Synthesis, Spectroscopy and Electrochemistry in Relation to DFT Computed Energies of Ferrocene- and Ruthenocene-Containing β-Diketonato Iridium(III) Heteroleptic Complexes. Structure of [(2-Pyridylphenyl)2Ir(RcCOCHCOCH3]

 , , , and
, , , and
Abstract
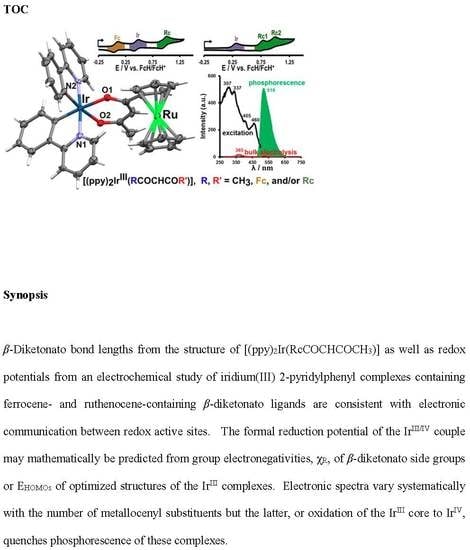
1. Introduction
2. Results and Discussion
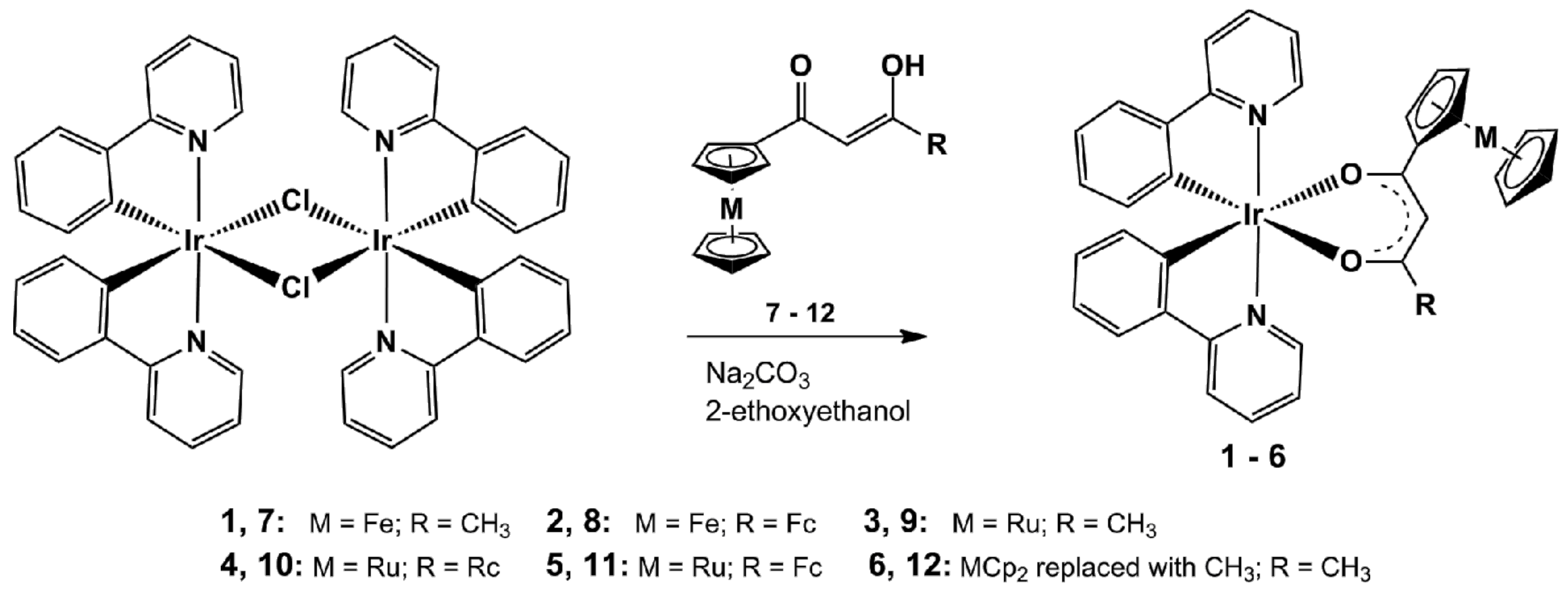
2.1. Synthesis
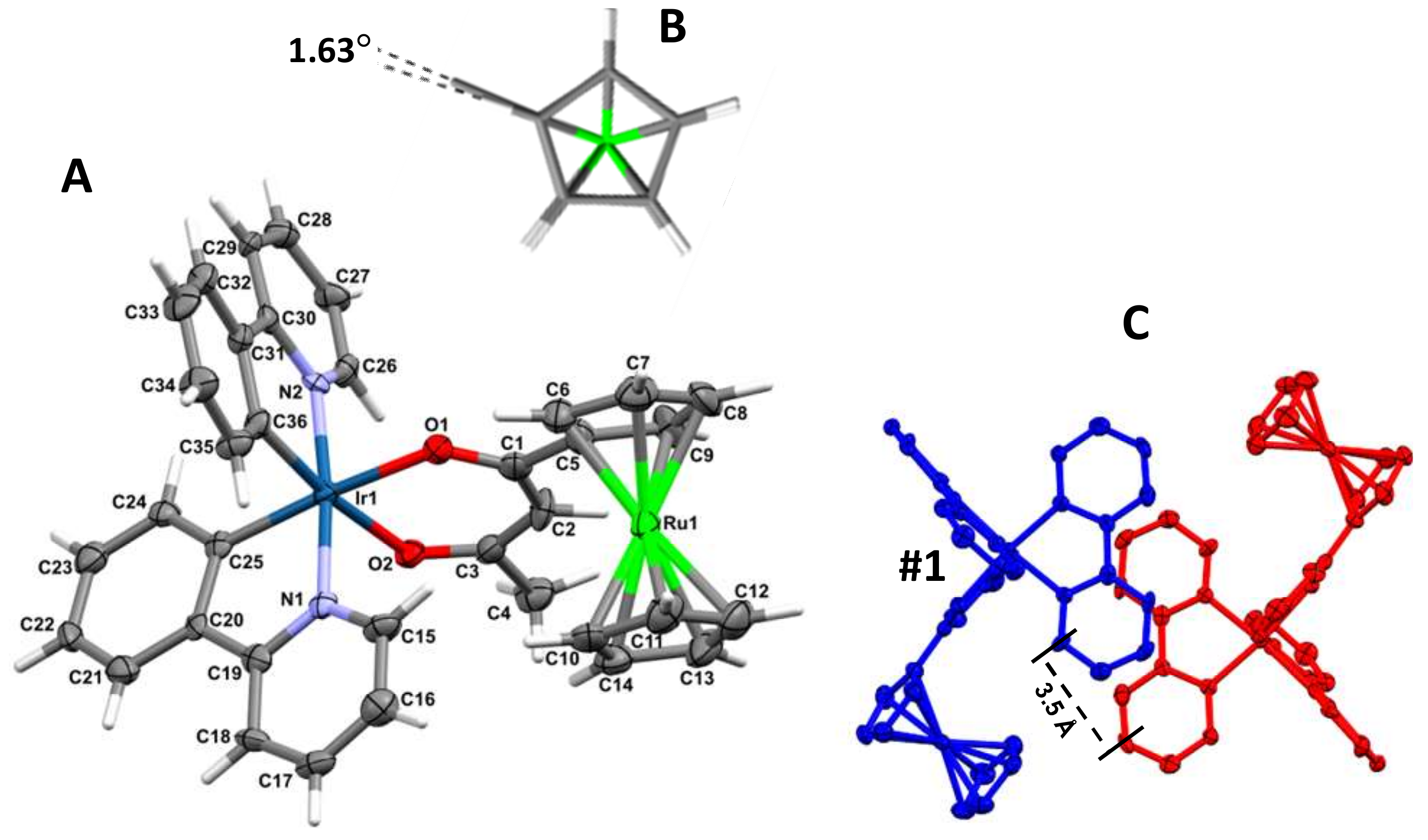
2.2. Single Crystal X-Ray Structure of 3
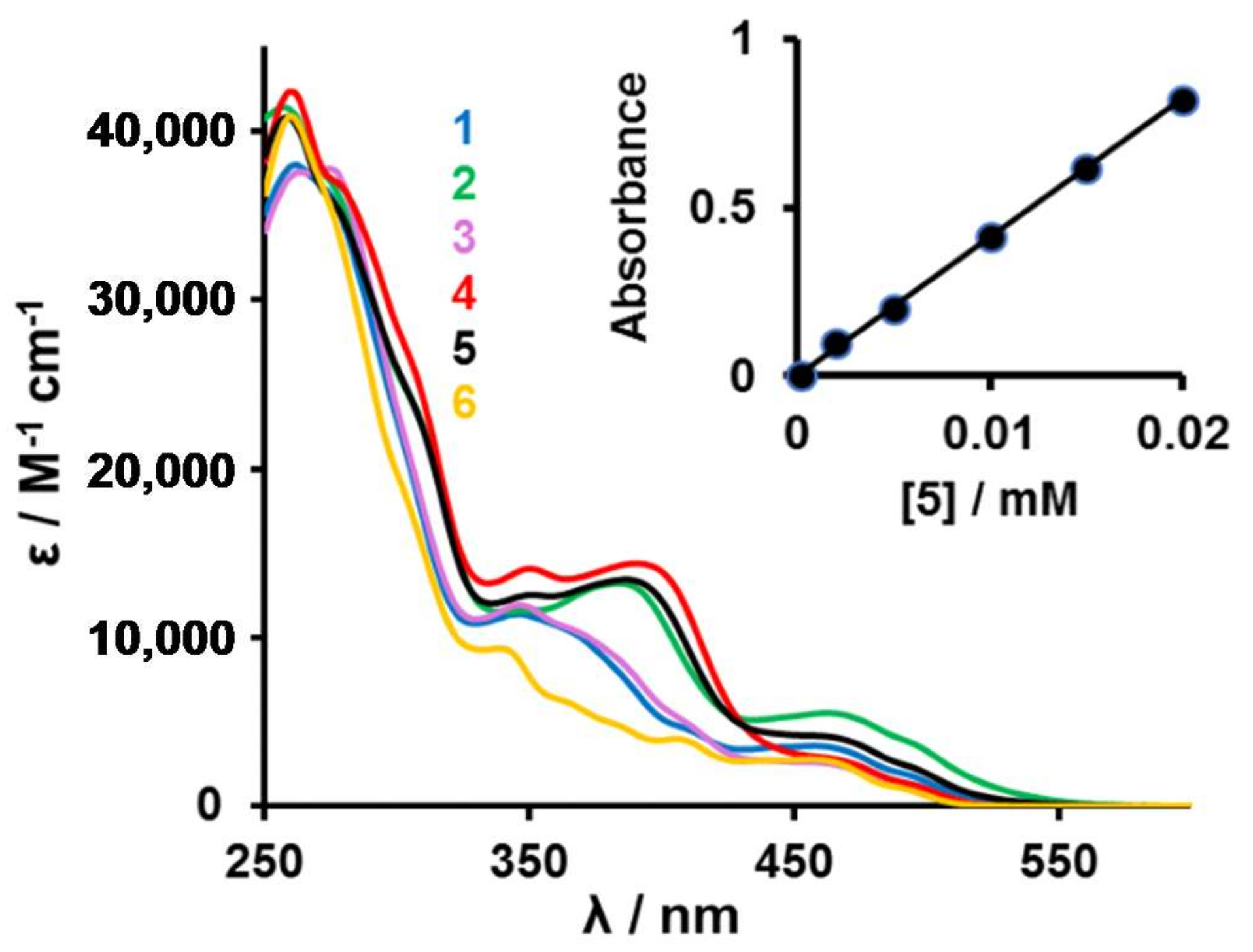
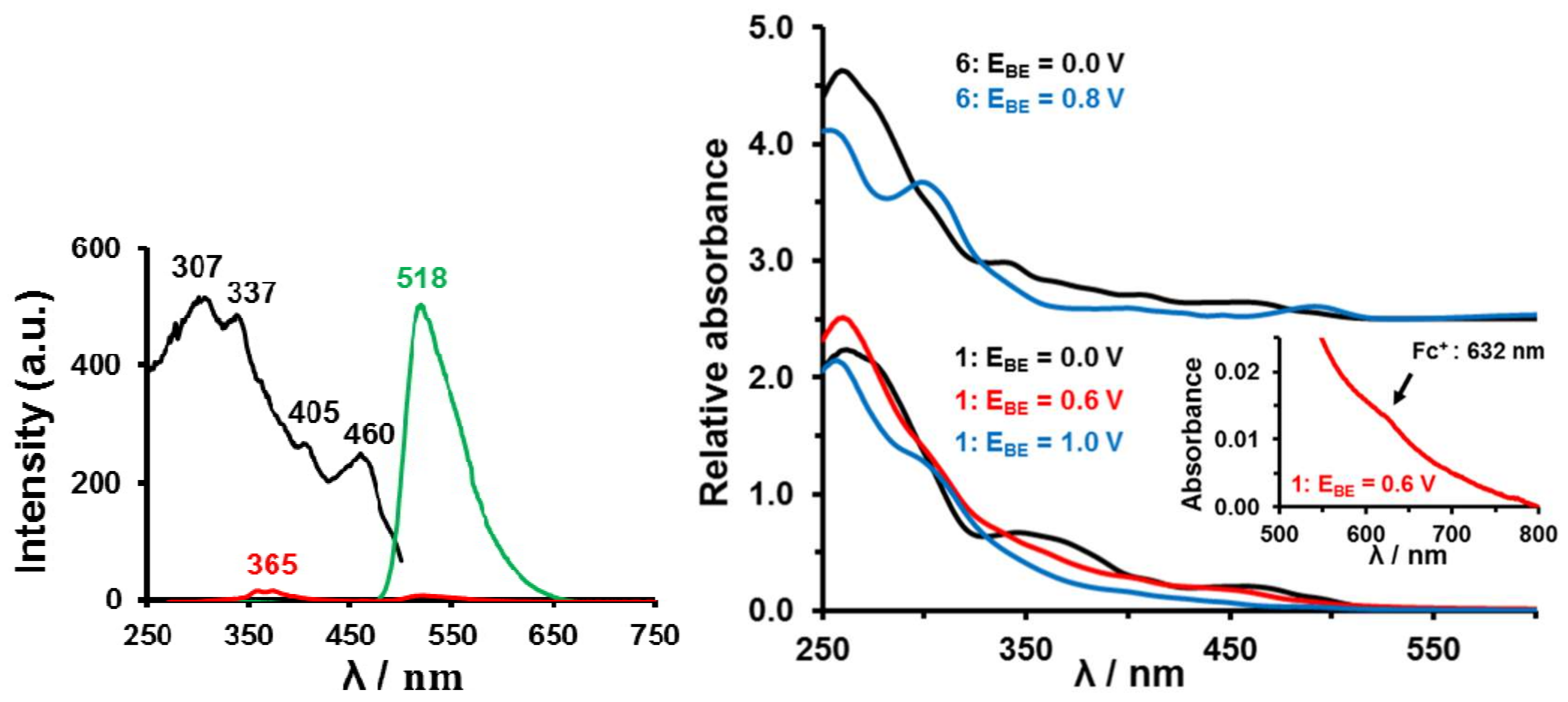
2.3. Spectroscopic Studies
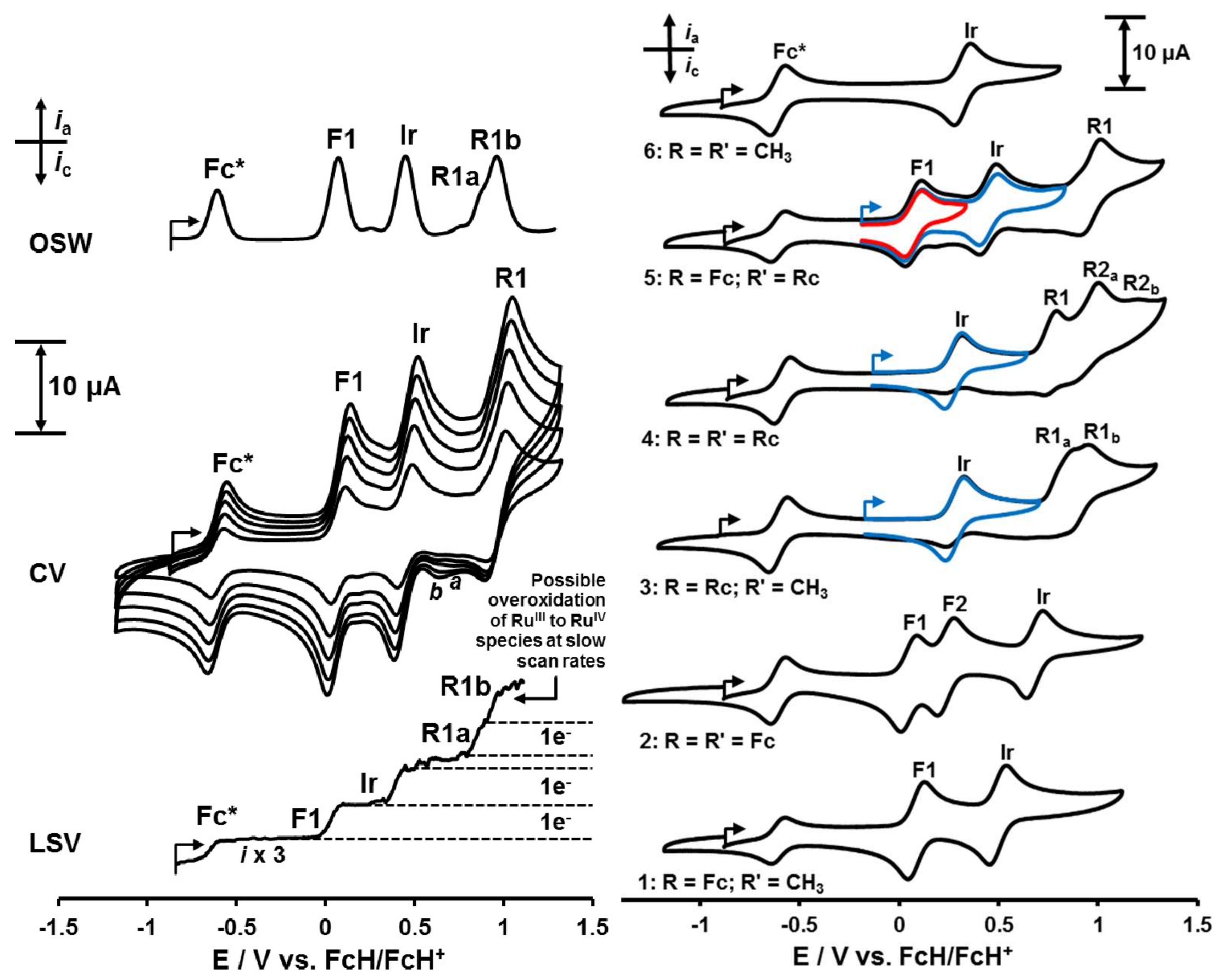
2.4. Electrochemistry
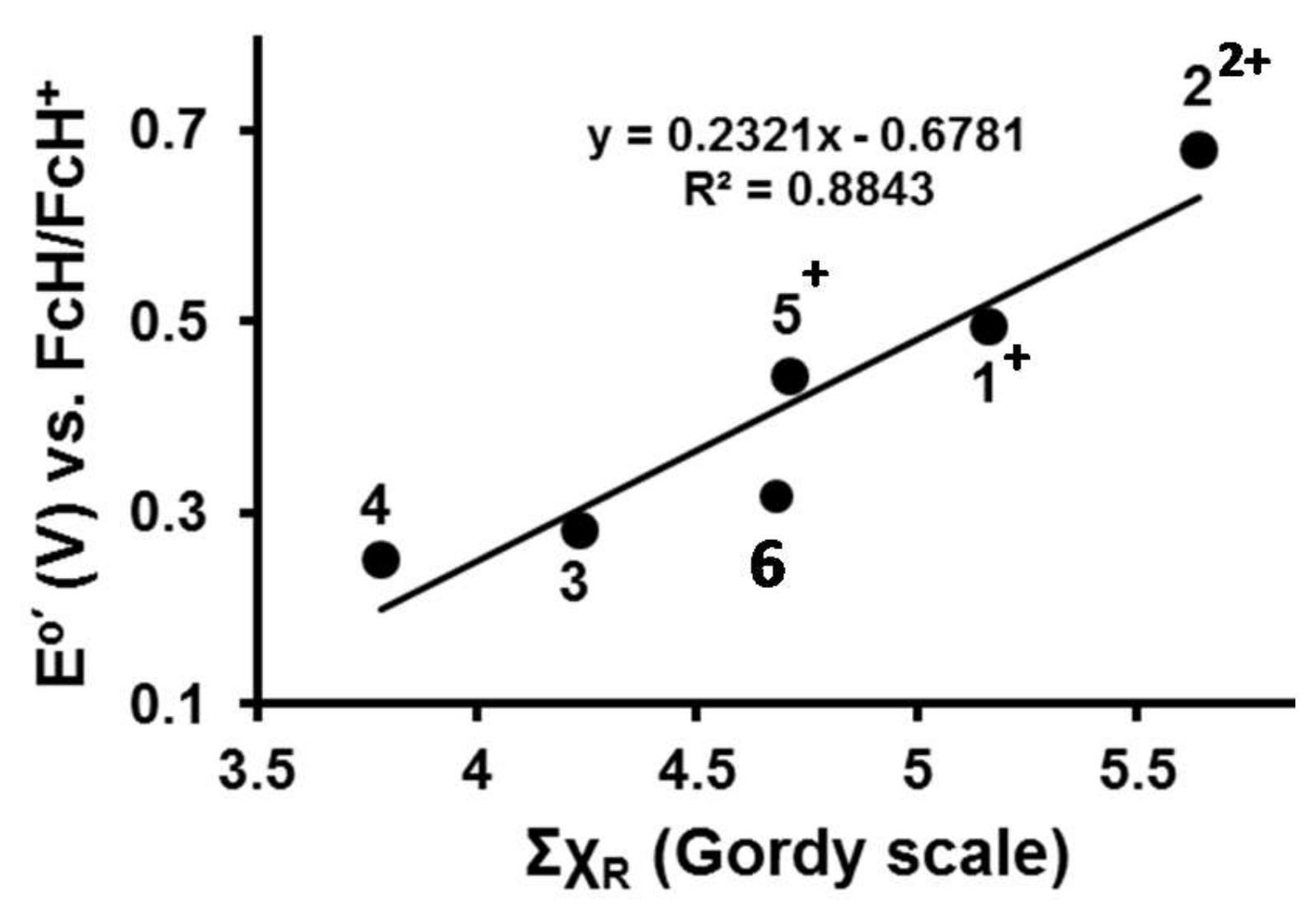
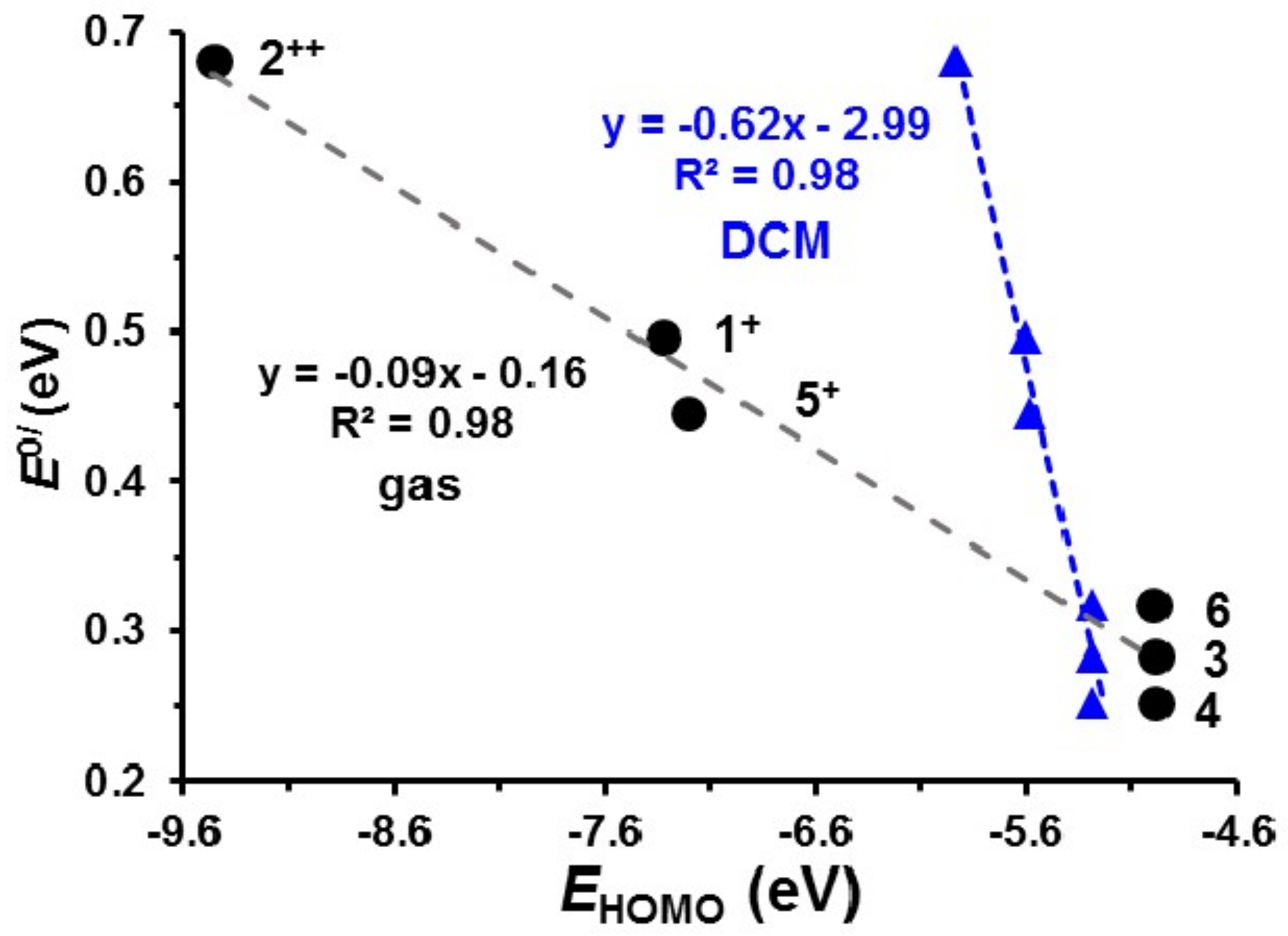
2.5. Quantification of the Relationship between ΣχR and Eo′
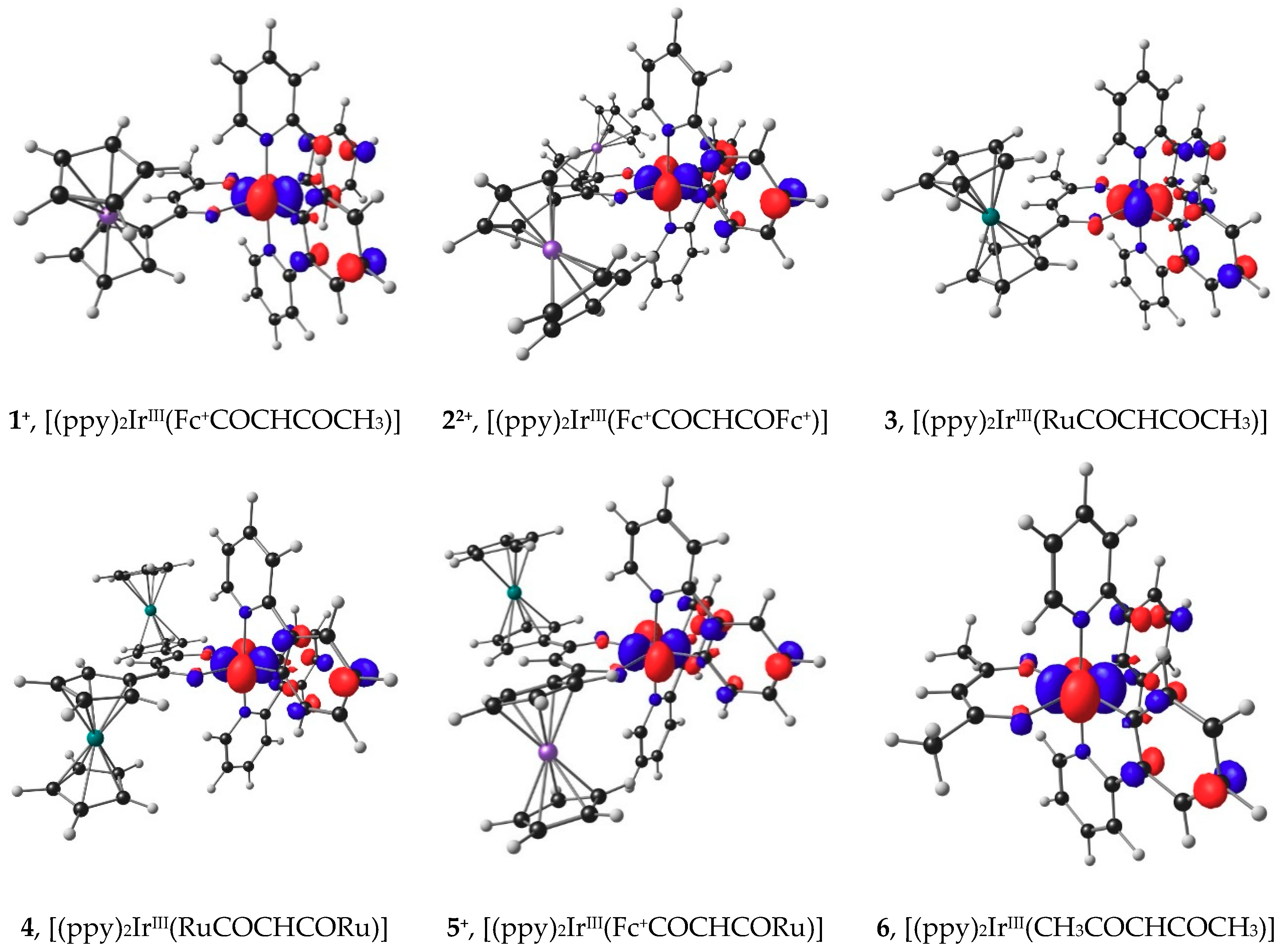
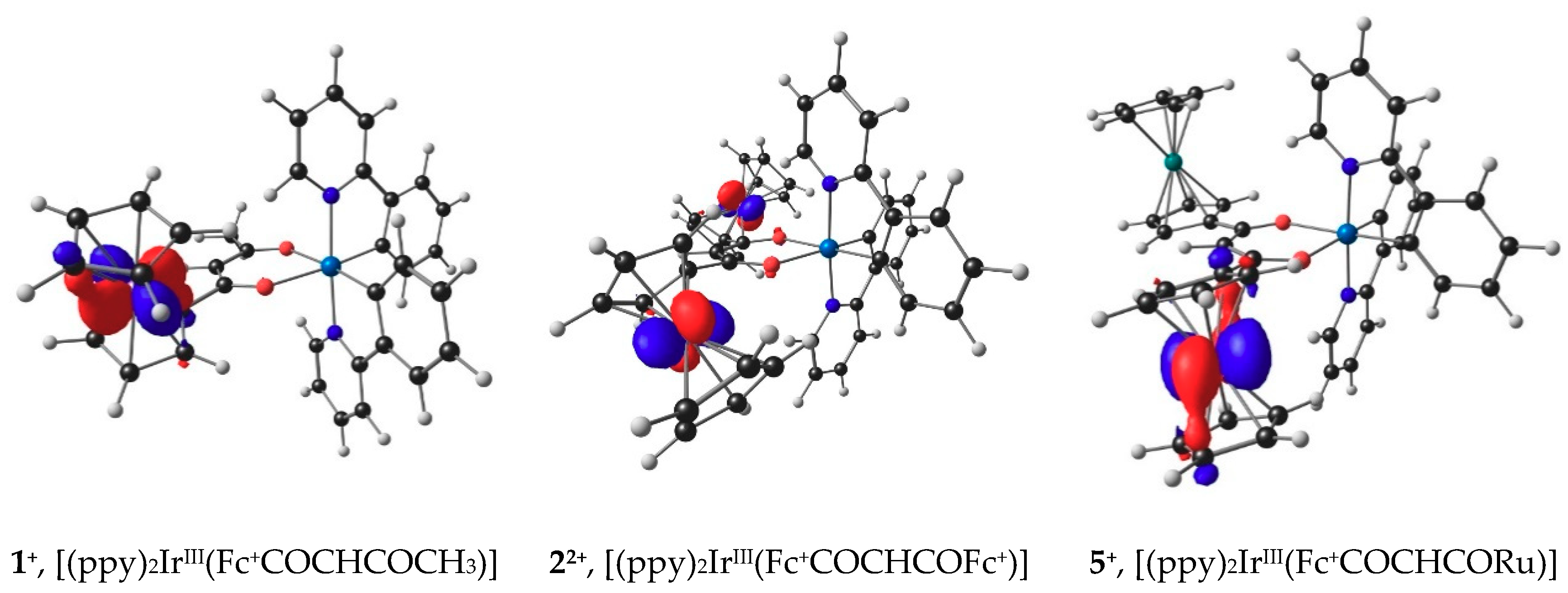
2.6. DFT view of Iridium Oxidation
2.7. Phosphorescence and Spectroelectrochemistry
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.3. Characterization Data
3.4. Crystal Structure Determination of 3
3.5. Electrochemistry
3.6. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Jones, J.H. The Cativa™ Process for the Manufacture of Acetic Acid. Platin. Met. Rev. 2000, 44, 94–105. [Google Scholar]
- Sunley, G.J.; Watson, D.J. High productivity methanol carbonylation catalysis using iridium—The Cativa™ process for the manufacture of acetic acid. Catal. Today 2000, 58, 293–307. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Van der Ham, C.J.M.; Diaz-Morales, O.; Verhoefen, M.W.G.M.; Longo, A.; Banerjee, D.; Niemantsverdriet, J.W.; Reek, J.N.H.; Feiters, M.C. Early stages of catalyst aging in the iridium mediated water oxidation reaction. Phys. Chem. Chem. Phys. 2016, 18, 10931–10940. [Google Scholar] [CrossRef]
- Sapountzi, F.M.; Gracia, J.M.; Westrate, C.J.; Fredriksson, H.O.A.; Niemantsverdriet, J.W. Electrocatalysts for the generation of hydrogen, oxygen and synthesis gas. Prog. Energy Combust. Sci. 2017, 58, 1–35. [Google Scholar] [CrossRef]
- Bennett, M.A.; Mitchell, T.R.B. γ-Carbon-bonded 2,4-pentanedionato complexes of trivalent iridium. Inorg. Chem. 1976, 15, 2936–2938. [Google Scholar] [CrossRef]
- Collins, J.E.; Castellani, M.; Rheingold, A.L.; Miller, E.J.; Geiger, W.E.; Rieger, A.L.; Rieger, P.H. Synthesis, characterization, and molecular structure of bis (tetraphenylcyclopentdienyl) rhodium (II). Organometallics 1995, 14, 1232–1238. [Google Scholar] [CrossRef]
