
Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques

 ,
, 
Abstract
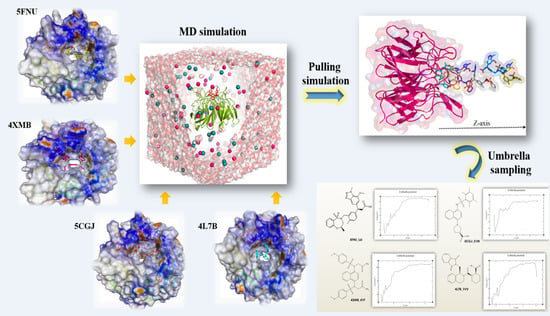
1. Introduction
2. Results
2.1. Ligand–Protein Interactions Analysis
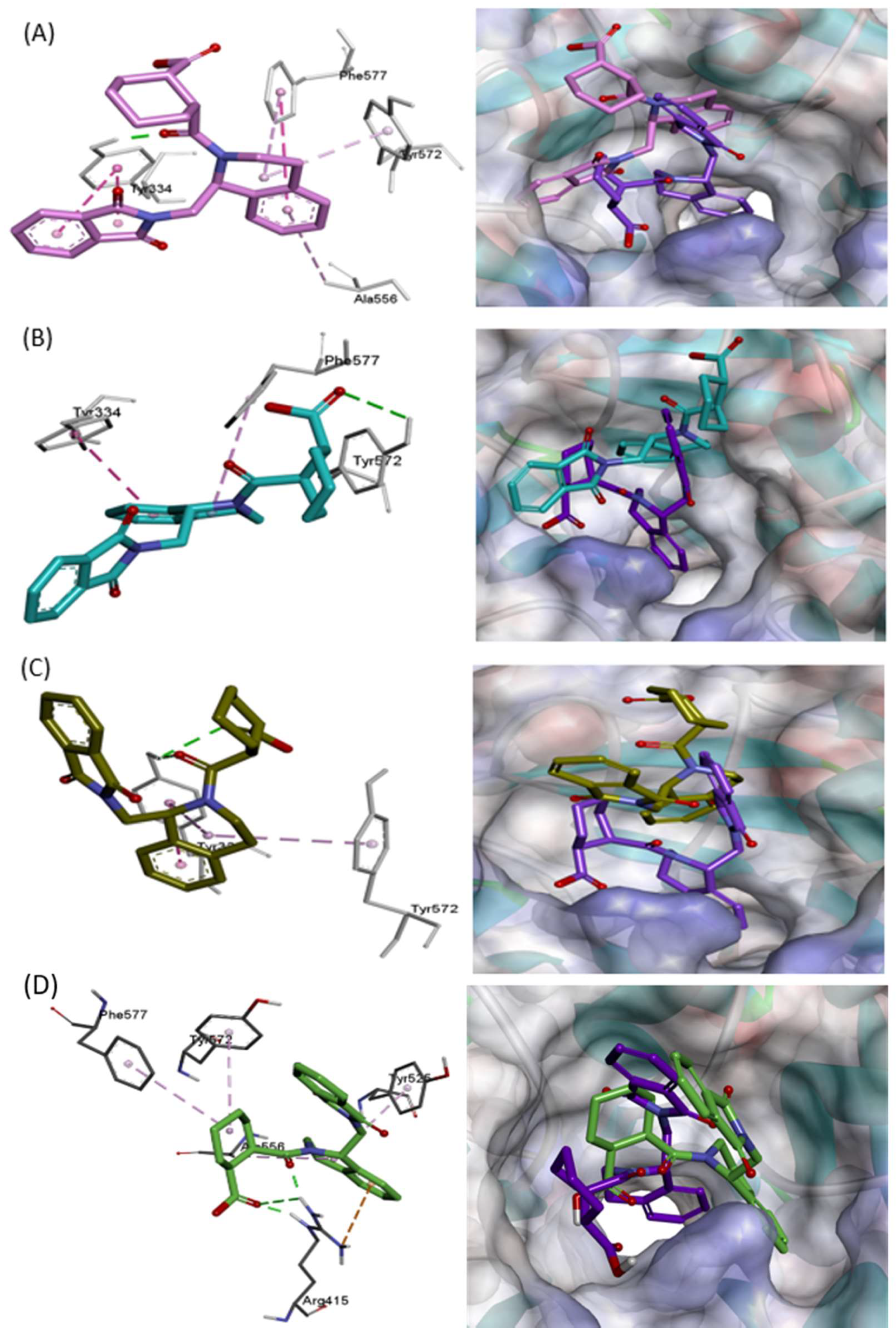
2.1.1. Interaction before MD Simulation
2.1.2. Docking Study
2.2. MD Simulation Results
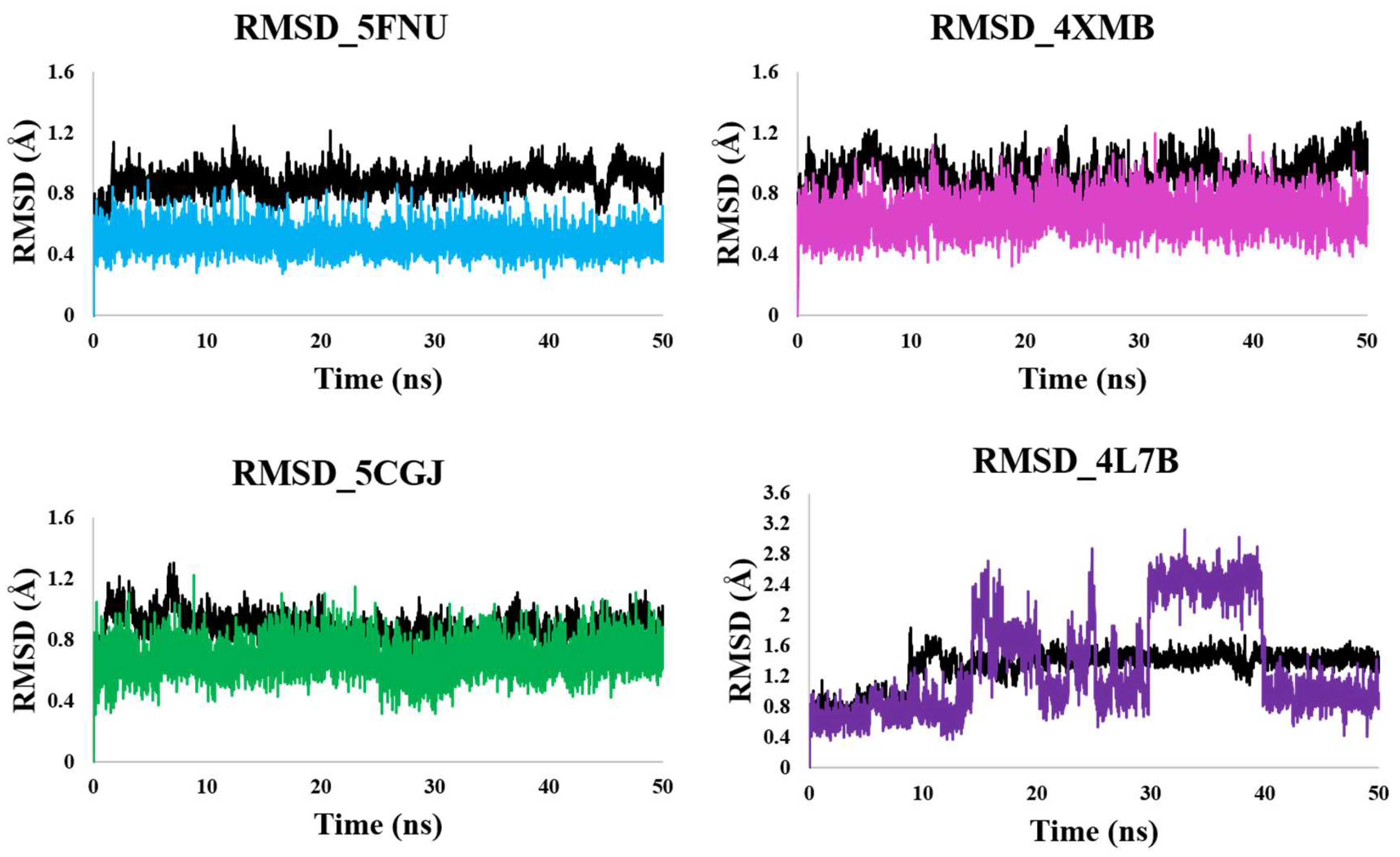
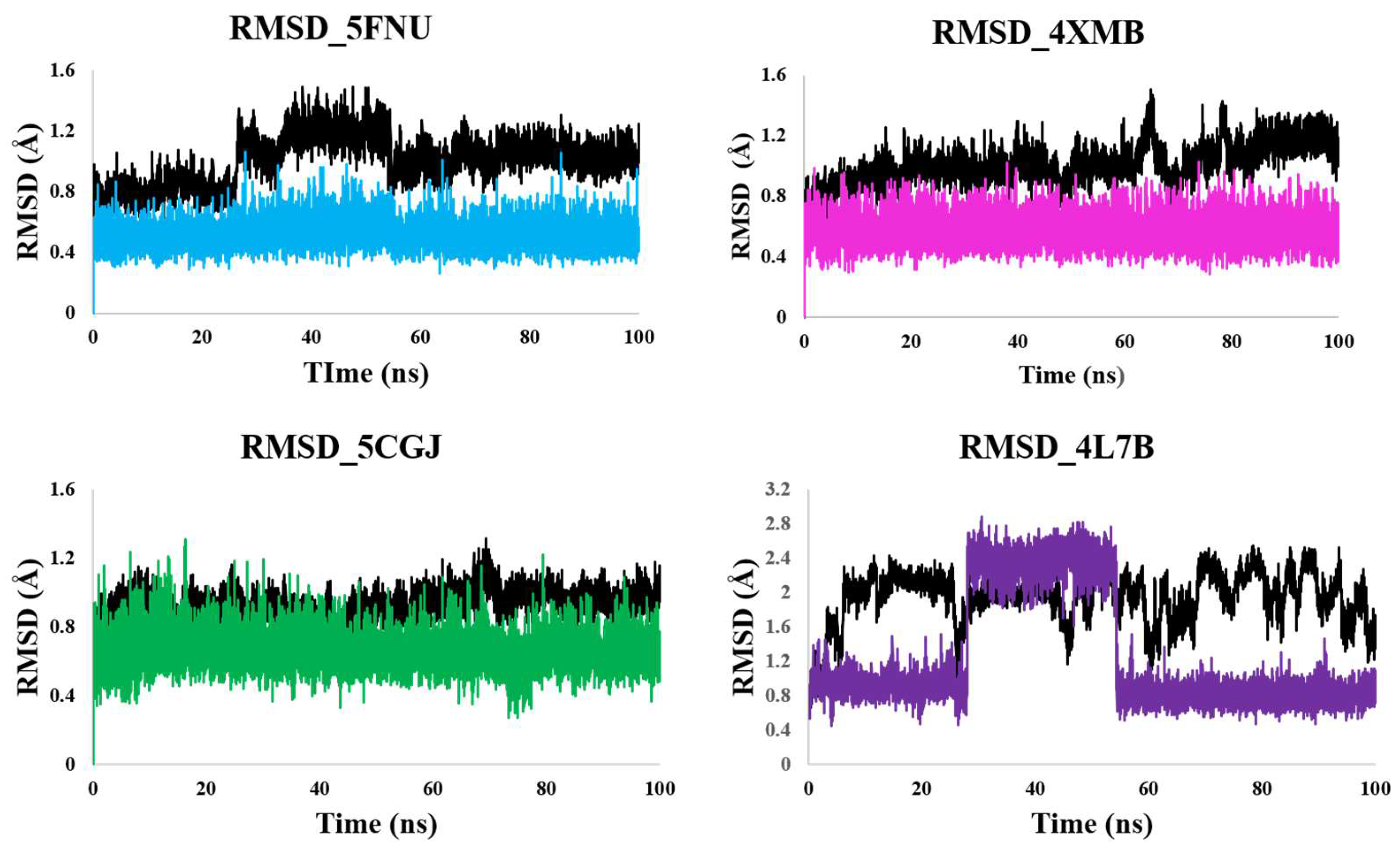
2.2.1. RMSD Calculation
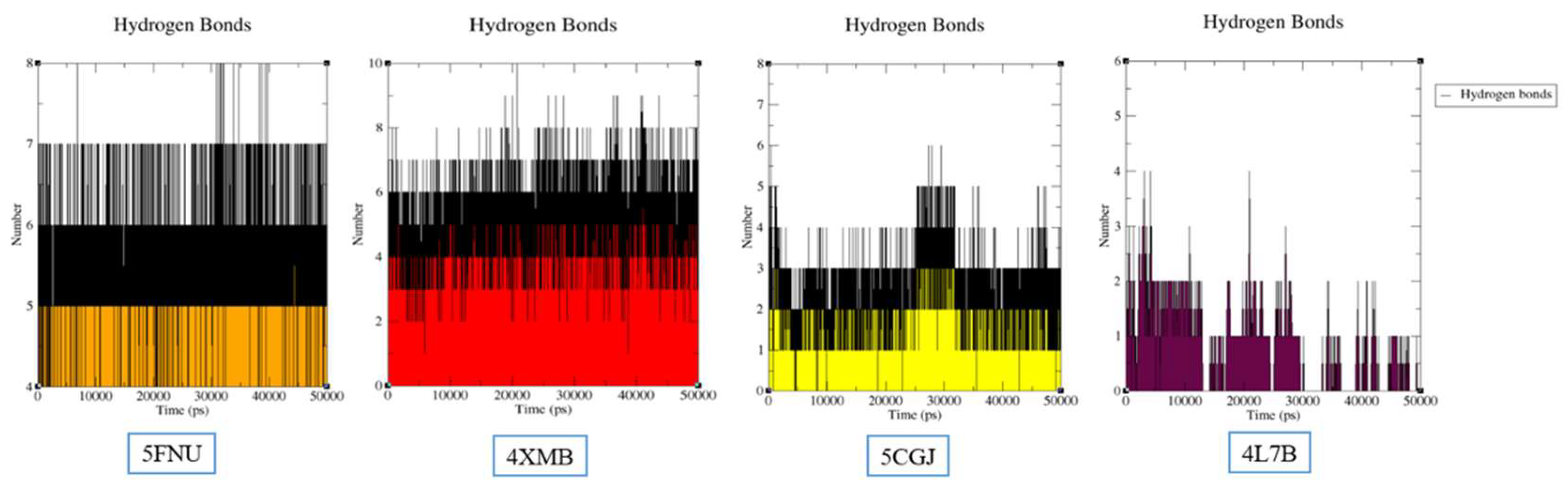
2.2.2. Hydrogen Bond Analysis
2.2.3. Residue Interaction Energy
5FNU
4XMB
5CGJ
4L7B
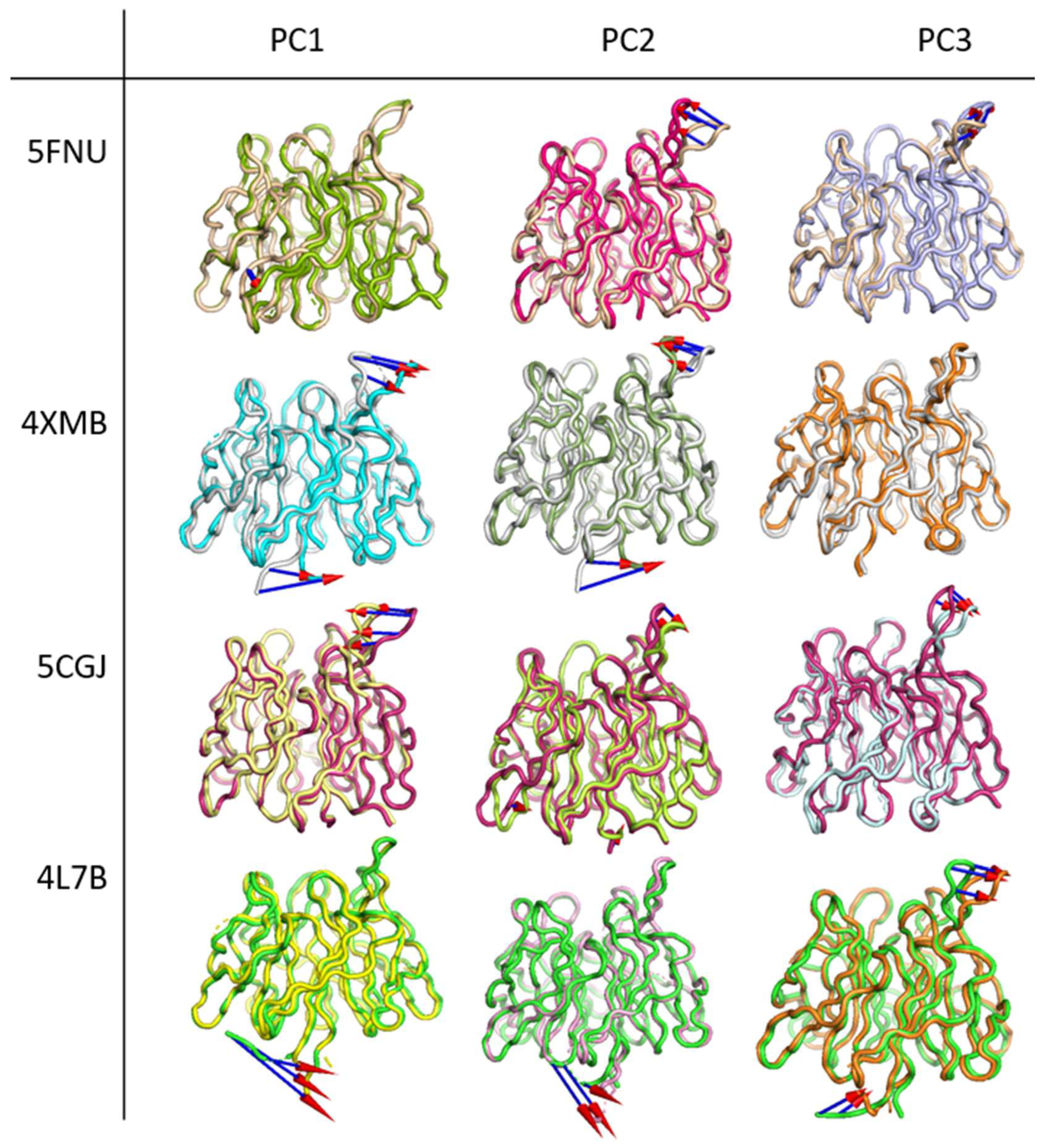
2.2.4. Principal Component Analysis
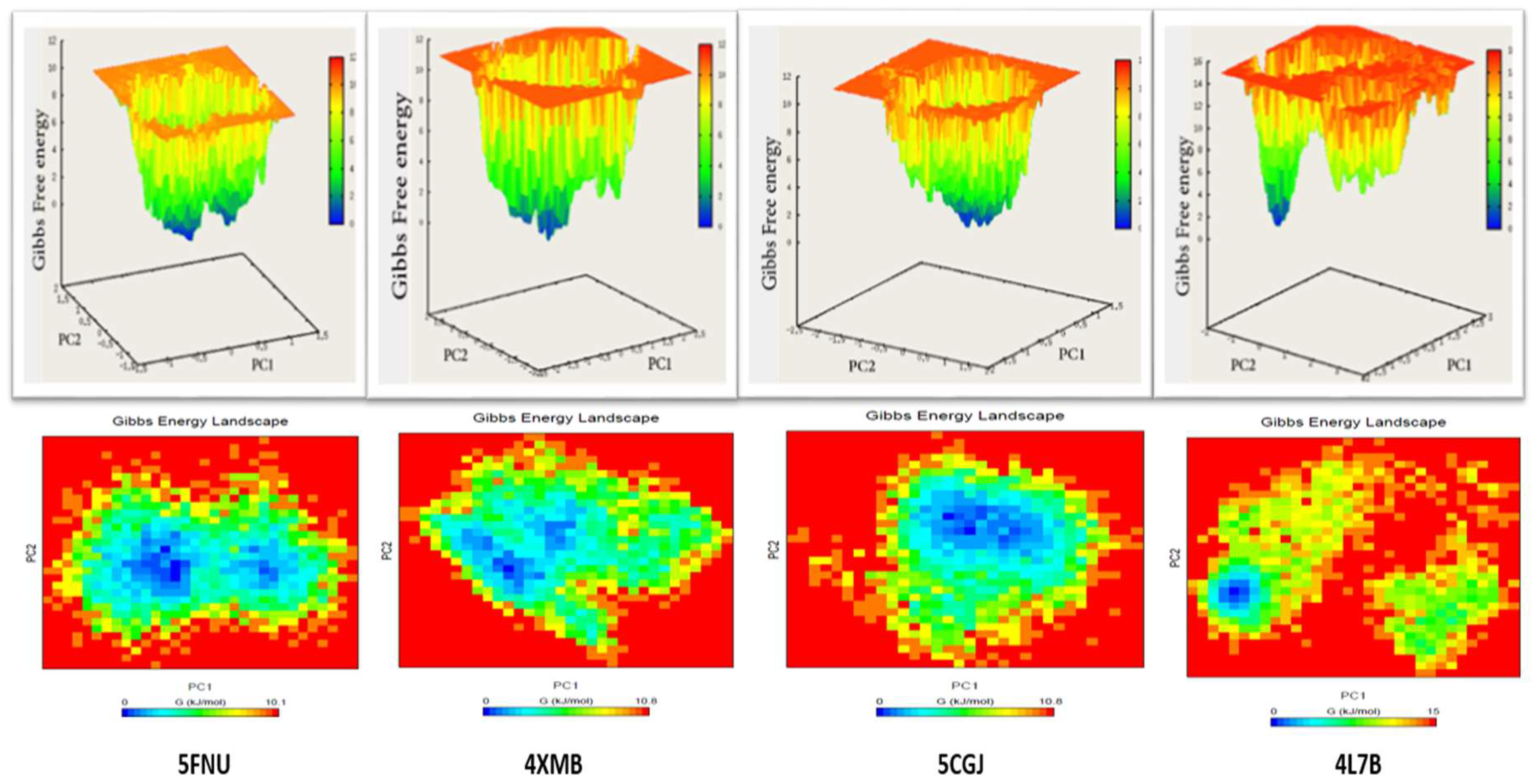
2.2.5. Free Energy Landscape
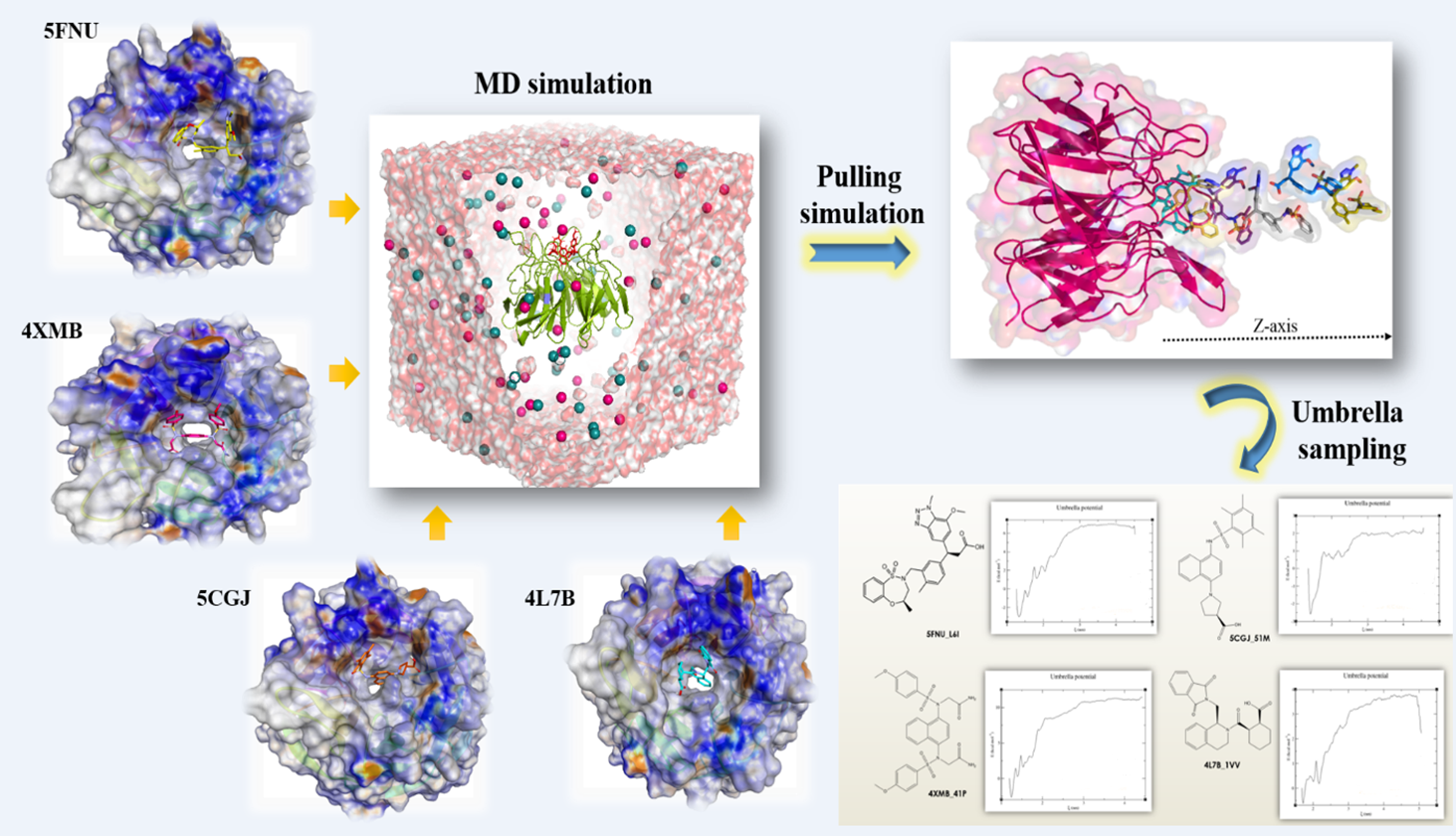
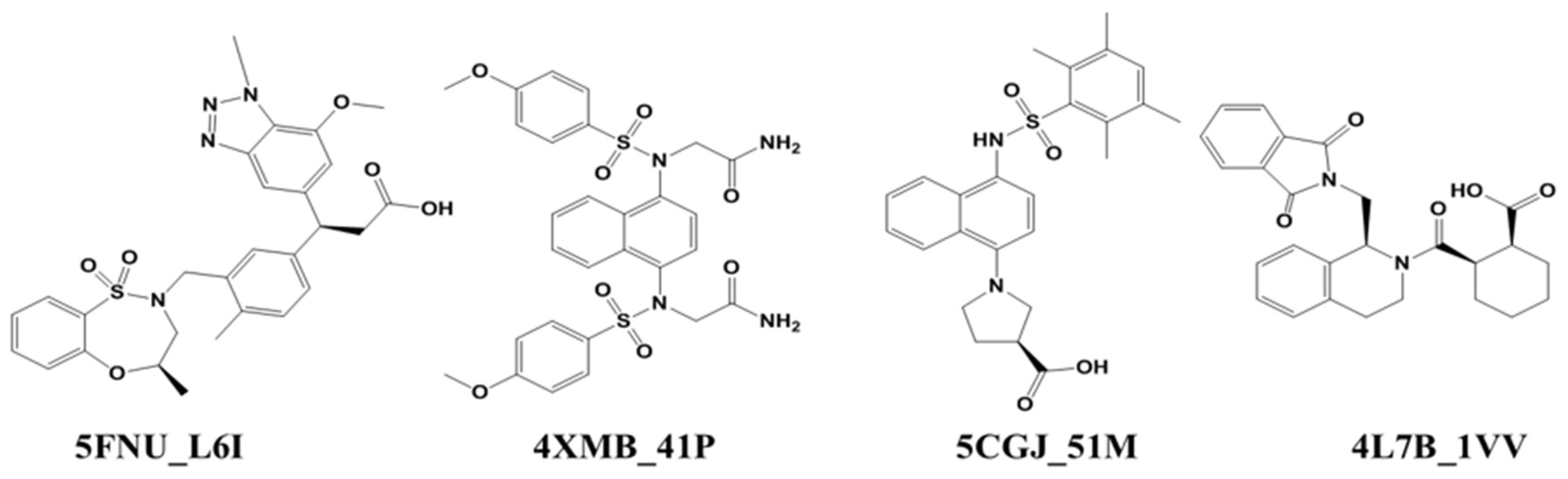
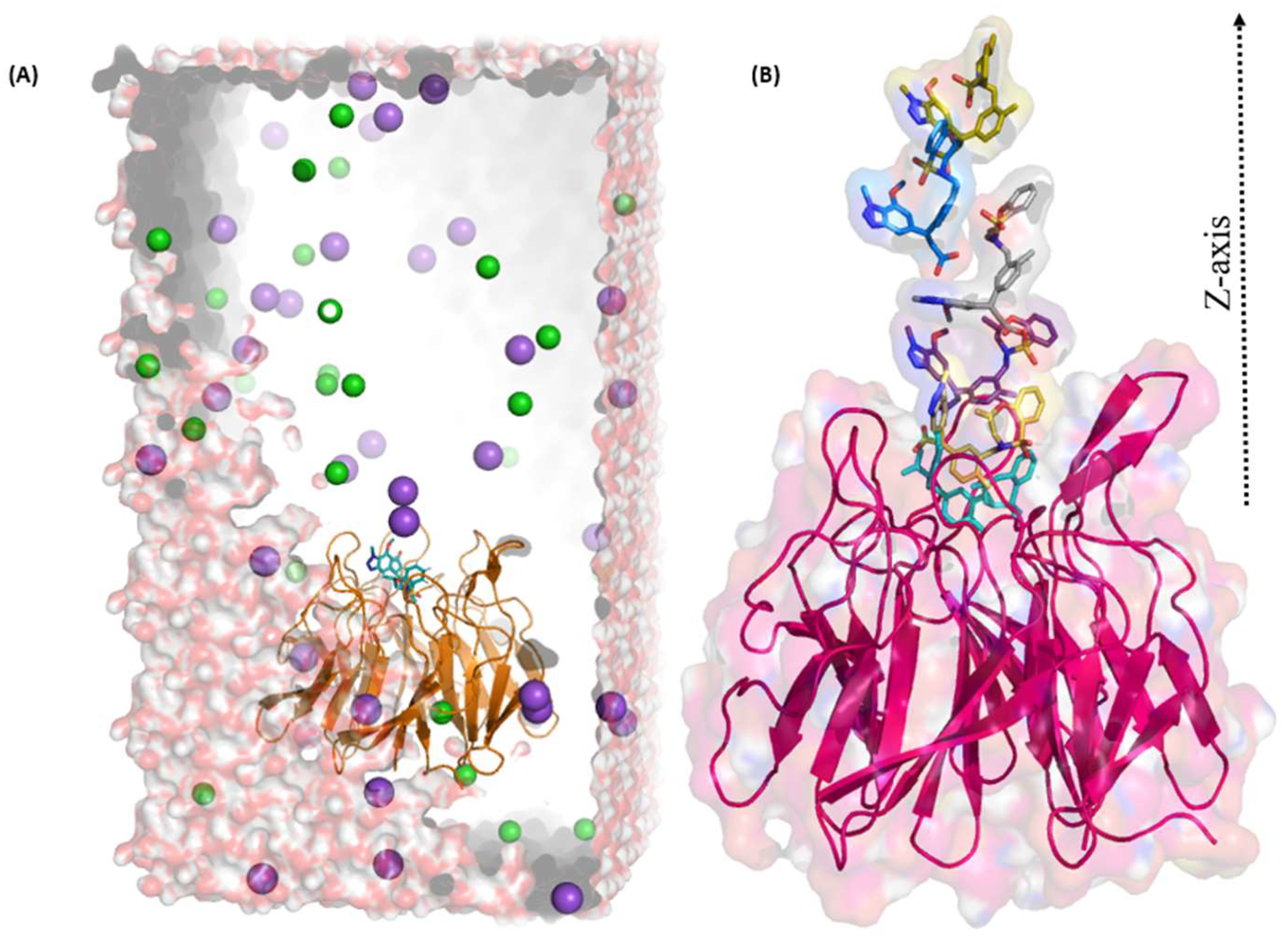
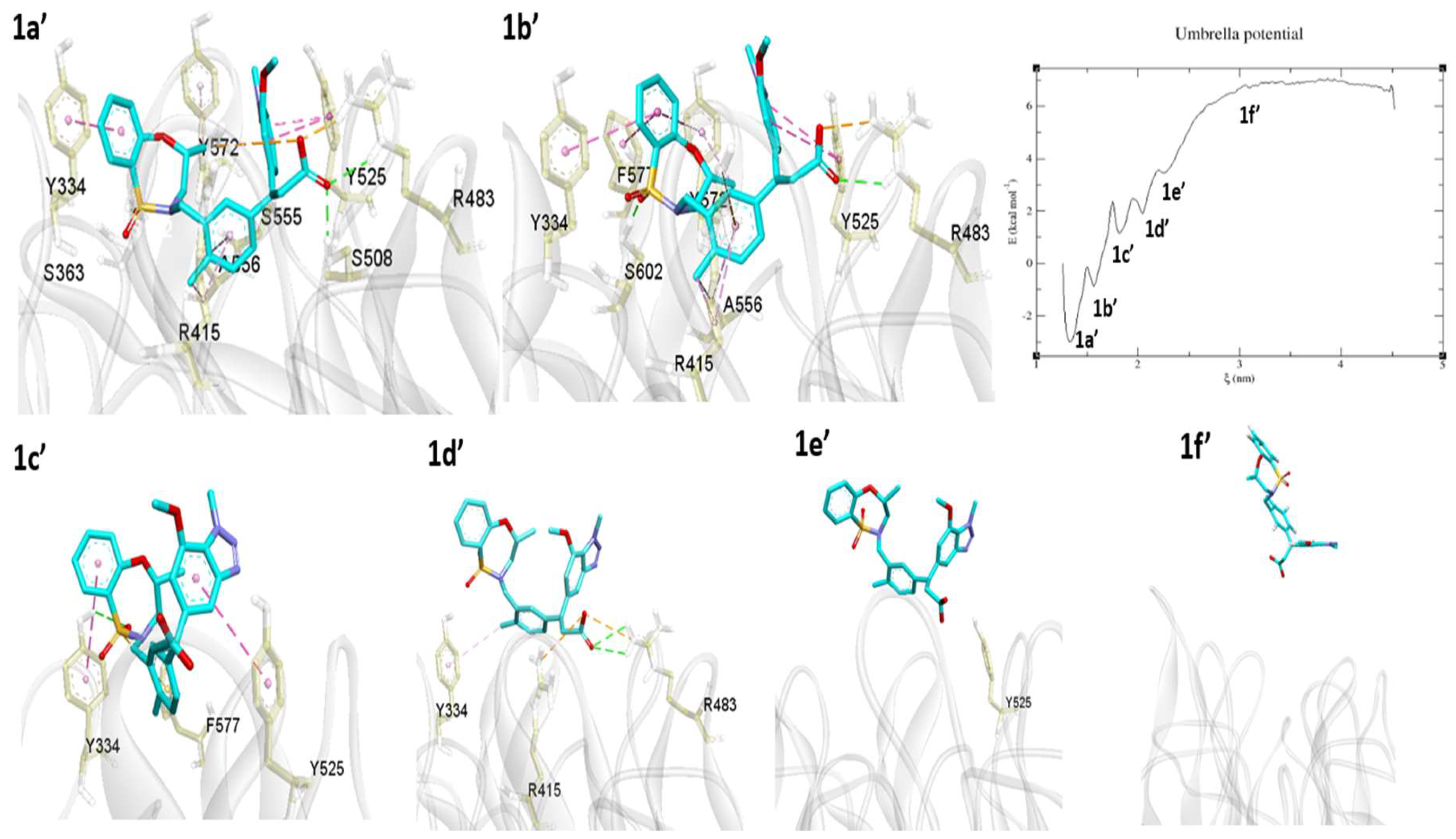
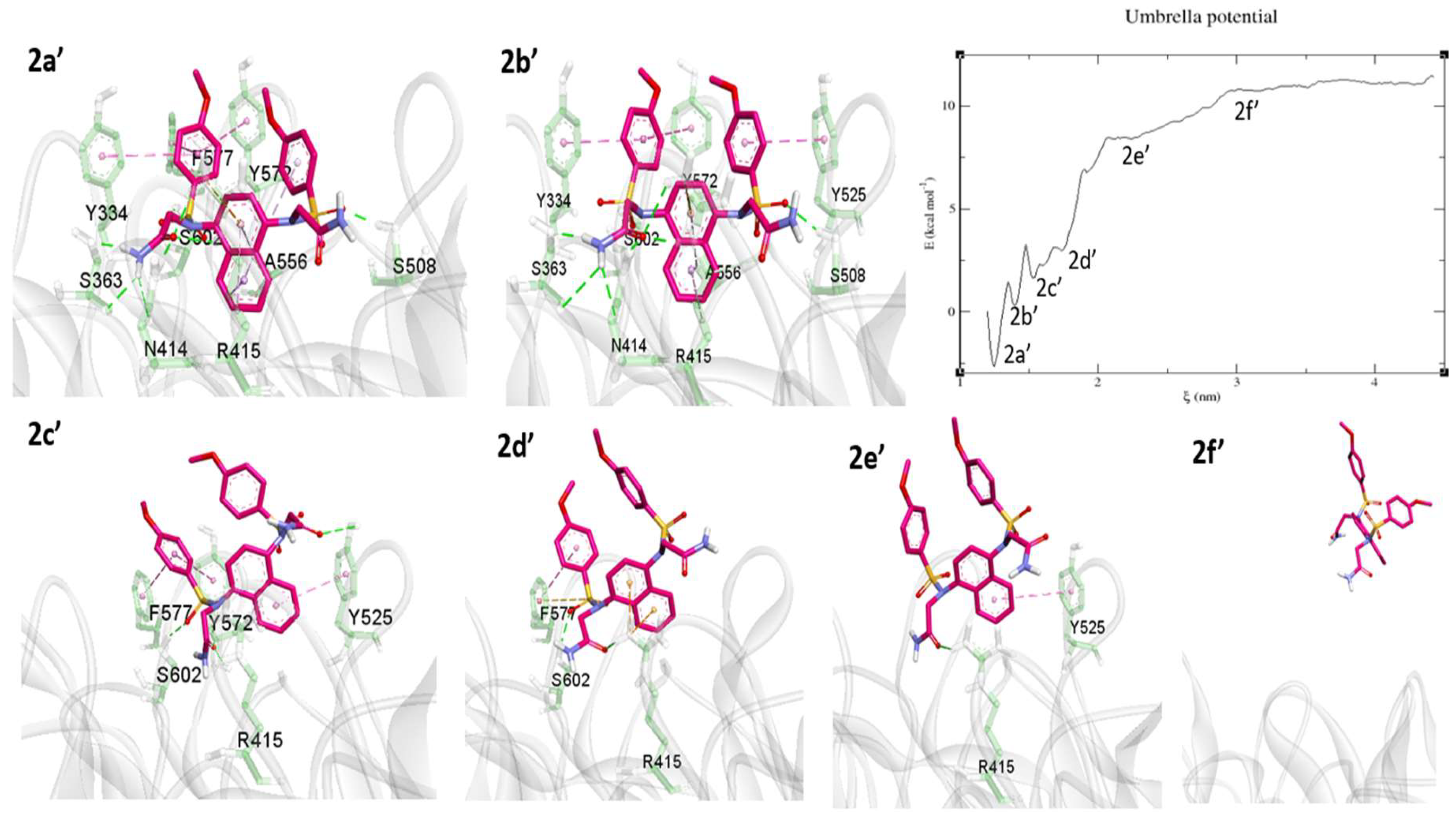
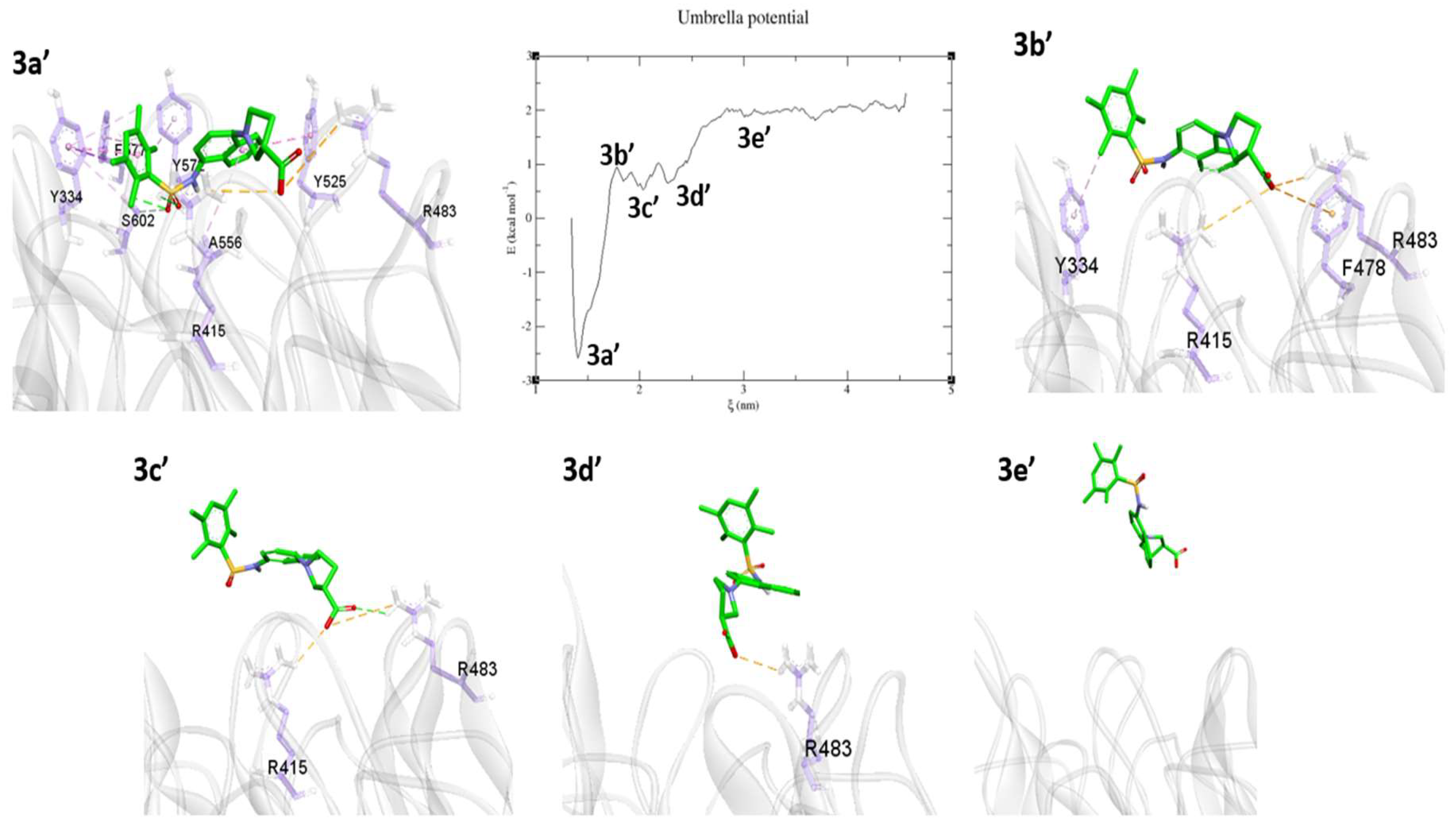
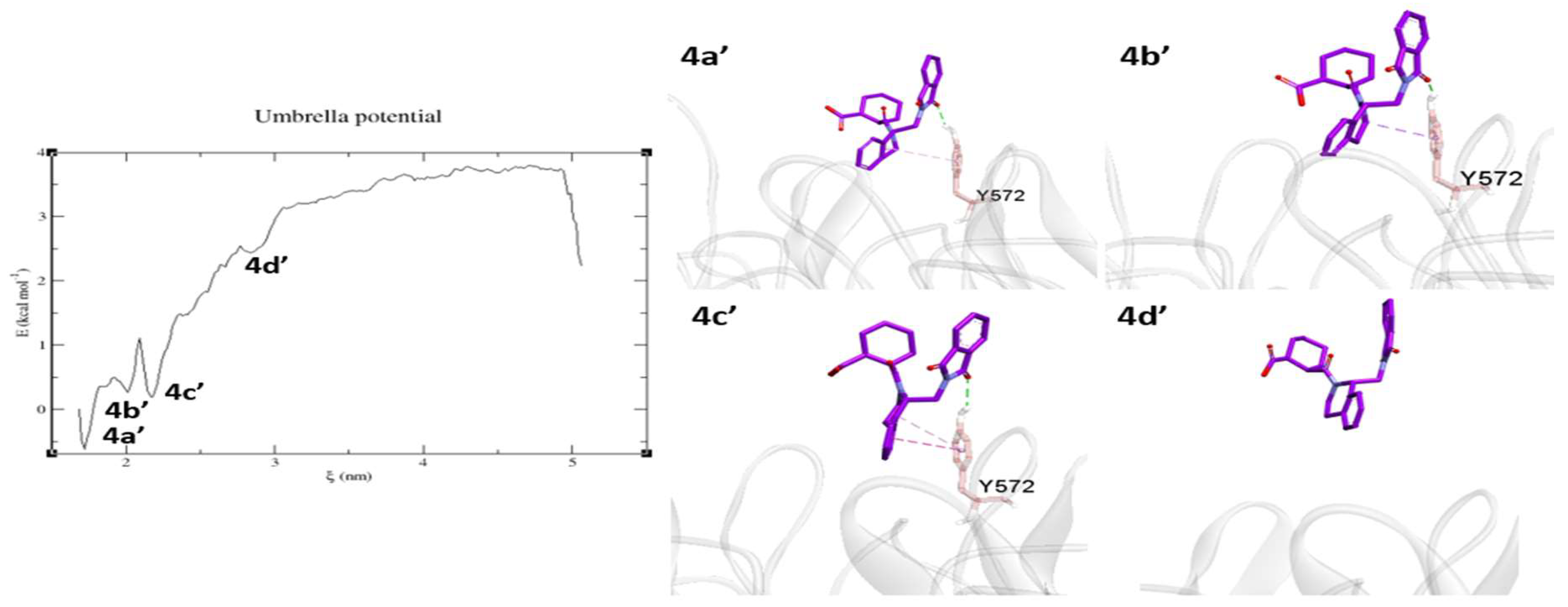
2.3. Umbrella Sampling (US) Results
3. Discussion
4. Materials and Methods
4.1. Protein–Ligand Complex Selection
4.2. Molecular Docking Study
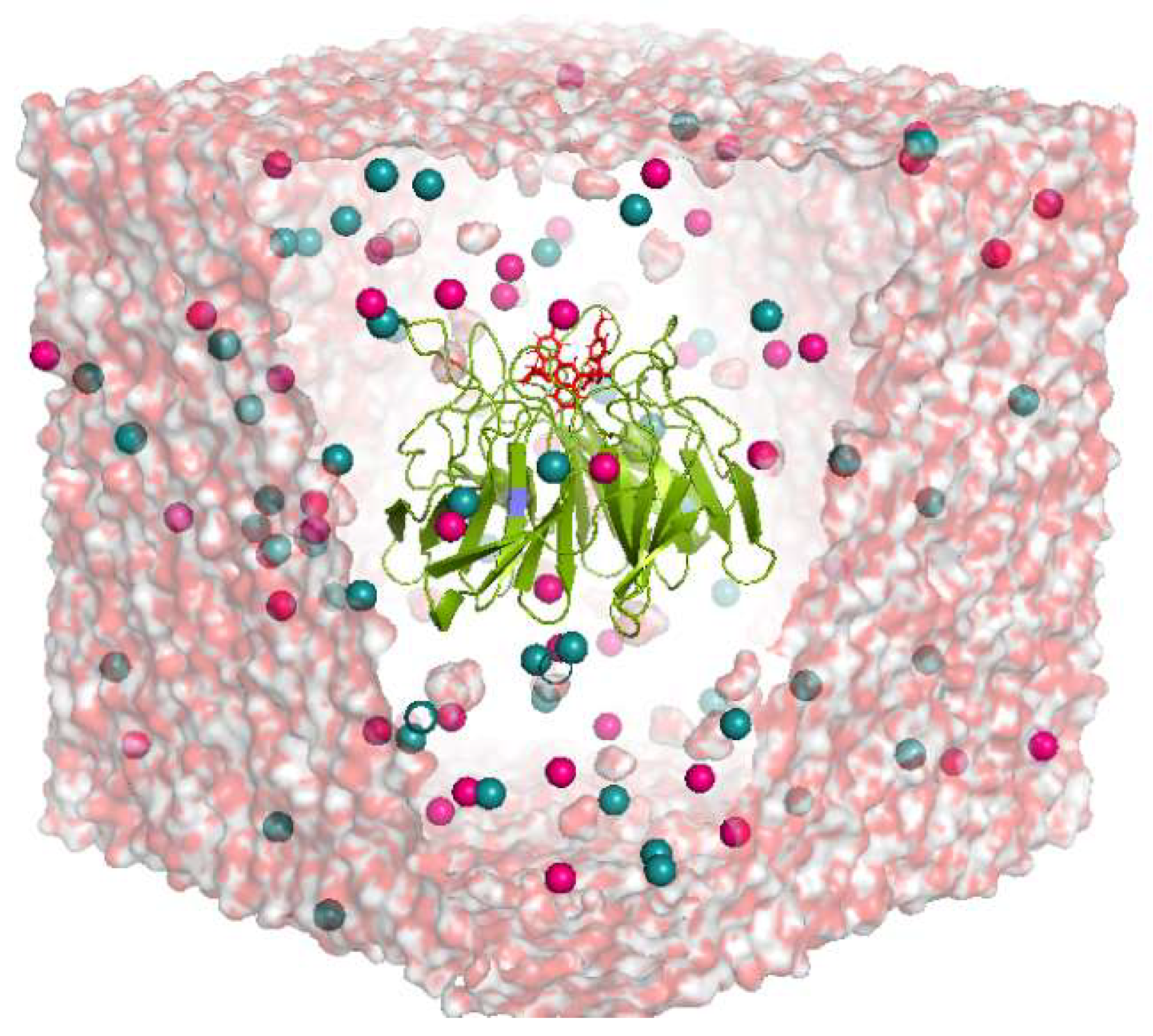
4.3. Molecular Dynamic (MD) Simulation
4.4. Umbrella Sampling (US) Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular dynamic |
| US | Umbrella sampling |
| PPI | Protein–Protein interactions |
| RMSD | Root mean square deviation |
| PMF | Potential of mean force |
| Coul-SR | Coulombic potential short-range |
| LJ-SR | Lennard–Jones potential short-range |
| PCA | Principal component analysis |
| H-bond | Hydrogen bond |
| ns | Nanosecond |
| µs | Microsecond |
