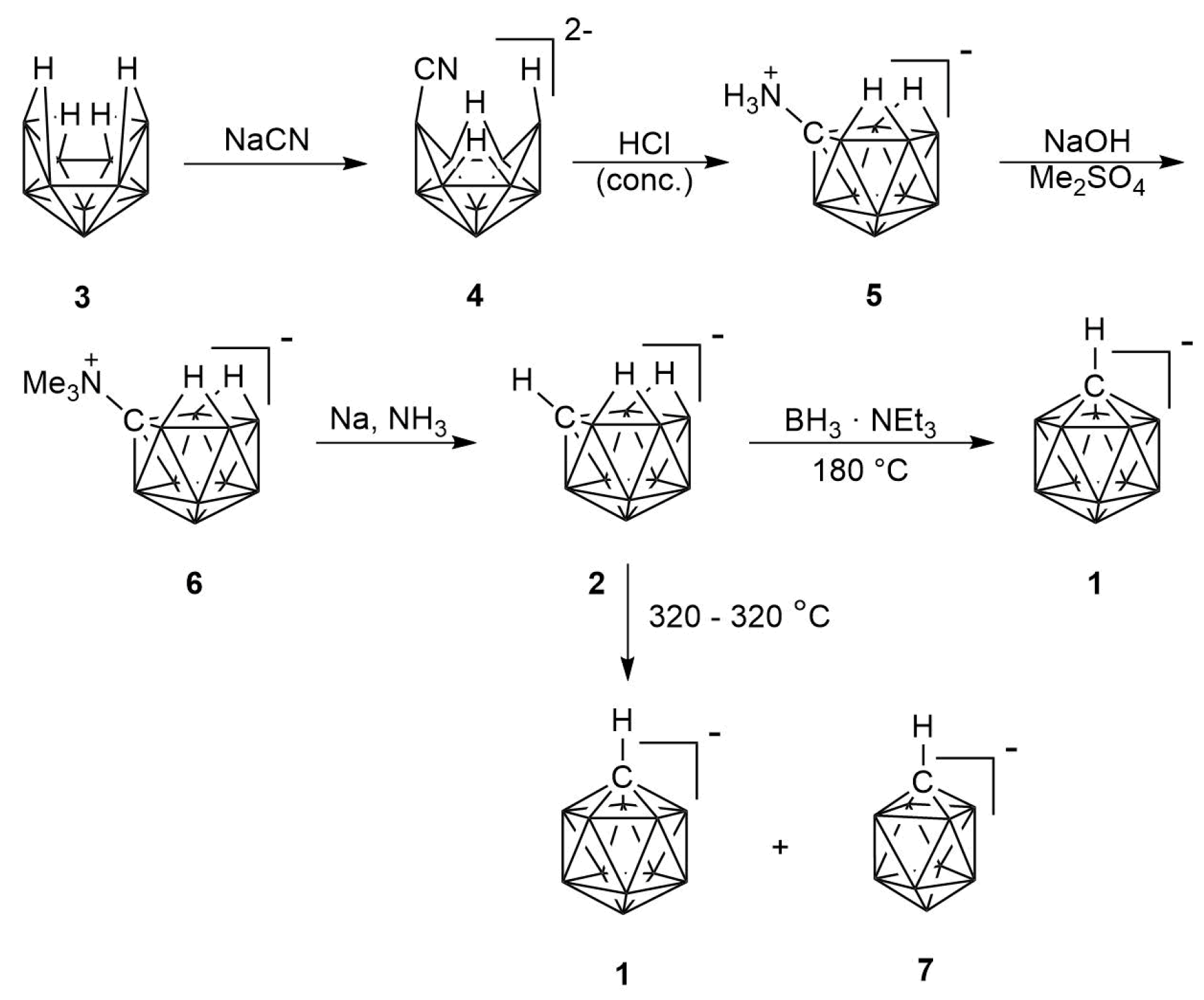
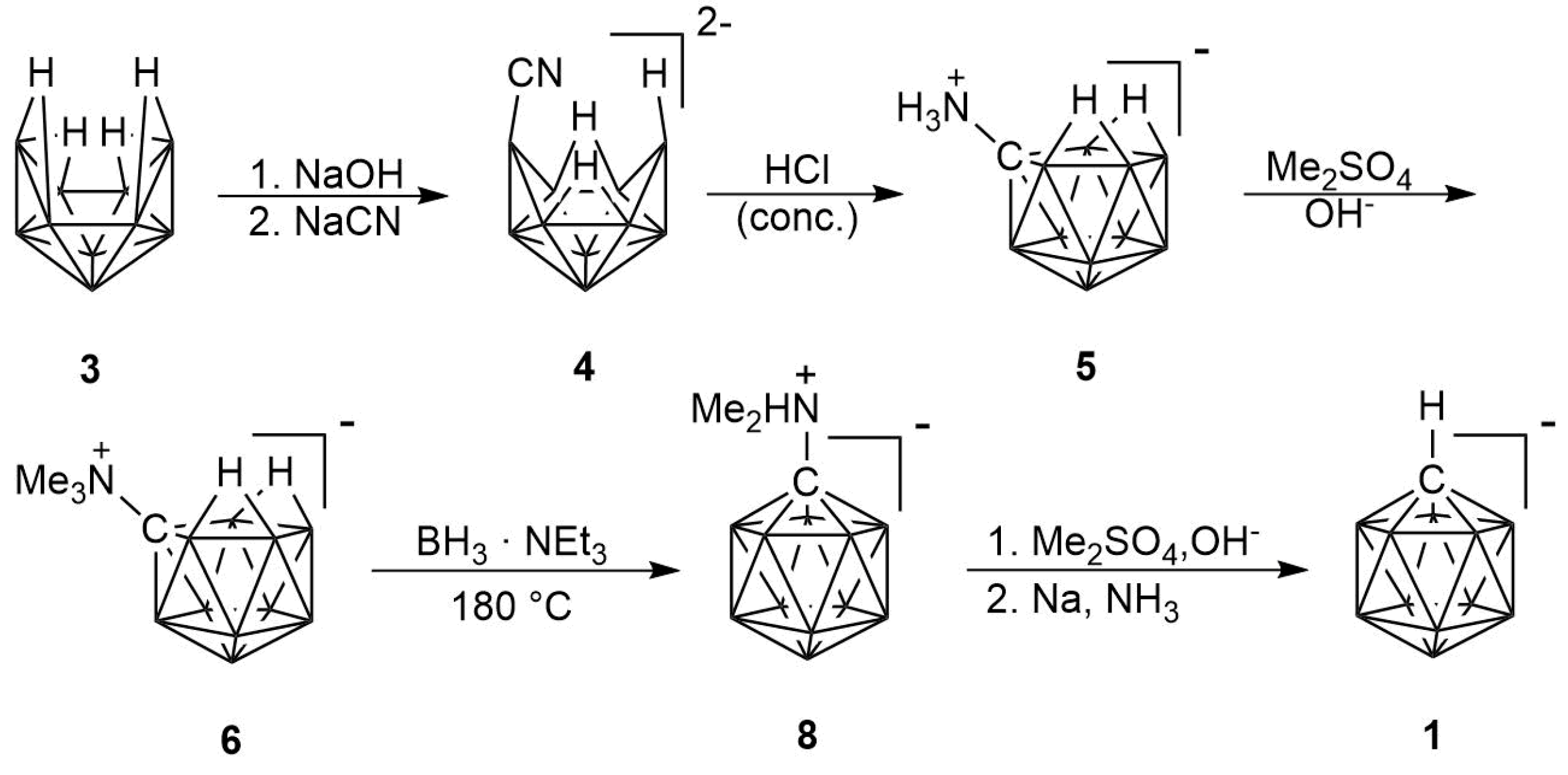
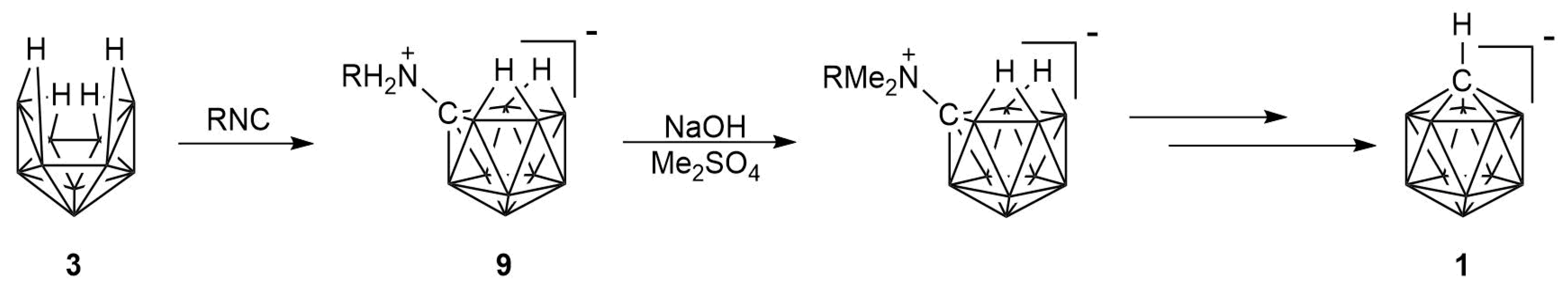
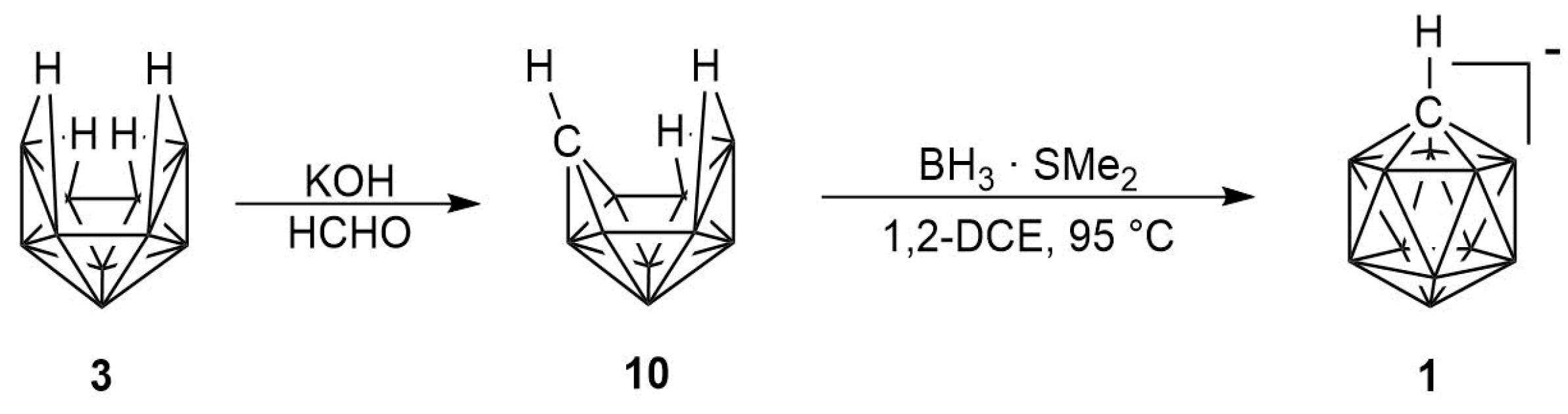
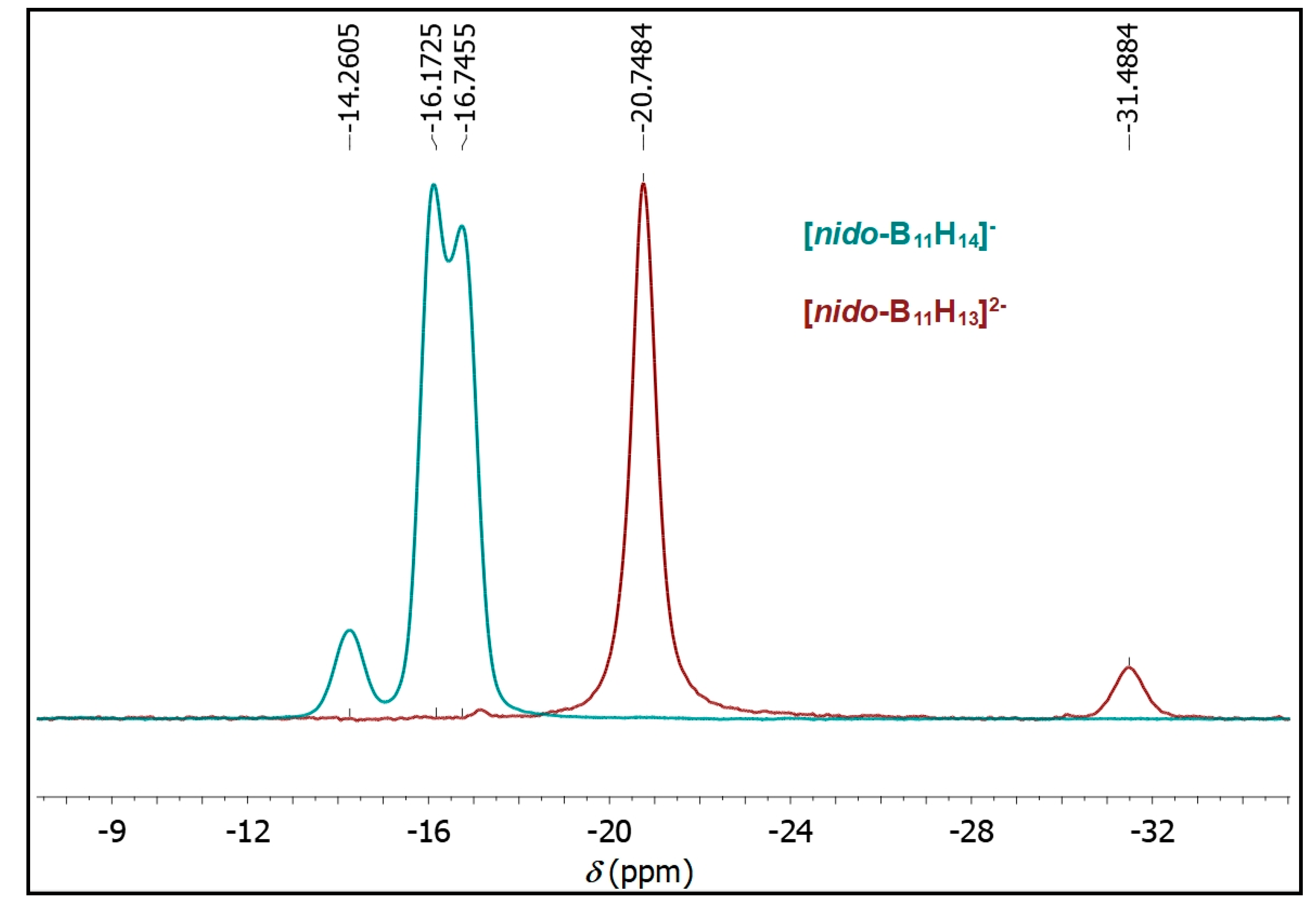
2. Results
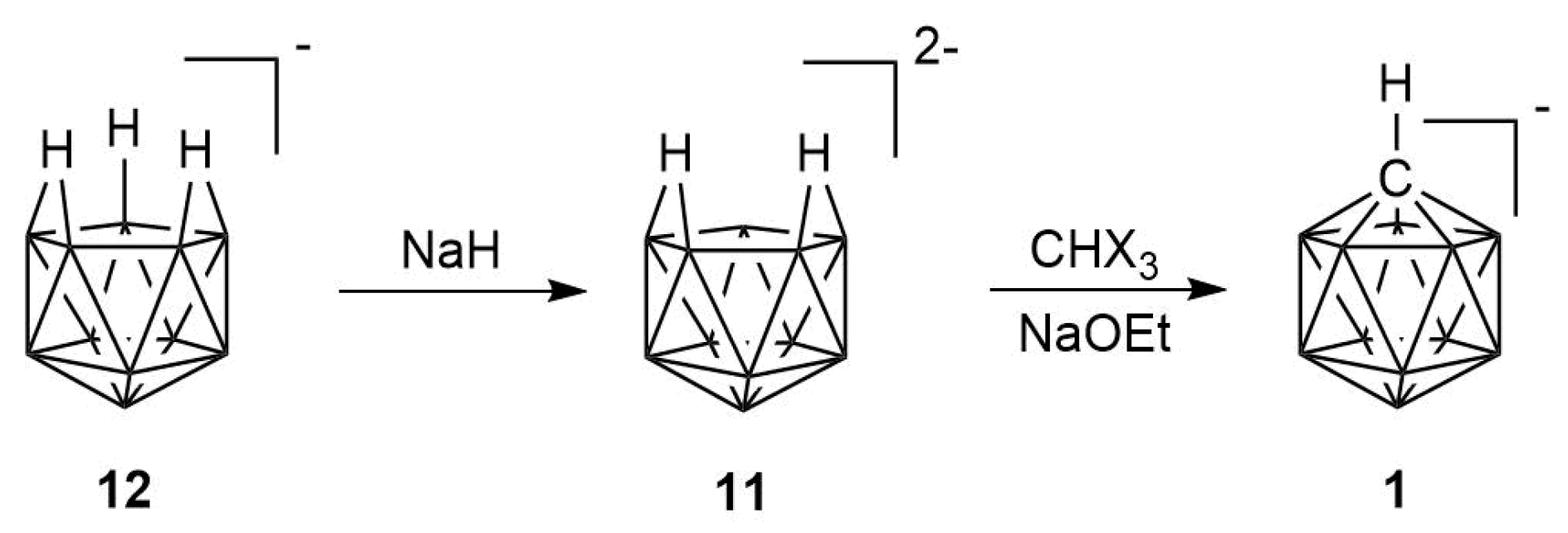
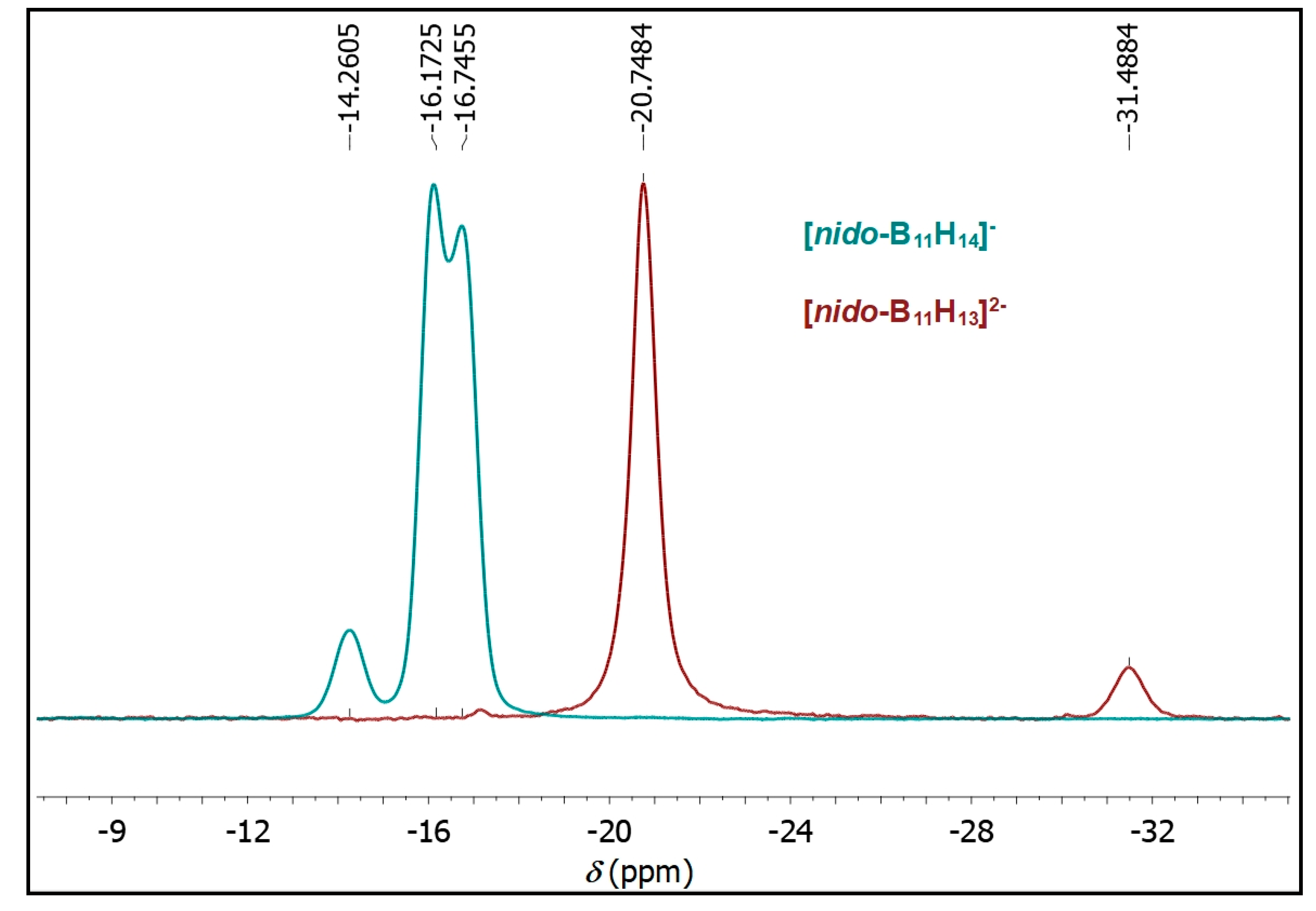
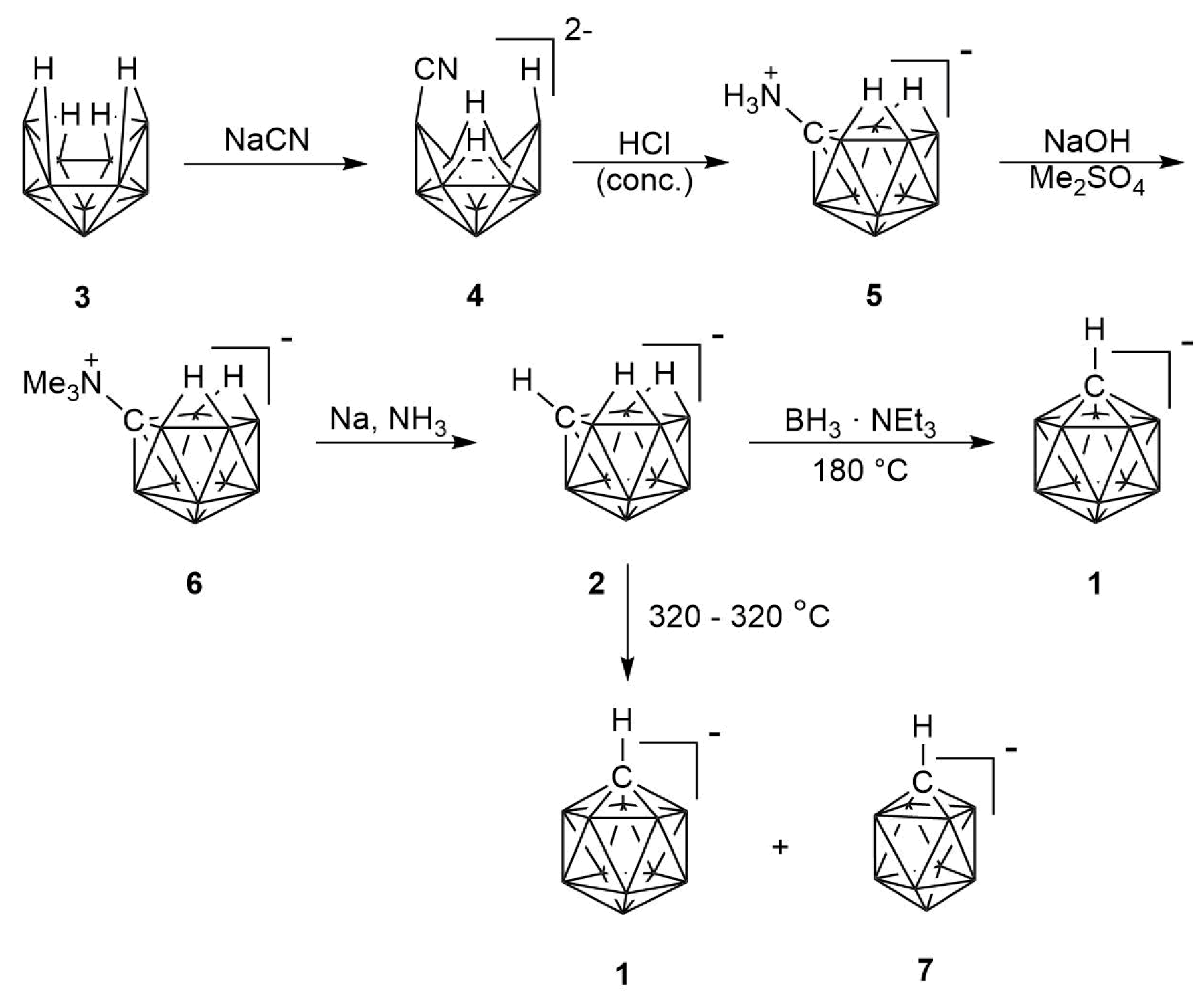
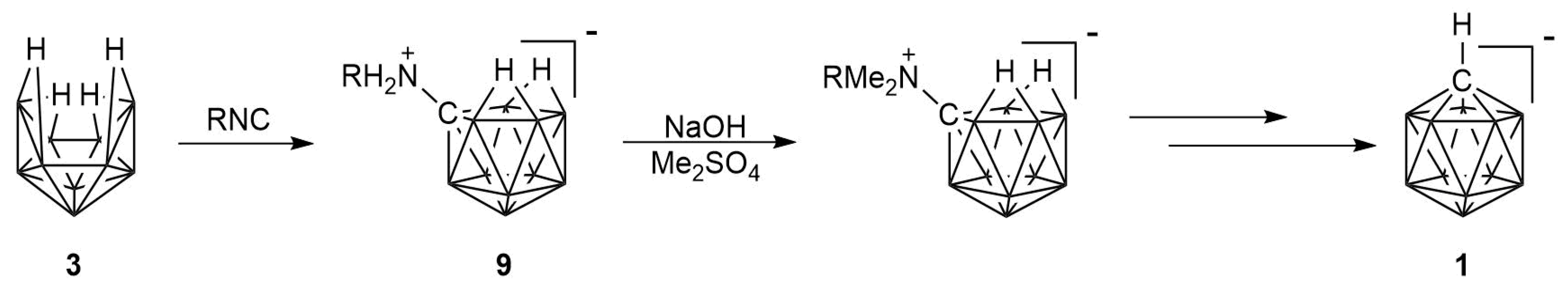
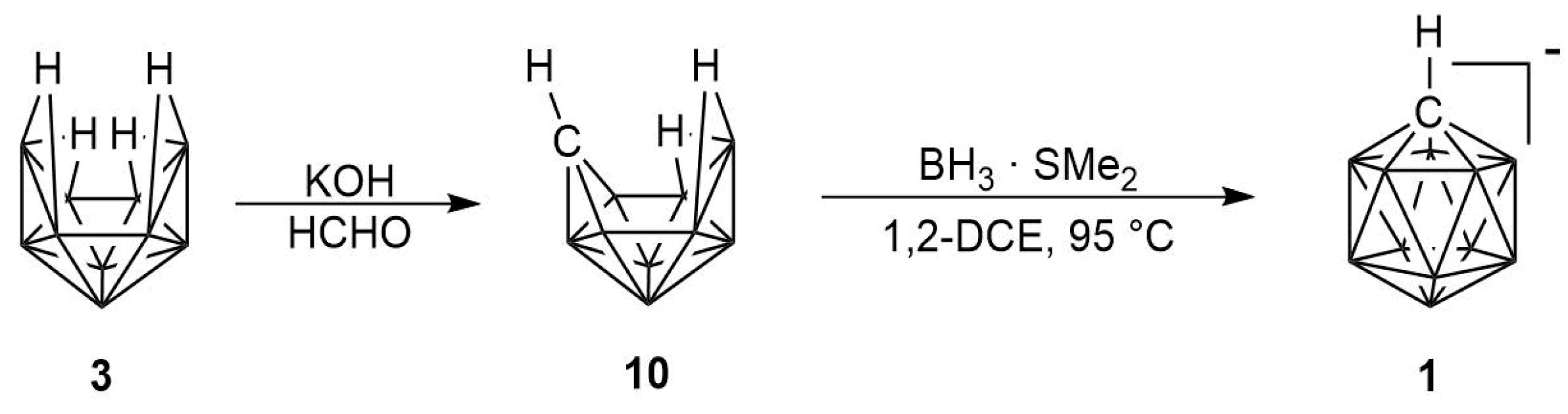
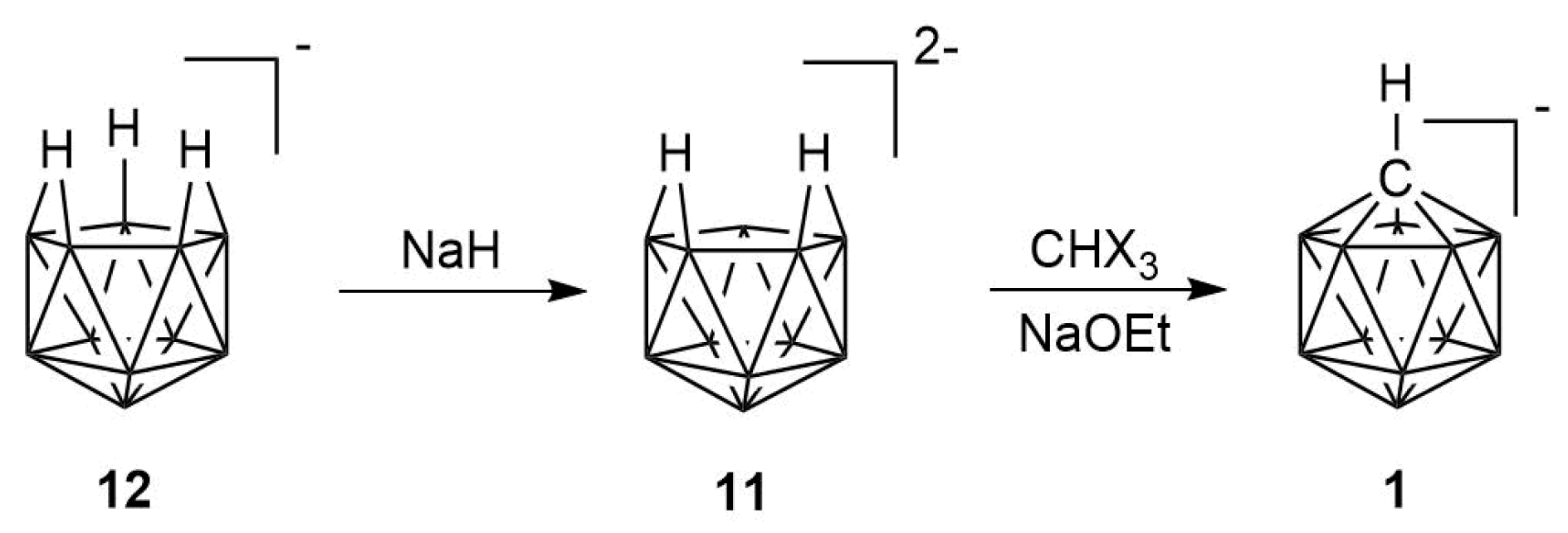
2.1. Synthesis
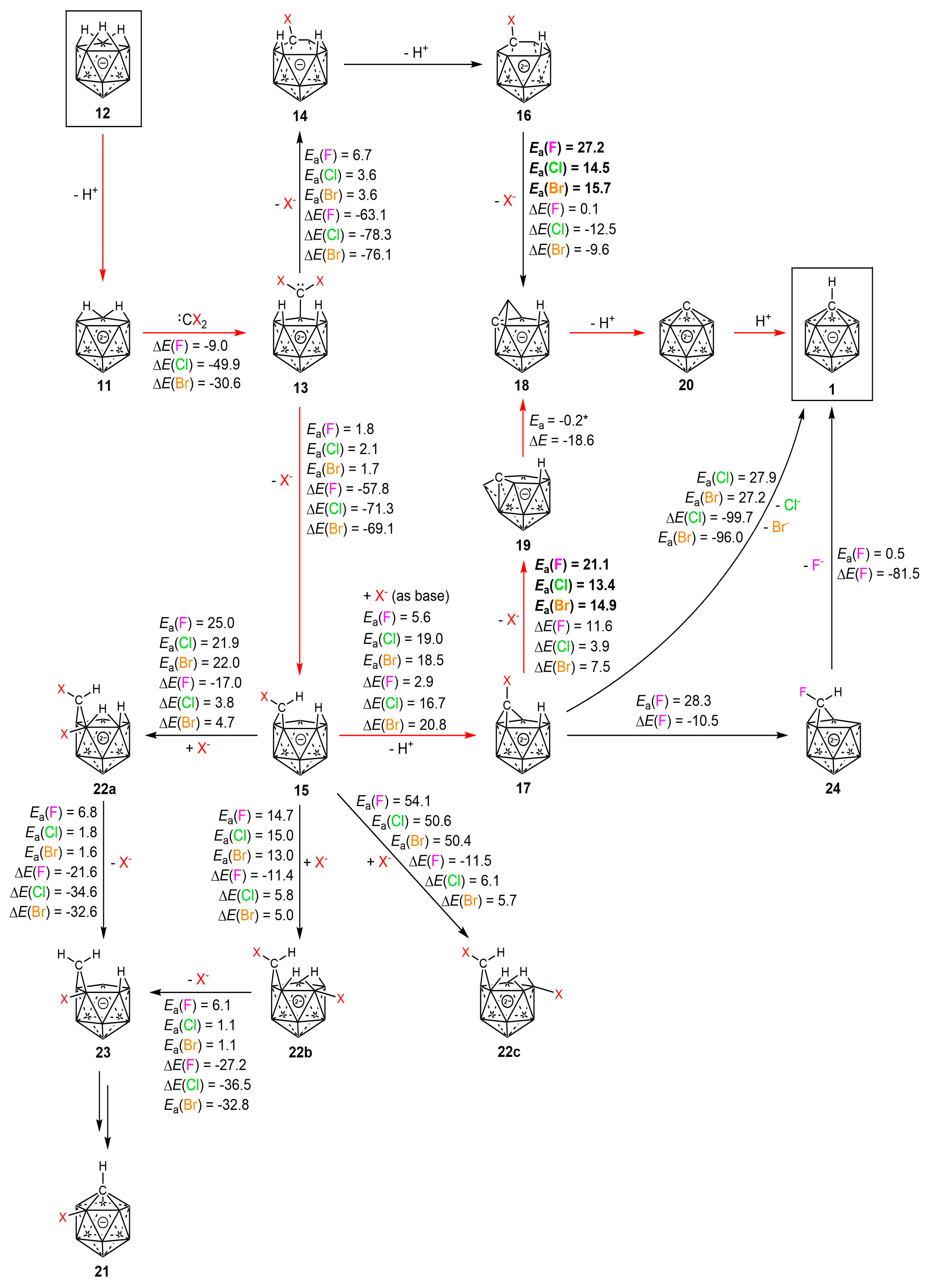
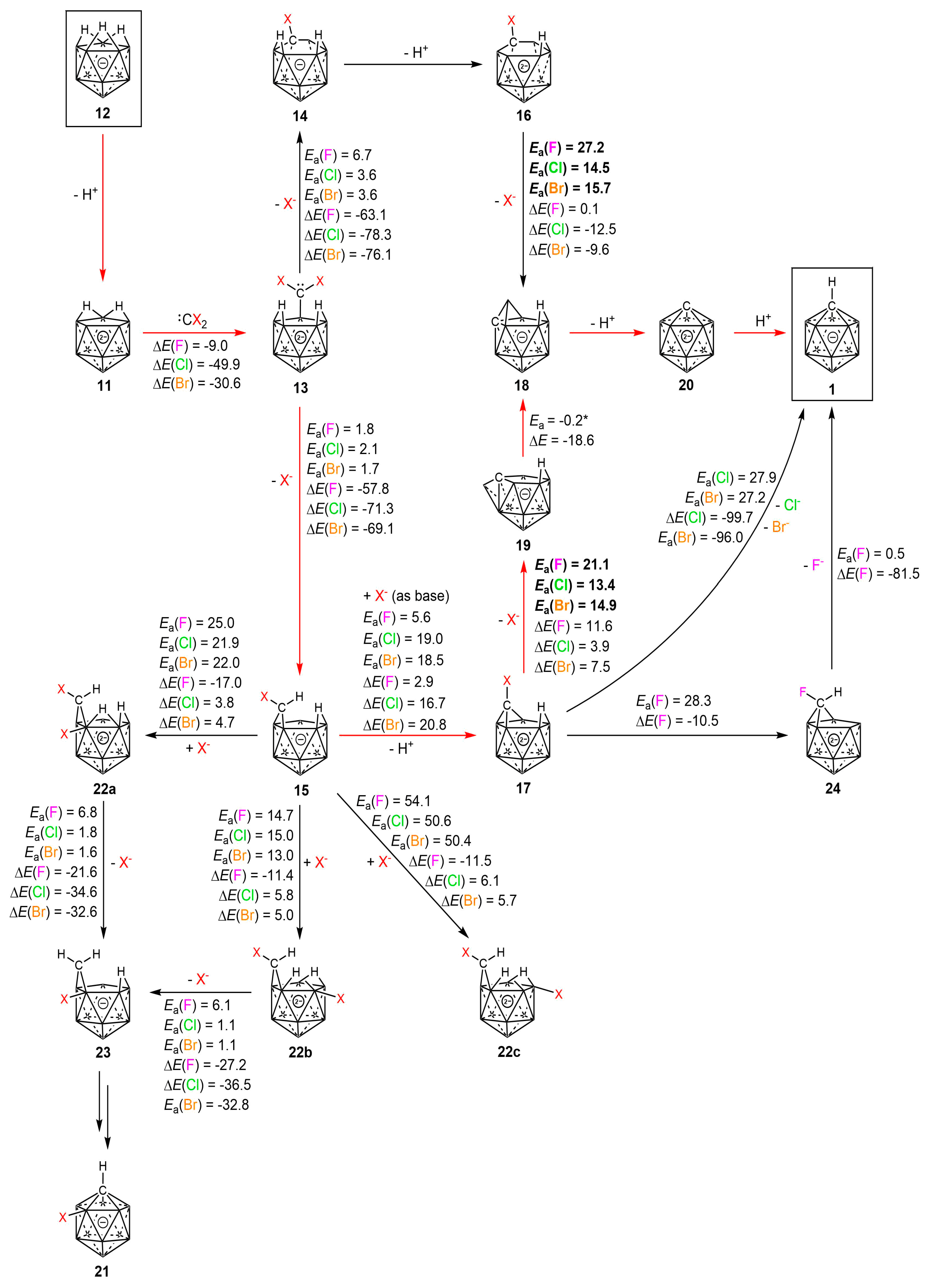
2.2. Mechanism
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Calculations
4.3. Procedures
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Douvris, C.; Michl, J. Update 1 of: Chemistry of the carba-closo-dodecaborate(-) anion, CB11H12−. Chem. Rev. 2013, 113, PR179–PR233. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: London, UK, 2016. [Google Scholar]
- Reed, C.A. H+, CH3+, and R3Si+ Carborane reagents: When triflates fail. Acc. Chem. Res. 2010, 43, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Küppers, T.; Bernhardt, E.; Willner, H. [Me3Si][R-CB11F11]− synthesis and properties. Angew. Chem. Int. Ed. 2007, 46, 6346–6349. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.; Stepp, B.; Douvris, C.; Valášek, M.; Štursa, J.; Klíma, J.; Piqueras, M.-C.; Crespo, R.; Ludvík, J.; Michl, J. Measured and calculated oxidation potentials of 1-X-12-Y-CB11Me10– anions. Inorg. Chem. 2012, 51, 5128–5137. