
New Molecular Insights into the Inhibition of Dipeptidyl Peptidase-4 by Natural Cyclic Peptide Oxytocin

 , , , , and
, , , , and
Abstract
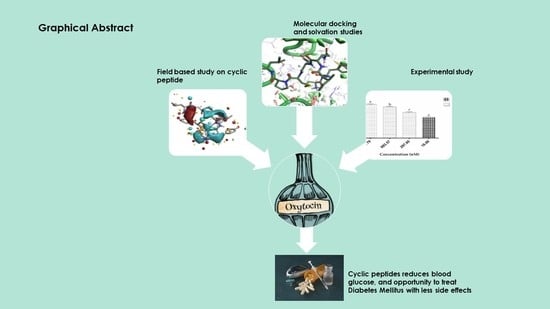
1. Introduction
2. Results and Discussion
2.1. Computational Studies
2.1.1. Activity Atlas and Field-Based Model Studies
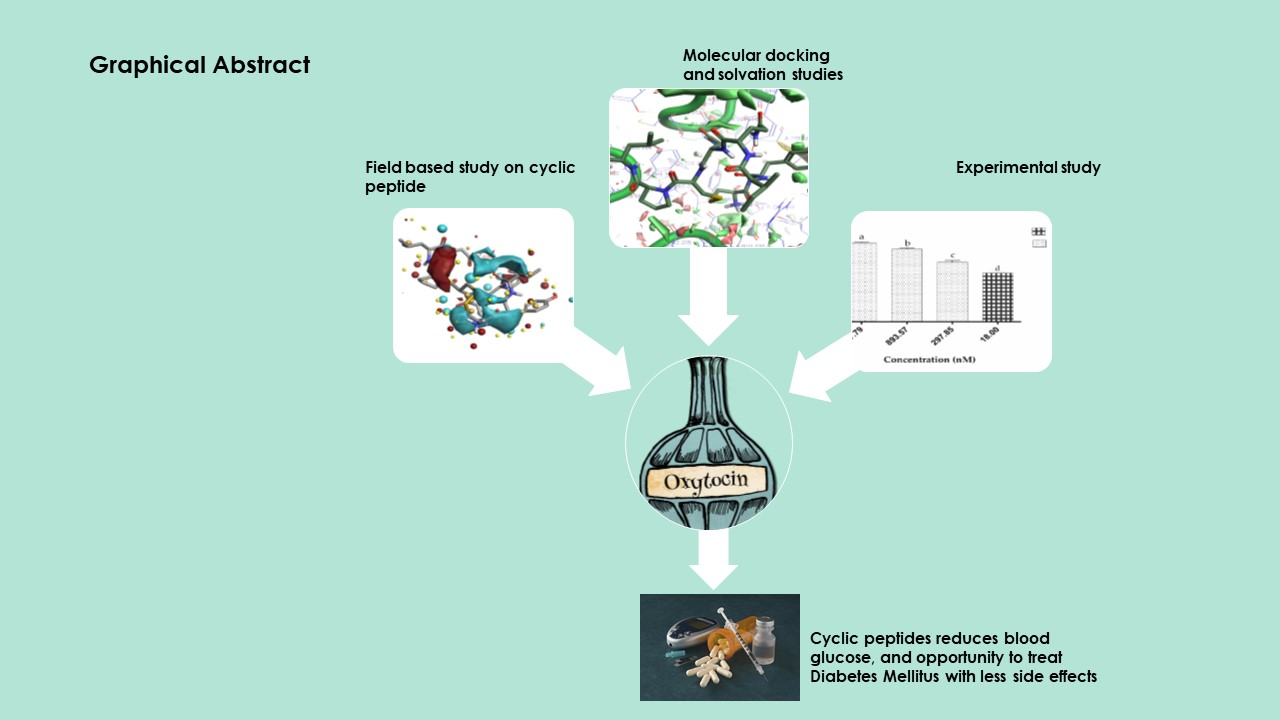
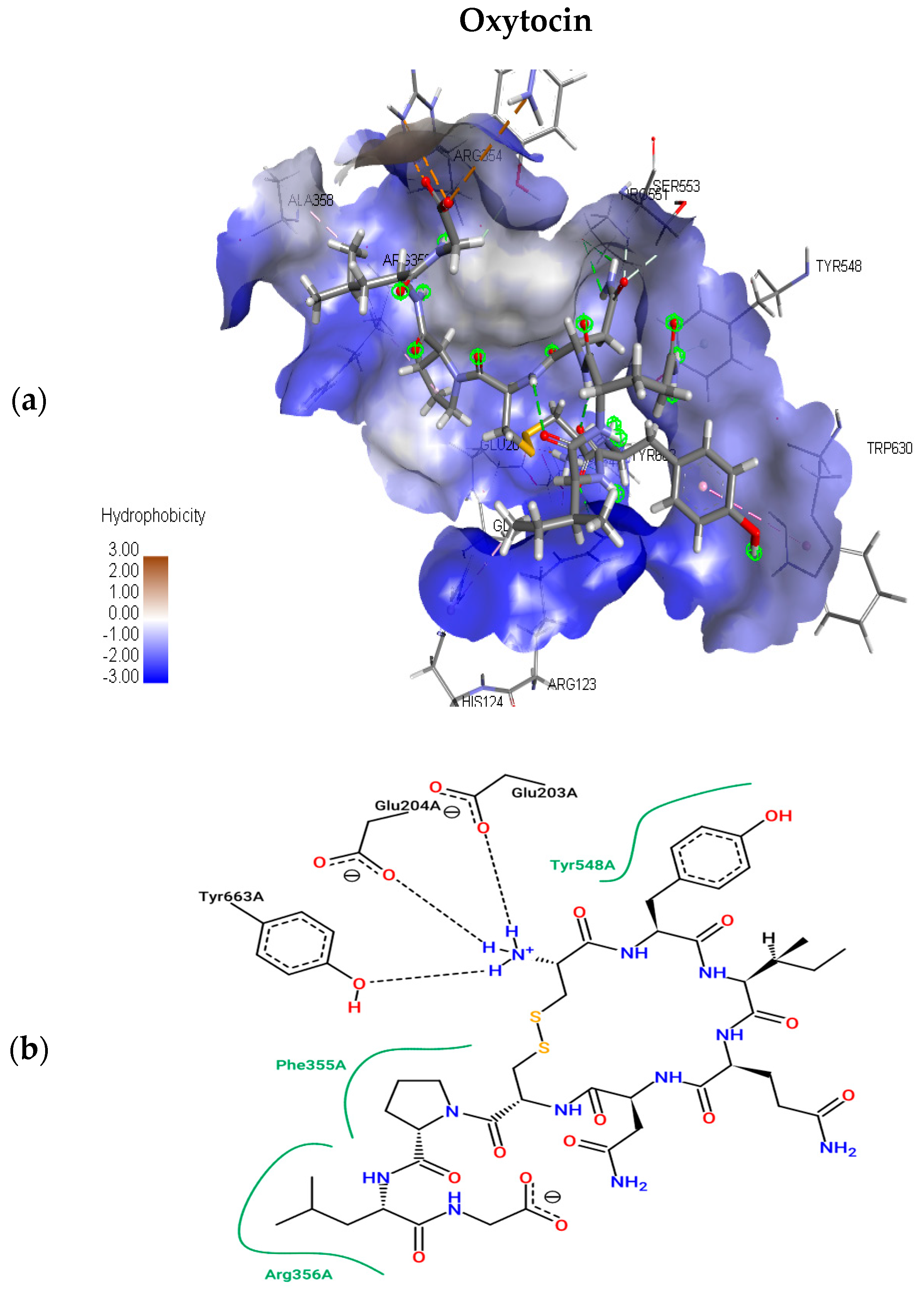
2.1.2. Molecular Docking Simulations
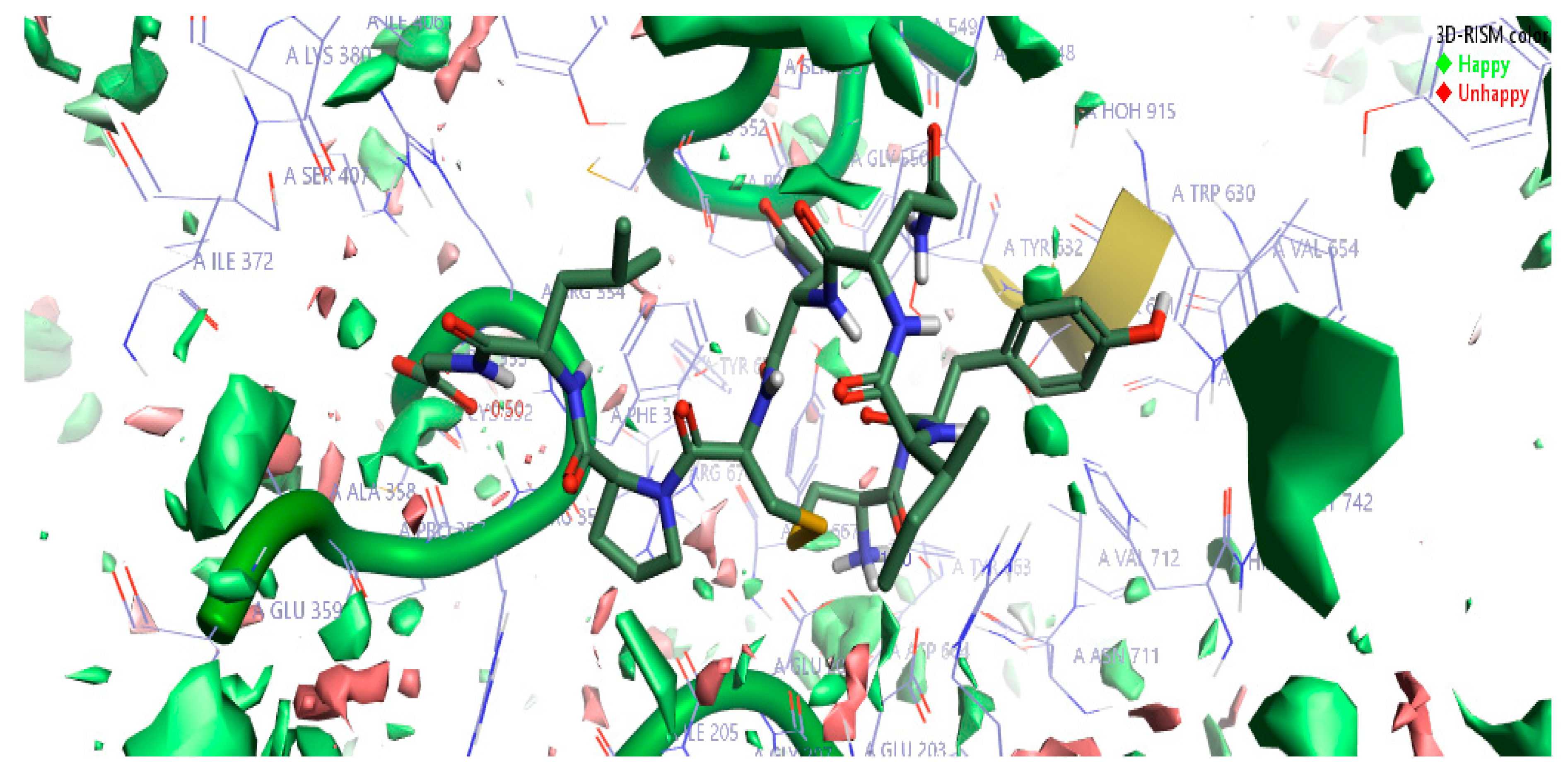
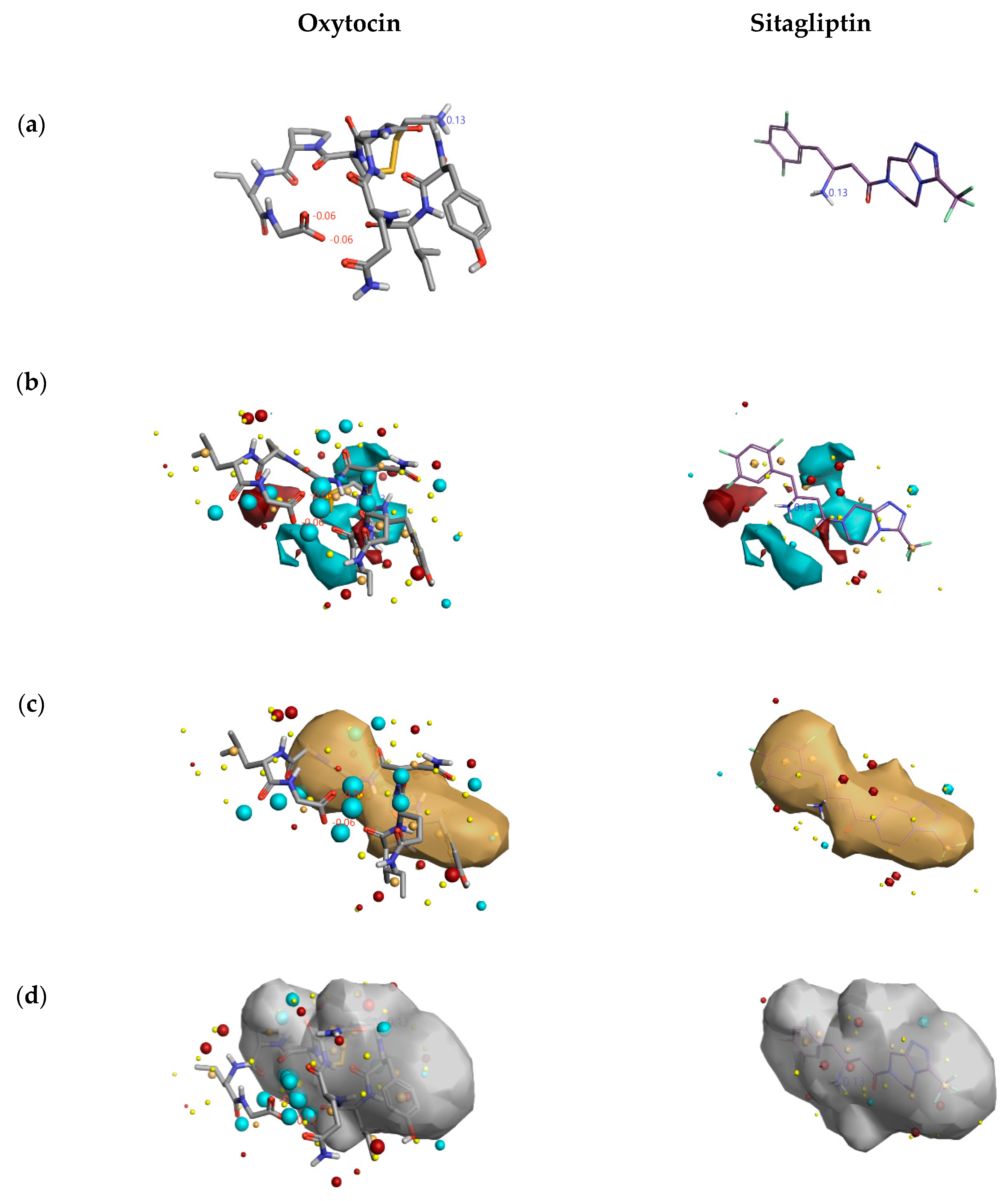
2.1.3. 3D-RISM Analysis of Oxytocin-DPP4 Complex
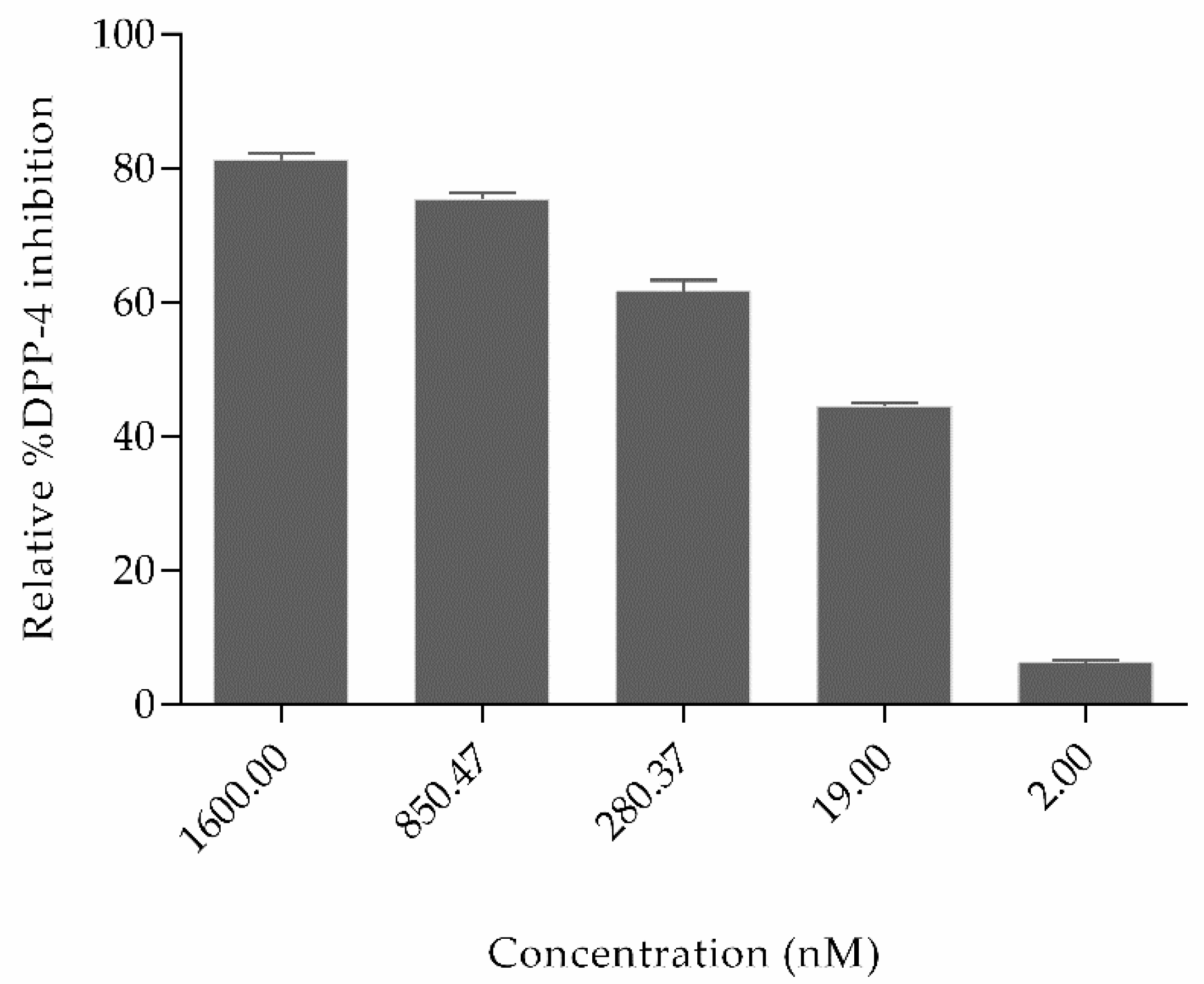
2.2. Experimental Studies
3. Materials and Methods
3.1. Materials
3.2. Computational Methodology
3.2.1. Field-Based and Activity Atlas Model Study
3.2.2. OXT-DPP4 Molecular Docking Simulations
3.2.3. 3D-RISM Analysis