| nm | Nanometer |
| ps | Picosecond |
| nM | Nanomolar |
| µM | Micromolar |
| NVT | Constant number of atoms, volume, and temperature |
| NPT | Constant number of atoms, pressure, and temperature |
References
- Marcotte, D.; Zeng, W.; Hus, J.C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorgan. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, synthesis, and evaluation of triazole derivatives that induce Nrf2 dependent gene products and inhibit the Keap1–Nrf2 protein–protein interaction. J. Med. Chem. 2015, 58, 7186–7194. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of potent Keap1–Nrf2 protein–protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of Keap1–Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Nakajima, M.; Yuasa, A.; Obata, R.; Takahashi, K.; Ohe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Imamura, R.; et al. Synthesis of Keap1-phosphorylated p62 and Keap1-Nrf2 protein-protein interaction inhibitors and their inhibitory activity. Bioorganic Med. Chem. Lett. 2016, 26, 5956–5959. [Google Scholar] [CrossRef] [PubMed]
- Kalthoff, S.; Ehmer, U.; Freiberg, N.; Manns, M.P.; Strassburg, C.P. Interaction between oxidative stress sensor Nrf2 and xenobiotic-activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP-glucuronosyltransferase 1A10. J. Biol. Chem. 2010, 285, 5993–6002. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1: Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.-M.; Li, J.; Johnson, J.A. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal. 2009, 11, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Wu, R.; Guo, Y.; Kong, A.