- Han, F.-Q.; Han, C.-M.; Xu, H. Recent progress in functionalized electrophosphorescent iridium (III) complexes. Chin. Chem. Lett. 2016, 27, 1193–1200. [Google Scholar] [CrossRef]
- Kwong, R.C.; Sibley, S.; Dubovoy, T.; Balbo, M.A.; Forrest, S.R.; Thompson, M.E. Efficient, saturated red organic light emitting devices based on phosphorescent platinum (II) porphyrins. Chem. Mater. 1999, 11, 3709–3713. [Google Scholar] [CrossRef]
- Tung, Y.-L.; Wu, P.-C.; Liu, C.-S.; Chi, Y.; Yu, J.-K.; Hu, J.-K.; Chou, P.-T.; Peng, S.-M.; Lee, G.-H.; Tao, Y.; et al. Efficient red phosphorescent osmium (II) complexes for OLED applications. Organometallics 2004, 23, 3745–3748. [Google Scholar] [CrossRef]
- Ragni, R.; Maiorano, V.; Pugliese, M.; Maggiore, A.; Orselli, E.; Babudri, F.; Gigli, G.; De Cola, L.; Farinola, G.M. A highly fluorinated iridium complex as a blue-green emitting component for white electroluminescence. Synth. Met. 2017, 227, 148–155. [Google Scholar] [CrossRef]
- Holder, E.; Langeveld, B.M.W.; Schubert, U.S. New trends in the use of transition metal-ligand complexes for applications in electroluminescent devices. Adv. Mater. 2005, 17, 1109–1121. [Google Scholar] [CrossRef]
- Baldo, M.A.; O’Brien, D.F.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M.E.; Forrest, S.R. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 1998, 395, 151–154. [Google Scholar] [CrossRef]
- Du Plessis, W.C.; Erasmus, J.J.C.; Lamprecht, G.J.; Conradie, J.; Cameron, T.S.; Aquino, M.A.S.; Swarts, J.C. Cyclic voltammetry of ferrocene-containing β-diketones as a tool to obtain group electronegativities. The structure of 3-ferrocenoyl-1,1,1-trifluoro-2-hydroxyprop-2-ene. Can. J. Chem. 1999, 77, 378–386. [Google Scholar] [CrossRef]
- Swarts, J.C.; Vosloo, T.G.; Cronje, S.J.; Du Plessis, W.C.; Van Rensburg, C.E.J.; Kreft, E.; Van Lier, J.E. Cytotoxicity of a series of ferrocene-containing beta-diketones. Anticancer Res. 2008, 28, 2781–2784. [Google Scholar] [PubMed]
- Kemp, K.C.; Fourie, E.; Conradie, J.; Swarts, J.C. Ruthenocene-containing β-diketones: Synthesis, pKa′ values, keto-enol isomerization kinetics, and electrochemical aspects. Organometallics 2008, 27, 353–362. [Google Scholar] [CrossRef]
- Kemp, K.C.; Nell, M.J.; Van Rensburg, C.E.J.; Swarts, J.C. Cytotoxicity of ruthenocene-containing β-diketones. Anticancer Res. 2012, 32, 2915–2918. [Google Scholar]
- Bryant, J.R.; Taves, J.E.; Mayer, J.M. Oxidations of hydrocarbons by manganese (III) tris (hexafluoroacetylacetonate). Inorg. Chem. 2002, 41, 2769–2776. [Google Scholar] [CrossRef]