3.3. Experimental Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Heutinck, K.M.; Berge, I.J.T.; Hack, C.E.; Hamann, J.; Rowshani, A.T. Serine proteases of the human immune system in health and disease. Mol. Immunol. 2010, 47, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Di Nardo, A.; Bardan, A.; Murakami, M.; Ohtake, T.; Coda, A.; A Dorschner, R.; Bonnart, C.; Descargues, P.; Hovnanian, A.; et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat. Med. 2007, 13, 975–980. [Google Scholar] [CrossRef]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The discovery of α1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- Vivithanaporn, P.; Asahchop, E.L.; Acharjee, S.; Baker, G.B.; Power, C. HIV protease inhibitors disrupt astrocytic glutamate transporter function and neurobehavioral performance. AIDS 2016, 30, 543–552. [Google Scholar] [CrossRef]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Livi, G.P.; et al. Neuroprotective role of the reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Boil. 2004, 24, 9848–9862. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, Z.; Wallace, M.B.; Stafford, J.A.; Kaldor, S.W.; Kassel, D.B.; Navre, M.; Shi, L.; Skene, R.J.; Asakawa, T.; et al. Discovery of Alogliptin: A potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J. Med. Chem. 2007, 50, 2297–2300. [Google Scholar] [CrossRef]
- Luckett, S.; Garcia, R.; Barker, J.; Konarev, A.; Shewry, P.; Clarke, A.; Brady, R. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Boil. 1999, 290, 525–533. [Google Scholar] [CrossRef]
- Quimbar, P.; Malik, U.; Sommerhoff, C.P.; Kaas, Q.; Chan, L.Y.; Huang, Y.-H.; Grundhuber, M.; Dunse, K.; Craik, D.J.; Anderson, M.A.; et al. High-affinity cyclic peptide matriptase inhibitors. J. Boil. Chem. 2013, 288, 13885–13896. [Google Scholar] [CrossRef]