-N.T. Regulation of Keap1–Nrf2 signaling: The role of epigenetics. Curr. Opin. Toxicol. 2016, 1, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y.; You, Q.D. The Keap1–Nrf2–ARE pathway as a potential preventive and therapeutic target: An update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Inoyama, D.; Kong, A.N.T.; Beamer, L.J.; Hu, L. Kinetic analyses of Keap1–Nrf2 interaction and determination of the minimal Nrf2 peptide sequence required for Keap1 binding using surface plasmon resonance. Chem. Biol. Drug Des. 2011, 78, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Yuan, Z.W.; Jiang, Y.L.; Chen, Z.Y.; You, Q.D.; Jiang, Z.Y. A systematic molecular dynamics approach to the study of peptide Keap1–Nrf2 protein–protein interaction inhibitors and its application to p62 peptides. Mol. Biosyst. 2016, 12, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.; Bertrand, H.C.; Tsujita, T.; Naz, S.; El-Bakry, A.; Laoruchupong, J.; Hayes, J.D.; Wells, G. Peptide inhibitors of the Keap1–Nrf2 protein–protein interaction. Free Radic. Biol. Med. 2012, 52, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Wells, G. Peptide and small molecule inhibitors of the Keap1–Nrf2 protein–protein interaction. Biochem. Soc. Trans. 2015, 43, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Miao, Z.; Sheng, C.; Zhang, W. Updated research and applications of small molecule inhibitors of Keap1-Nrf2 protein-protein interaction: A review. Curr. Med. Chem. 2014, 21, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J. Discovery of a small-molecule inhibitor and cellular probe of Keap1–Nrf2 protein–protein interaction. Bioorganic Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.G.; Jain, A.D.; Speltz, T.E.; Moore, T.W. Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorganic Med. Chem. Lett. 2015, 25, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Zhang, J.T.; Yang, G.J.; Liu, H.; Han, Q.B.; Ma, D.L. Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. Int. J. Mol. Sci. 2019, 20, 4445. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.D.; Potteti, H.; Richardson, B.G.; Kingsley, L.; Luciano, J.P.; Ryuzoji, A.F.; Lee, H.; Krunic, A.; Mesecar, A.D.; Reddy, S.P. Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur. J. Med. Chem. 2015, 103, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.G.; Jain, A.D.; Potteti, H.R.; Lazzara, P.R.; David, B.P.; Tamatam, C.R.; Choma, E.; Skowron, K.; Dye, K.; Siddiqui, Z.; et al. Replacement of a Naphthalene Scaffold in Kelch-like ECH-Associated Protein 1 (KEAP1)/Nuclear factor (erythroid-derived 2)-like 2 (NRF2) Inhibitors. J. Med. Chem. 2018, 61, 8029–8047. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. Aabc 2015, 8, 37. [Google Scholar] [PubMed]
- Kothandan, G.; Gadhe, C.G.; Cho, S.J. Theoretical Characterization of Galanin Receptor Type 3 (G al3) and Its Interaction with Agonist (GALANIN) and Antagonists (SNAP 37889 and SNAP 398299): An in Silico Analysis. Chem. Biol. Drug Des. 2013, 81, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Plazinski, W.; Knys-Dzieciuch, A. The ‘order-to-disorder’conformational transition in CD44 protein: An umbrella sampling analysis. J. Mol. Graph. Model. 2013, 45, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Gadhe, C.G.; Kim, M.