- Lamprecht, G.J.; Swarts, J.C.; Conradie, J.; Leipoldt, J.G. Structure of carbonyl[(ferrocenecarbonyl)trifluoroacetonato-κO,κO]triphenylphosphinerhodium (I). Acta Cryst. 1993, C49, 82–84. [Google Scholar] [CrossRef]
- Erasmus, E.; Conradie, J.; Muller, A.; Swarts, J.C. Synthesis, crystal structure and electrochemistry of tetrahedral mono-β-diketonato titanocenyl complexes. Inorg. Chim. Acta 2007, 360, 2277–2283. [Google Scholar] [CrossRef]
- Imai, H.; Yoshikatsu, Y. Syntheses of Acetoacetylferrocene and Its Metal Chelates. Nippon Kagaku Zasshi 1970, 91, 452–457. [Google Scholar] [CrossRef]
- Atim, S.; Yang, L.; Nesterov, V.; Wang, X.; Richmond, M.G. Synthesis of the labile rhenium (I) complexes fac-Re(CO)3(L)[k2-O,O-FcC(O)CHC(O)Me] (where Fc = ferrocenyl; L = THF, H2O, alkyne) and alkyne addition to the diketonate ligand. J. Organomet. Chem. 2018, 874, 87–100. [Google Scholar] [CrossRef]
- Obuah, C.; Ainooson, M.K.; Darkwa, J. Effects of electrochemical properties of ferrocenylpyrazolylnickel (II) and palladium (II) compounds on their catalytic activities in ethylene oligomerisation reactions. RSC Adv. 2018, 8, 5362–5371. [Google Scholar] [CrossRef]
- Koroteev, P.S.; Dobrokhotova, Z.V.; Ilyukhin, A.B.; Efimov, N.N.; Rouzières, M.; Kiskin, M.A.; Clérac, R.; Novotortsev, V.M. Synthesis, structure, and physical properties of new rare earth ferrocenoylacetonates. Dalton Trans. 2016, 45, 6405–6417. [Google Scholar] [CrossRef] [PubMed]
- Koroteev, P.S.; Dobrokhotova, Z.V.; Ilyukhin, A.B.; Efimov, N.N.; Novotortsev, V.M. Synthesis, structure, and magnetic properties of lanthanide ferrocenoylacetonates with nitrate and 2,2′-bipyridine ligands. J. Coord. Chem. 2016, 69, 2723–2735. [Google Scholar] [CrossRef]
- Buitendach, B.E.; Gągor, A.; Swarts, J.C. Electrochemical Evidence of Intramolecular Electronic Communication in Zr and Hf Phthalocyanines Bearing Ferrocene-Containing β-Diketonato Axial Ligands: Structure of [PcHf(FcCOCHCOC6H5)2]. Inorg. Chem. 2013, 52, 10245–10257. [Google Scholar] [CrossRef]
- Gericke, H.J.; Muller, A.J.; Swarts, J.C. Electrochemical illumination of intramolecular communication in ferrocene-containing tris-β-diketonato aluminum (III) complexes; cytotoxicity of Al(FcCOCHCOCF3)3. Inorg. Chem. 2012, 51, 1552–1561. [Google Scholar] [CrossRef]
- Buitendach, B.E.; Erasmus, E.; Landman, M.; Niemantsverdriet, J.W.; Swarts, J.C. Consequences of electron-density manipulations on the X-ray photoelectron spectroscopic properties of ferrocenyl-β-diketonato complexes of manganese (III). Structure of [Mn(FcCOCHCOCH3)3]. Inorg. Chem. 2016, 55, 1992–2000. [Google Scholar] [CrossRef]