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, M.F.; Maderna, A. Applications of radiolabeled boron clusters to the diagnosis and treatment of cancer. Chem. Rev. 1999, 99, 3421–3434. [Google Scholar] [CrossRef]
- Kaszynski, P. Four Decades of Organic Chemistry of closo-Boranes: A Synthetic Toolbox for Constructing Liquid Crystal Materials. A Review. Collect. Czechoslov. Chem. Commun. 1999, 64, 895–926. [Google Scholar] [CrossRef]
- Ringstrand, B.; Jankowiak, A.; Johnson, L.E.; Pociecha, D.; Kaszynski, P.; Górecka, E. Anion-driven mesogenicity: A comparative study of ionic liquid crystals based on the [closo-1-CB9H10]− and [closo-1-CB11H12]− clusters. J. Mater. Chem. 2012, 22, 4874–4880. [Google Scholar] [CrossRef]
- Pecyna, J.; Ringstrand, B.; Domagala, S.; Kaszynski, P.; Wozniak, K. Synthesis of 12-pyridinium derivatives of the [closo-1-CB11H12]− anion. Inorg. Chem. 2014, 53, 12617–12626. [Google Scholar] [CrossRef]
- Larsen, A.S.; Holbrey, J.D.; Tham, F.S.; Reed, C.A. Designing ionic liquids: Imidazolium melts with inert carborane anions. J. Am. Chem. Soc. 2000, 122, 7264–7272. [Google Scholar] [CrossRef]
- Dymon, J.; Wibby, R.; Kleingardner, J.; Tanski, J.M.; Guzei, I.A.; Holbrey, J.D.; Larsen, A.S. Designing ionic liquids with boron cluster anions: Alkylpyridinium and imidazolium [nido-C2B9H11] and [closo-CB11H12] carborane salts. Dalton Trans. 2008, 2999. [Google Scholar] [CrossRef]