-h. Insights into the binding modes of CC chemokine receptor 4 (CCR4) inhibitors: A combined approach involving homology modelling, docking, and molecular dynamics simulation studies. Mol. Biosyst. 2015, 11, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.G.; Wixted, W.E.; Coyle, J.E.; Griffiths-Jones, C.; Hearn, K.; McMenamin, R.; Norton, D.; Rich, S.J.; Richardson, C.; Saxty, G. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: Nuclear factor erythroid 2-related factor 2 (KEAP1: NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 2016, 59, 3991–4006. [Google Scholar] [CrossRef] [PubMed]
- Winkel, A.F.; Engel, C.K.; Margerie, D.; Kannt, A.; Szillat, H.; Glombik, H.; Kallus, C.; Ruf, S.; Güssregen, S.; Riedel, J. Characterization of RA839, a noncovalent small molecule binder to Keap1 and selective activator of Nrf2 signaling. J. Biol. Chem. 2015, 290, 28446–28455. [Google Scholar] [CrossRef] [PubMed]
- Jnoff, E.; Albrecht, C.; Barker, J.J.; Barker, O.; Beaumont, E.; Bromidge, S.; Brookfield, F.; Brooks, M.; Bubert, C.; Ceska, T. Binding mode and structure–activity relationships around direct inhibitors of the Nrf2–Keap1 complex. ChemMedChem 2014, 9, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Nagai, T.; Srivastava, A.; Miyashita, O.; Tama, F. Role of computational methods in going beyond X-ray crystallography to explore protein structure and dynamics. Int. J. Mol. Sci. 2018, 19, 3401. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, M.