- Joubert, C.C.; Van As, L.; Jakob, A.; Speck, M.J.; Lang, H.; Swarts, J.C. Intramolecular electronic communication in ferrocene-based β-diketonato copper (II) complexes as observed by an electrochemical study. Polyhedron 2013, 55, 80–86. [Google Scholar] [CrossRef]
- Conradie, J.; Swarts, J.C. Oxidative addition of CH3I and CO migratory insertion in a series of ferrocene-containing carbonyl phosphine betadiketono rhodium (I) complexes. Organometallics 2009, 28, 1018–1026. [Google Scholar] [CrossRef]
- Vosloo, T.G.; du Plessis, W.C.; Swarts, J.C. Kinetics of substitution of ferrocenyl-containing β-diketonatoligands by phenanthroline fromβ-diketonato-1,5-cyclooctadienerhodium (I) complexes. Inorg. Chim. Acta 2002, 331, 188–193. [Google Scholar] [CrossRef]
- Conradie, J. A DFT study of the reactivity of β-diketonato-1,5-cyclo-octadieneiridium(I) complexes. Polyhedron 2013, 51, 164–167. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Kwong, R.; Tsyba, I.; Bortz, M.; Mui, B.; Thompson, M.E. Synthesis and Characterization of Phosphorescent Cyclometalated Iridium Complexes. Inorg. Chem. 2001, 40, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Chepelin, O.; Ujma, J.; Wu, X.; Slawin, A.M.Z.; Pitak, M.B.; Coles, S.J.; Michel, J.; Jones, A.C.; Barran, P.E.; Lusby, P.J. Luminescent, Enantiopure, Phenylatopyridine Iridium-Based Coordination Capsules. J. Am. Chem. Soc. 2012, 134, 19334–19337. [Google Scholar] [CrossRef] [PubMed]
- Takayasu, S.; Suzuki, T.; Shinozaki, K. Intermolecular Interactions and Aggregation of fac-Tris(2-phenylpyridinato-C2,N)iridium (III) in Nonpolar Solvents. J. Phys. Chem. B 2013, 117, 9449–9456. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Davies, J.E.; Galloy, J.J.; Johnson, O.; Kennard, O.; Macrae, C.F.; Mitchell, E.M.; Mitchell, G.F.; Smith, J.M.; Watson, D.G. The development of versions 3 and 4 of the Cambridge Structural Database System. J. Chem. Inf. Comput. Sci. 1991, 31, 187–204. [Google Scholar] [CrossRef]
- Du Plessis, W.C.; Davis, W.L.; Cronje, S.J.; Swarts, J.C. Structural, thermodynamic and kinetic consequences of a spectroscopic study of the equilibrium between isomeric forms of ferrocene-containing β-diketones. Inorg. Chim. Acta 2001, 314, 97–104. [Google Scholar] [CrossRef]
- Von Chrzanowski, L.S.; Lutz, M.; Spek, A.L. γ-Tris(2,4-pentanedionato-κ2O,O′) aluminium (III) at 110 K. Acta Cryst. 2006, E62, 3318–3320. [Google Scholar] [CrossRef]