- Knoth, W.H. 1-B9H9CH− and B11H11CH−. J. Am. Chem. Soc. 1967, 89, 1274–1275. [Google Scholar] [CrossRef]
- Knoth, W.H. B10H12CNH3, B9H9CH−, B11H11CH−, and metallomonocarboranes. Inorg. Chem. 1971, 10, 598–605. [Google Scholar] [CrossRef]
- Plešek, J.; Jelínek, T.; Drdáková, E.; Heřmánek, S.; Stibr, B. A convenient preparation of 1-CB11H12− and its C-amino derivatives. Collect. Czechoslov. Chem. Commun. 1984, 49, 1559–1562. [Google Scholar] [CrossRef]
- Goeta, A.E.; Hughes, A.K.; Batsanov, A.S.; Fox, M.A.; Howard, J.A.K.; Malget, J.M. A convenient cyanide-free “one-pot” synthesis of nido-Me3N-7-CB10H12 and nido-7-CB10H13−. J. Chem. Soc. Dalton Trans. 2002, 2624. [Google Scholar] [CrossRef]
- Franken, A.; Bullen, N.J.; Jelínek, T.; Thornton-Pett, M.; Teat, S.J.; Clegg, W.; Kennedy, J.D.; Hardie, M.J. Structural chemistry of halogenated monocarbaboranes: the extended structures of Cs [1-HCB9H4Br5], Cs[1-HCB11H5Cl6] and Cs[1-HCB11H5Br6]. New J. Chem. 2004, 28, 1499–1505. [Google Scholar] [CrossRef]
- Franken, A.; King, B.T.; Rudolph, J.; Rao, P.; Noll, B.C.; Michl, J. Preparation of [closo-CB11H12]− by dichlorocarbene insertion into [nido-B11H14]−. Collect. Czech. Chem. Commun. 2001, 66, 1238–1249. [Google Scholar] [CrossRef]
- Dunks, G.B.; Ordonez, K.P. A one-step synthesis of B11H14− ion from NaBH4. Inorg. Chem. 1978, 17, 1514–1516. [Google Scholar] [CrossRef]