-D.; Wei, H.; Leuenberger, M.; Lochner, M.; Thompson, A.J. The binding orientations of structurally-related ligands can differ; A cautionary note. Neuropharmacology 2017, 119, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Shatsky, M.; Wolfson, H.J.; Nussinov, R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 2002, 11, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.L.; Dill, K.A. Binding of small-molecule ligands to proteins: “What you see” is not always “what you get”. Structure 2009, 17, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes; BIOVIA. Discovery Studio Modeling Environment; Release 2018; Dassault Systèmes: San Diego, CA, USA, 2018. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Satoh, M.; Saburi, H.; Tanaka, T.; Matsuura, Y.; Naitow, H.; Shimozono, R.; Yamamoto, N.; Inoue, H.; Nakamura, N.; Yoshizawa, Y. Multiple binding modes of a small molecule to human Keap1 revealed by X-ray crystallography and molecular dynamics simulation. FEBS Open Bio. 2015, 5, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Cheng, I.-C.; Chen, Y.-J.; Ku, C.-W.; Huang, Y.-W.; Yang, C.-N. Structural and dynamic characterization of mutated Keap1 for varied affinity toward Nrf2: A molecular dynamics simulation study. J. Chem. Inf. Model. 2015, 55, 2178–2186. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
Sample Availability: Not available. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Structure | Ligand Name | Resolution | Activity (IC50) FP Assay | MD Simulation (ns) | |
|---|---|---|---|---|---|
| First half | Second half | ||||
| 4L7B | 1VV | 2.41Å | 2.3 µM | 50 | 100 |
| 5CGJ | 51M | 3.36Å | 0.14 µM | 50 | 100 |
| 4XMB | 41P | 2.43Å | 61 nM | 50 | 100 |
| 5FNU | L6I | 1.78Å | 15 nM | 50 | 100 |