- Dulatas, L.T.; Brown, S.N.; Ojomo, E.; Noll, B.C.; Cavo, M.J.; Holt, P.B.; Wopperer, M.M. Intermetallic Communication in Titanium (IV) Ferrocenyldiketonates. Inorg. Chem. 2009, 48, 10789–10799. [Google Scholar] [CrossRef]
- Woisetschlager, O.E.; Geisbauer, A.; Polborn, K.; Beck, W.Z. Kohlenwasserstoffverbrückte Metallkomplexe. XLIX Koordinationschemie von bis (ferrocenyl) substituierten 1,3-Diketonaten mit Ruthenium, Rhodium, Iridium und Palladium. Anorg. Allg. Chem. 2000, 626, 766–774. [Google Scholar] [CrossRef]
- Bell, W.; Crayston, J.A.; Glidewell, C.; Mazid, M.A.; Hursthause, M.B. The constitution of a ferrocenyl diketone: Solution and solid state NMR spectroscopy. Crystal structure of 1-ferrocenyl-3-hydroxybut-2-en-1-one. J. Organomet. Chem. 1992, 434, 115–121. [Google Scholar] [CrossRef]
- Woisetschläger, O.E.; Geisbauer, A.; Polborn, K.; Sünkel, K.; Beck, W.Z. Spacer-verbrückte Bis-, Tris-und Tetrakis (ferrocenyl)-1,3-Diketone. Anorg. Allg. Chem. 1999, 625, 2164–2168. [Google Scholar] [CrossRef]
- Chang, C.-J.; Yang, C.-H.; Chen, K.; Chi, Y.; Shu, C.-F.; Ho, M.-L.; Yeh, Y.-S.; Chou, P.-T. Color tuning associated with heteroleptic cyclometalated Ir (III) complexes: Influence of the ancillary ligand. Dalton Trans. 2007, 36, 1881–1890. [Google Scholar] [CrossRef]
- Swarts, J.C.; Nafady, A.; Roudebush, J.H.; Trupia, S.; Geiger, W.E. One-Electron Oxidation of Ruthenocene: Reactions of the Ruthenocenium Ion in Gentle Electrolyte Media. Inorg. Chem. 2009, 48, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Barrière, F.; Geiger, W.E. Use of Weakly Coordinating Anions to Develop an Integrated Approach to the Tuning of ΔE1/2 Values by Medium Effects. J. Am. Chem. Soc. 2006, 128, 3980–3989. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.J.; Chambrier, I.; White, G.F.; Fourie, E.; Swarts, J.C. Electrochemical and EPR studies of two substituted bis-cadmium tris-phthalocyanine complexes: Elucidation of unexpectedly different free-radical character. Dalton Trans. 2009, 7, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, P.T.; Heineman, W.R. Cyclic Voltammetry. J. Chem. Educ. 1983, 60, 702–706. [Google Scholar] [CrossRef]
- Fourie, E.; Swarts, J.C.; Chambrier, I.; Cook, M.J. Electrochemical and spectroscopic detection of self-association of octa-alkyl phthalocyaninato cadmium compounds into dimeric species. Dalton Trans. 2009, 7, 1145–1154. [Google Scholar] [CrossRef]