- Toom, L.; Kütt, A.; Leito, I. Simple and scalable synthesis of the carborane anion CB11H12. Dalton Trans. 2019, 48, 7499–7502. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Prakash, G.K.S. Silicon-based reagents for difluoromethylation and difluoromethylenation reactions. Synthesis 2017, 49, 3394–3406. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Krishnamoorthy, S.; Ganesh, S.K.; Kulkarni, A.; Haiges, R.; Olah, G.A. N-difluoromethylation of imidazoles and benzimidazoles using the Ruppert-Prakash reagent under neutral conditions. Org. Lett. 2013, 16, 54–57. [Google Scholar] [CrossRef]
- Rempala, P.; Michl, J. A Proposed Mechanism of [closo-CB11H12]− Formation by dichlorocarbene insertion into [nido-B11H14]−. A computational study by density functional theory. Collect. Czechoslov. Chem. Commun. 2003, 68, 644–662. [Google Scholar] [CrossRef]
- Howard, J.L.; Schotten, C.; Alston, S.T.; Browne, D.L. Preparation of difluoromethylthioethers through difluoromethylation of disulfides using TMS-CF2H. Chem. Commun. 2016, 52, 8448–8451. [Google Scholar] [CrossRef] [PubMed]
- Kosobokov, M.D.; Dilman, A.D.; Levin, V.V.; Struchkova, M.I. Difluoro(trimethylsilyl)acetonitrile: Synthesis and fluoroalkylation reactions. J. Org. Chem. 2012, 77, 5850–5855. [Google Scholar] [CrossRef] [PubMed]
- Tsymbal, A.V.; Kosobokov, M.D.; Levin, V.V.; Struchkova, M.I.; Dilman, A.D. Nucleophilic bromodifluoromethylation of iminium ions. J. Org. Chem. 2014, 79, 7831–7835. [Google Scholar] [CrossRef]
- Koidan, G.N.; Marchenko, A.P.; Pinchuk, A.M. Binary triamidophosphite-haloform system as a new source of trihalomethanide ions. J. Gen. Chem. USSR (Engl. Transl.). 1990, 60, 622–623. [Google Scholar]