| Residue | 5FNU (L6I) (IC50 = 15 nM) | 4XMB (41P) (IC50 = 61 nM) | 5CGJ (51M) (IC50 = 0.14 µM) | 4L7B (1VV) (IC50 = 0.75 µM) | ||||
|---|---|---|---|---|---|---|---|---|
| LJ-SR kJ/mol | Coul –SR kJ/mol | LJ-SR kJ/mol | Coul-SR kJ/mol | LJ-SR kJ/mol | Coul-SR kJ/mol | LJ-SR kJ/mol | Coul-SR kJ/mol | |
| Tyr 334 | −22.08 | −5.96 | −15.32 | −4.14 | −21.41 | −3.35 | −17.44 | −18.71 |
| Ser 363 | −7.42 | −10.45 | −7.73 | −22.68 | −5.54 | −6.43 | −2.09 | 0.41 |
| Gly 364 | −6.53 | −3.44 | −9.31 | −1.29 | −6.64 | −0.07 | −1.39 | 0.49 |
| Arg 380 | −2.01 | 1.01 | −6.81 | 3.63 | −1.30 | 0.55 | −5.10 | −2.19 |
| ASN 382 | −0.75 | 0.067 | −0.03 | −0.02 | −1.82 | 0.36 | −3.25 | −0.92 |
| Asn 414 | −1.73 | 0.32 | −4.15 | −22.40 | −1.70 | 0.11 | −1.07 | −4.11 |
| Arg 415 | −25.36 | 4.019 | −24.87 | −43.66 | −29.077 | 9.19 | −7.44 | −10.12 |
| Ile 461 | −5.343 | −0.55 | −7.22 | −2.52 | −6.113 | −0.005 | −8.80 | −0.07 |
| Gly 462 | −5.74 | 1.01 | −6.79 | −1.32 | −6.46 | −0.04 | −0.84 | 0.37 |
| Phe 477 | −8.20 | −2.55 | −4.47 | 0.67 | −8.63 | −4.16 | −0.03 | 0.03 |
| Arg 483 | 9.59 | −173.22 | −5.48 | 5.75 | −1.85 | −39.09 | −0.39 | 0.182 |
| Ser 508 | −3.72 | −59.40 | −7.66 | −18.12 | −10.13 | −11.05 | −0.54 | 0.037 |
| Gly 509 | −6.27 | 1.20 | −7.95 | 0.25 | −8.24 | 0.75 | −0.98 | 0.311 |
| Tyr 525 | −34.91 | −9.86 | −19.39 | −4.24 | −8.88 | −0.47 | −1.69 | −0.28 |
| Gly 530 | −3.72 | −29.59 | −6.67 | −2.14 | −0.52 | 0.44 | −0.57 | −0.188 |
| Ser 555 | −6.00 | −13.48 | −9.13 | −2.91 | −5.11 | 0.70 | −1.53 | −0.34 |
| Ala 556 | −11.81 | 1.34 | −15.60 | −4.18 | −14.91 | −1.13 | −3.28 | −0.01 |