- Gericke, H.J.; Barnard, N.I.; Erasmus, E.; Swarts, J.C.; Cook, M.J.; Aquino, M.A.S. Solvent and Electrolyte Effects in Enhancing the Identification of Intramolecular Electronic Communication in a Multi Redox-Active Diruthenium Tetraferrocenoate Complex, a Triple-Sandwiched Dicadmium Phthalocyanine and a Ruthenocene-Containing β-Diketone. Inorg. Chim. Acta 2010, 363, 2222–2232. [Google Scholar] [CrossRef]
- Cohen, S.G. Gordy Scale Group Electronegativities, χR, Are Empirical Numbers that Express the Combined Tendency of a Group of Atoms, Like R = CF3 or ferrocenyl (Fc), to Attract Electrons (Including Those in a Covalent Bond) as a Function of the Number of Valence Electrons, n, and the Covalent Radius, r (Å), of Groups as Discussed in Reference 62 and also by Wells, P.R. In Progress in Physical Organic Chemistry; John Wiley & Sons, Inc.: New York, NY, USA, 1968; Volume 6, pp. 111–145. [Google Scholar]
- Kagarise, R.E. Relationship Between the Electronegativities of Adjacent Substituents and the Stretching Frequency of the Carbonyl Group. J. Am. Chem. Soc. 1955, 77, 1377–1379. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Hill, M.G.; Lamanna, W.M.; Mann, K.R. Tetrabutylammonium tetrakis [3,5-bis(trifluoromethyl)phenyl]borate as a noncoordinating electrolyte: Reversible 1e- oxidations of ruthenocene, osmocene, and Rh2 (TM4)42+ (TM4 = 2,5-diisocyano-2,5-dimethylhexane). Inorg. Chem. 1991, 30, 4687–4690. [Google Scholar] [CrossRef]
- Conradie, J. A Frontier orbital energy approach to redox potentials. J. Phys. Conf. Ser. 2015, 633, 12045–12050. [Google Scholar] [CrossRef]
- Ferreira, H.; Conradie, M.M.; Conradie, J. Electrochemical properties of a series of Co (II) complexes, containing substituted phenanthrolines. Electrochim. Acta 2018, 292, 489–501. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.-E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly phosphorescent bis-cyclometalated iridium complexes: Synthesis, photophysical characterization, and use in organic light emitting diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Traverso, O.; Rossi, R.; Magon, L.; Cinquantini, A.; Kemp, T.J. The quenching of excited uranyl ion by d 6 metallocenes. J. Chem. Soc. Dalton Trans. 1978, 6, 569–572. [Google Scholar] [CrossRef]
- Erasmus, J.J.C.; Lamprecht, G.J.; Swarts, J.C.; Roodt, A.; Oskarsson, Å. (E)-1,3-Diferrocenyl-2-buten-1-one-Water (4/1). Acta Cryst. 1996, C52, 3000–3002. [Google Scholar] [CrossRef]