- Wu, N.; Wahl, B.; Woodward, S.; Lewis, W. 1,4-Addition of TMSCCl3 to nitroalkenes: Efficient reaction conditions and mechanistic understanding. Chem. - A Eur. J. 2014, 20, 7718–7724. [Google Scholar] [CrossRef]
- Kaleta, J.; Akdag, A.; Crespo, R.; Piqueras, M.-C.; Michl, J. Evidence for an intermediate in the methylation of CB11H12− with methyl triflate: Comparison of electrophilic substitution in cage boranes and in arenes. ChemPlusChem 2013, 78, 1174–1183. [Google Scholar] [CrossRef]
- Grimmer, S.; Ehrlich, S.; Goerigk, L. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Marenich, V.A.; Cramer, J.C.; Truhlar, G.D. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. A salt of the [closo-1-CB11H12- anion can probably be purchased from Boron Specialties LLC. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Carbene Source | Solvent | Volume [mL] | Temp [°C] | Yield of 1 [%] |
|---|---|---|---|---|---|
| 1 | CHCl2P(O)(OEt)2 | THF | 20 | rt | 65 |
| 2 | CHCl[P(O)(OEt)2]2 | THF | 20 | rt | 50 b |
| 3 | CN2[P(O)(OEt)2]2 | THF | 20 | rt | 0 c |
| 4 | CCl3COONa | Diglyme | 10 | 150 | 0 c |
| 5 | ClF2COONa | Diglyme | 10 | 165 | 0 c |
| 6 | CF3COOLi | Diglyme | 10 | 165 | 0 c |
| No. | TMS-CF3 (equiv) | Solvent | Volume [mL] | Initiator [mol%] | Temp [°C] | Yield of 1 [%] |
|---|---|---|---|---|---|---|
| 1 | 2.5 | DME | 20 | TBAB [5] | 85 | 67 |