| Tyr 572 | −14.14 | −4.34 | −16.75 | −1.79 | −10.79 | −0.60 | −13.48 | −2.27 |
| Phe 577 | −3.03 | −1.63 | −8.85 | −1.12 | −7.69 | 0.78 | −8.41 | −1.38 |
| Ser 602 | −4.40 | −29.12 | −10.02 | −17.11 | −9.37 | −21.23 | −3.26 | −3.46 |
| GLY 603 | −8.22 | −0.26 | −9.38 | −0.64 | −9.59 | 0.17 | −1.84 | 0.39 |
| Total | −171.79 | −334.88 | −203.58 | −139.98 | −175.77 | −74.57 | −83.42 | −41.82 |
| Total LJ_SR and Coul-SR | −506.67 | −343.56 | −250.34 | −125.24 | ||||
| Protein Structure | Ligand Name | Resolution | Year | Activity (IC50) | Binding Free Energy (kcal/mol) |
|---|---|---|---|---|---|
| 4L7B | 1VV | 2.41 Å | 2013 | 0.75µM | −4.35 |
| 5CGJ | 51M | 3.36 Å | 2015 | 0.14µM | −4.55 |
| 4XMB | 41P | 2.43 Å | 2015 | 61nM | −13.48 |
| 5FNU | L6I | 1.78 Å | 2016 | 15nM | −9.80 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Londhe, A.M.; Gadhe, C.G.; Lim, S.M.; Pae, A.N. Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques. Molecules 2019, 24, 4085. https://doi.org/10.3390/molecules24224085
Londhe AM, Gadhe CG, Lim SM, Pae AN. Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques. Molecules. 2019; 24(22):4085. https://doi.org/10.3390/molecules24224085
Chicago/Turabian StyleLondhe, Ashwini Machhindra, Changdev Gorakshnath Gadhe, Sang Min Lim, and Ae Nim Pae. 2019. "Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques" Molecules 24, no. 22: 4085. https://doi.org/10.3390/molecules24224085
APA StyleLondhe, A. M., Gadhe, C. G., Lim, S. M., & Pae, A. N. (2019). Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques. Molecules, 24(22), 4085. https://doi.org/10.3390/molecules24224085