- LeSuer, R.J.; Buttolph, C.; Geiger, W.E. Comparison of the Conductivity Properties of the Tetrabutylammonium Salt of Tetrakis (pentafluorophenyl) borate Anion with Those of Traditional Supporting Electrolyte Anions in Nonaqueous Solvents. Anal. Chem. 2004, 76, 6395–6401. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEXII and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. Shelxt—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Ruiz Aranzaes, J.; Daniel, M.-C.; Astruc, D. Metallocenes as references for the determination of redox potentials by cyclic voltammetry: Permethylated iron and cobalt sandwich complexes, inhibition by polyamine dendrimers and the role of hydroxyl-containing ferrocenes. Can. J. Chem. 2006, 84, 288–299. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| empirical formula | C36.33H29.66Cl0.66IrN2O2Ru | absorption coeff. (mm−1) | 4.893 |
| molecular weight | 843.12 | θ range for data collection (deg) | 2.304–25.349 |
| crystal size (mm3) | 0.348 × 0.334 × 0.058 | index ranges | −17 ≤ h ≤ 17 |
| temperature (K) | 150(2) | −11 ≤ k ≤ 11 | |
| wavelength (Å) | 0.71073 | −26 ≤ l ≤ 26 | |
| crystal system | monoclinic | no. of reflections collected | 65183 |
| space group | P21/n | no. of independent reflections | 5368 |
| unit cell dim. (Å; deg) | a = 14.5533(18); α = 90 | completeness to θ = 25.00° | 96.1% |
| b = 9.6595(12); β = 95.707(4) | refinement method | full-matrix least-squares on F2 | |
| c = 22.053(3); γ = 90 | data/restraints/parameters | 5368/0/408 | |
| volume (Å3) | 3084.8(7) | goodness of fit on F2 | 1.168 |
| Z | 4 | final R indices [I > 2σ(I)] | R1 = 0.0500; wR2 = 0.1189 |
| density (calc.) (g cm−3) | 1.815 | R indices (all data) | R1 = 0.0554; wR2 = 0.1219 |
| F(000) | 1640 | largest diff. peak and hole (e Å−3) | 1.58 and −0.93 |
| Complex (χR) | λmax/nm (ε/M−1 cm−1) | Complex (χR) | λmax/nm (ε/M−1 cm−1) |
|---|---|---|---|
| 1: R = Fc (χFc = 1.87) R′ = CH3 (χCH3 = 2.34) | 262 (38016); 346 (11374); 372 (sh,b 9010); 457 (3575) | 4: R = Rc (χRc = 1.99), R′ = Rc (χRc+ = 2.87) | 260 (42334); 350 (14083); 390 (14390); 462 (2721) |
| 2: R = Fc (χFc = 1.87), R′ = Fc (χFc+ = 2.82)a | 256 (41428); 350 (11589); 384 (13205); 462 (5520) | 5: R = Fc (χFc = 1.87), a R′ = Rc (χ(Rc): 1.89) | 258 (40825); 351 (12518); 386 (13375); 465 (4076) |
| 3: R = Rc χRc = 1.99), R′ = CH3 (χCH3 = 2.34) | 262 (37568); 346 (11938); 371 (sh,b 9010); 462 (2548) | 6: R = R′ = CH3 (χCH3 = 2.34) | 260 (40897); 339 (9358); 405 (3979); 456 (2778) |
| Wave | Epa/V | ΔEp/V | Eo′/V | ipa/μA | ipc/ipa | Wave | Epa/V | ΔEp/V | Eo′/V | ipa/μA | ipc/ipa |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Decamethylferrocene, Fc* | |||||||||||
| Fc* | −0.570 | 0.077 | −0.608 | 1.80 | 1.00 | 4, [(ppy)2Ir(RcCOCHCORc)] | |||||