| 2 | 2.5 | DME | 10 | TBAB [5] | 85 | 0 b |
| 3 | 2.5 | THF | 20 | TBAB [5] | 66 | 78 |
| 4 | 2.5 | THF | 10 | TBAB [5] | 66 | 75 |
| 5 | 4.0 | DME | 20 | TBAB [5] | 85 | 85 |
| 6 | 2.5 | Diglyme | 10 | TBAB [5] | 120 | 0 b |
| 7 | 4.0 | DME | 10 | TBAF [5] | 85 | 0 b |
| 8 | 2.5 | DME | 10 | TBAB [5] | 70 | 0 c |
| No. | TMS-CF3 (equiv) | Solvent | Volume [mL] | Initiator [mol%] | Temp [°C] | Yield of 1 [%] |
|---|---|---|---|---|---|---|
| 1 | 2.5 | THF | 10 | LiCl [10]/TBAB [5] | 66 | 94 b |
| 2 | 2.5 | DME | 10 | LiCl [10]/TBAB [5] | 85 | 0 c |
| 3 | 2.5 | DME | 10 | LiCl [10]/TBAB [10] | 70 | 0 c |
| 4 | 2.5 | THF | 20 | LiCl [10]/TBAB [5] | 66 | 76 |
| 5 | 2.5 | DME | 20 | LiCl [10]/TBAB [5] | 85 | 0 c |
| 6 | 2.5 | THF | 10 | LiCl [10]/TBAB [10] | 66 | 67 d |
| 7 | 3.5 | THF | 10 | LiCl [10]/TBAB [5] | 66 | 68 |
| 8 | 2.5 | THF | 10 | LiCl [10]/TBAB [5] | 66 | 69 e |
| 9 | 2.5 | THF | 10 | LiCl [10]/TBAB [5] | 60 | 61 |
| 10 | 2.5 | THF | 10 | LiBr [10]/TBAB [5] | 66 | 62 |
| 11 | 2.5 | THF | 10 | LiI [10]/TBAB [5] | 66 | 50 |
| 12 | 3.5 | THF | 10 | LiBr [10]/TBAB [5] | 66 | 65 |
| 13 | 5.0 | THF | 10 | LiCl [10]/LiBr [10] | 66 | 76 |
| 14 | 2.5 | THF | 10 | LiCl [15] | 66 | 78 f |
| 15 | 5.0 | THF | 10 | LiCl [15] | 66 | 78 |
| 16 | 5.0 | THF | 10 | LiCl [25] | 66 | 68 |
| 17 | 5.0 | THF | 10 | LiCl [50] | 66 | 73 |
| 18 | 2.5 | THF | 10 | LiBr [15] | 66 | 72 |
| 19 | 3.5 | THF | 10 | LiBr [15] | 60 | 45 |
| 20 | 3.5 | THF | 10 | LiBr [50] | 66 | 74 |