| IrIII/IV | 0.296 | 0.088 | 0.252 | 5.56 | 0.20 | ||||||
| 1, [(ppy)2Ir(FcCOCHCOCH3)] | R1 | 0.771 | 0.067 | 0.737 | 5.74 | 0.13 | |||||
| F1 | 0.126 | 0.081 | 0.086 | 6.03 | 1.00 | R2a | 0.984 | 0.181 | 0.893 | 5.56 | 0.30 |
| IrIII/IV | 0.539 | 0.085 | 0.497 | 5.69 | 0.97 | R2b | 1.179 | - a | - a | 0.37 | - a |
| 2, [(ppy)2Ir(FcCOCHCOFc)] | 5, [(ppy)2Ir(FcCOCHCORc)] | ||||||||||
| F1 | 0.090 | 0.080 | 0.050 | 5.96 | 0.94 | F1 | 0.114 | 0.082 | 0.073 | 5.80 | 0.55 |
| F2 | 0.277 | 0.083 | 0.235 | 5.58 | 1.00 | IrIII/IV | 0.487 | 0.083 | 0.445 | 5.40 | 0.78 |
| IrIII/IV | 0.721 | 0.082 | 0.681 | 5.77 | 1.00 | R1 | 1.013 | 0.108 | 0.959 | 5.80 | 0.76 |
| 3, [(ppy)2Ir(RcCOCHCOCH3)] | 6, [(ppy)2Ir(CH3COCHCOCH3] | ||||||||||
| IrIII/IV | 0.327 | 0.087 | 0.283 | 5.68 | 0.32 | IrIII/IV | 0.359 | 0.081 | 0.318 | 5.94 | 1.00 |
| R1a | 0.889 | 0.083 | 0.847 | 6.91 | 0.04 | ||||||
| R1b | 0.951 | - a | - a | 2.76 | - a | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buitendach, B.E.; Conradie, J.; Malan, F.P.; Niemantsverdriet, J.W.; Swarts, J.C. Synthesis, Spectroscopy and Electrochemistry in Relation to DFT Computed Energies of Ferrocene- and Ruthenocene-Containing β-Diketonato Iridium(III) Heteroleptic Complexes. Structure of [(2-Pyridylphenyl)2Ir(RcCOCHCOCH3]. Molecules 2019, 24, 3923. https://doi.org/10.3390/molecules24213923
Buitendach BE, Conradie J, Malan FP, Niemantsverdriet JW, Swarts JC. Synthesis, Spectroscopy and Electrochemistry in Relation to DFT Computed Energies of Ferrocene- and Ruthenocene-Containing β-Diketonato Iridium(III) Heteroleptic Complexes. Structure of [(2-Pyridylphenyl)2Ir(RcCOCHCOCH3]. Molecules. 2019; 24(21):3923. https://doi.org/10.3390/molecules24213923
Chicago/Turabian StyleBuitendach, Blenerhassitt E., Jeanet Conradie, Frederick P. Malan, J. W. (Hans) Niemantsverdriet, and Jannie C. Swarts. 2019. "Synthesis, Spectroscopy and Electrochemistry in Relation to DFT Computed Energies of Ferrocene- and Ruthenocene-Containing β-Diketonato Iridium(III) Heteroleptic Complexes. Structure of [(2-Pyridylphenyl)2Ir(RcCOCHCOCH3]" Molecules 24, no. 21: 3923. https://doi.org/10.3390/molecules24213923
APA StyleBuitendach, B. E., Conradie, J., Malan, F. P., Niemantsverdriet, J. W., & Swarts, J. C. (2019). Synthesis, Spectroscopy and Electrochemistry in Relation to DFT Computed Energies of Ferrocene- and Ruthenocene-Containing β-Diketonato Iridium(III) Heteroleptic Complexes. Structure of [(2-Pyridylphenyl)2Ir(RcCOCHCOCH3]. Molecules, 24(21), 3923. https://doi.org/10.3390/molecules24213923