| 21 | 3.5 | THF | 10 | LiBr [100] | 66 | 77 |
| 22 | 3.5 | THF | 20 | LiBr [15] | 66 | 61 |
| 23 | 2.5 | THF | 10 | LiI [15] | 66 | 62 |
| 24 | 5.0 | THF | 10 | LiI [15] | 66 | 67 |
| 25 | 2.5 | THF | 10 | LiCl [5] | 66 | 60 |
| 26 | 2.5 | THF | 5 | LiCl [15] | 66 | 56 |
| 27 | 2.5 | THF | 5 | LiCl [5] | 66 | 40 |
| 28 | 2.5 | THF | 10 | LiCl [15] | 50 | 54 |
| 29 | 2.5 | THF | 10 | LiCl [15] | rt | 52 g |
| 30 | 2.5 | THF | 10 | - | 66 | 40 |
| 31 | 2.5 | Diglyme | 10 | LiCl [15] | 100 | 50 |
| 32 | 2.5 | Diglyme | 10 | LiCl [15] | 66 | 0 h |
| 33 | 2.5 | THF | 10 | LiCl [15] | 66 | 77 i |
| 34 | 2.5 | DME | 10 | LiCl [15] | 85 | 52 i |
| 35 | 2.5 | Diglyme | 10 | LiCl [15] | 100 | 25 i |
| 36 | 2.5 | THF | 10 | LiCl [15] | 66 | 56 j |
| 37 | 2.5 | THF | 200 | LiCl [15] | 66 | 77 k |
| No. | TMS-CF3 (equiv) | Solvent | Volume [mL] | Initiator [mol%] | Temp [°C] | Yield of 1 [%] |
|---|---|---|---|---|---|---|
| 1 | 2.5 | THF | 10 | KF [15] | 66 | 53 |
| 2 | 2.5 | THF | 10 | NaCl [15] | 66 | 65 |
| 3 | 2.5 | THF | 10 | KCl [15] | 66 | 54 |
| 4 | 2.5 | THF | 10 | CsCl [15] | 66 | 71 |
| 5 | 2.5 | THF | 10 | MgCl2 [15] | 66 | 64 |
| 6 | 2.5 | THF | 10 | ZnCl2[15] | 66 | 0 b |
| 7 | 2.5 | THF | 10 | NaI [15] | 66 | 60 |
| 8 | 2.5 | THF | 10 | KI [15] | 66 | 70 |
| 9 | 2.5 | THF | 10 | CsI [15] | 66 | 57 |
| 10 | 2.5 | THF | 10 | KBr [15] | 66 | 54 |
| 11 | 2.5 | THF | 10 | PdCl2 [15] | 66 | 76 c |
| 12 | 2.5 | THF | 10 | GdF3 [15] | 66 | 59 c |
| 13 | 2.5 | HF | 10 | InCl3 [15] | 66 | 65 c |
| No. | Reagent (equiv) | Solvent | Volume [mL] | Initiator | Temp [°C] | Yield of 1 [%] |
|---|---|---|---|---|---|---|
| 1 | TMS-CCl3 (2.5) | THF | 10 | LiCl | 66 | 0 b |
| 2 | TMS-CCl3 (2.5) | diglyme | 10 | LiCl | 120 | 0 b |
| 3 | TMS-CCl3 (2.5) | THF | 10 | LiCl | 66 | 21 c,d,e |
| 4 | TMS-CCl3 (3.5) | THF | 10 | LiCl | 66 | 5 c,f,g |
| 5 | TMS-CBr3 (2.5) | THF | 10 | LiCl | 66 | 0 b |
| 6 | TMS-CBr3 (2.5) | THF | 10 | LiCl | 66 | 40 c,d,h |
| 7 | TMS-CBr3 (3.5) | THF | 10 | LiCl | 66 | Traces c,f,i |
| 8 | TMS-CF2Br (2.5) | THF | 10 | LiCl | 66 | 71 c |
| 9 | TMS-CF2Cl (2.5) | THF | 10 | LiCl | 66 | 47 c |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecyna, J.; Rončević, I.; Michl, J. Insertion of Carbenes into Deprotonated nido-Undecaborane, B11H13(2-). Molecules 2019, 24, 3779. https://doi.org/10.3390/molecules24203779
Pecyna J, Rončević I, Michl J. Insertion of Carbenes into Deprotonated nido-Undecaborane, B11H13(2-). Molecules. 2019; 24(20):3779. https://doi.org/10.3390/molecules24203779
Chicago/Turabian StylePecyna, Jacek, Igor Rončević, and Josef Michl. 2019. "Insertion of Carbenes into Deprotonated nido-Undecaborane, B11H13(2-)" Molecules 24, no. 20: 3779. https://doi.org/10.3390/molecules24203779
APA StylePecyna, J., Rončević, I., & Michl, J. (2019). Insertion of Carbenes into Deprotonated nido-Undecaborane, B11H13(2-). Molecules, 24(20), 3779. https://doi.org/10.3390/molecules24203779