- Liu, T.; Liu, Y.; Kao, H.-Y.; Pei, D. Membrane permeable cyclic peptidyl inhibitors against human peptidylprolyl isomerase Pin1. J. Med. Chem. 2010, 53, 2494–2501. [Google Scholar] [CrossRef] [PubMed]
- Naumann, T.A.; Tavassoli, A.; Benkovic, S.J. Genetic selection of cyclic peptide dam methyltransferase inhibitors. ChemBioChem 2008, 9, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Nickeleit, I.; Zender, S.; Sasse, F.; Geffers, R.; Brandes, G.; Sörensen, I.; Steinmetz, H.; Kubicka, S.; Carlomagno, T.; Menche, D.; et al. Argyrin A reveals a critical role for the tumor suppressor protein p27kip1 in mediating antitumor activities in response to proteasome inhibition. Cancer Cell 2008, 14, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Hayouka, Z.; Hurevich, M.; Levin, A.; Benyamini, H.; Iosub, A.; Maes, M.; Shalev, D.E.; Loyter, A.; Gilon, C.; Friedler, A. Cyclic peptide inhibitors of HIV-1 integrase derived from the LEDGF/p75 protein. Bioorganic Med. Chem. 2010, 18, 8388–8395. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Boil. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Boddy, C.N.C.; Bräse, S.; Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. 1999, 38, 2096–2152. [Google Scholar] [CrossRef]
- Denning, D.W. Echinocandin antifungal drugs. Lancet 2003, 362, 1142–1151. [Google Scholar] [CrossRef]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef]
- Appetecchia, M.; Baldelli, R. Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumours, current aspects and new perspectives. J. Exp. Clin. Cancer Res. 2010, 29, 19. [Google Scholar] [CrossRef]
- Bryant, A.P.; Busby, R.W.; Bartolini, W.P.; Cordero, E.A.; Hannig, G.; Kessler, M.M.; Pierce, C.M.; Solinga, R.M.; Tobin, J.V.; Mahajan-Miklos, S.; et al. Linaclotide is a potent and selective guanylate cyclase C agonist that elicits pharmacological effects locally in the gastrointestinal tract. Life Sci. 2010, 86, 760–765. [Google Scholar] [CrossRef]
- MacDougall, I.C. New anemia therapies: Translating novel strategies from bench to bedside. Am. J. Kidney Dis. 2012, 59, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, T.; Bennett, C.L.; MacDougall, I.C. Peginesatide for the treatment of anemia due to chronic kidney disease - an unfulfilled promise. Expert Opin. Drug Saf. 2016, 15, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Young, T.S.; Young, D.D.; Ahmad, I.; Louis, J.M.; Benkovic, S.J.; Schultz, P.G. Evolution of cyclic peptide protease inhibitors. Proc. Natl. Acad. Sci. 2011, 108, 11052–11056. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Oberer, L.; Ino, T.; König, W.A.; Busch, M.; Weckesser, J. Cyanopeptolins, new depsipeptides from the cyanobacterium Microcystis sp. pcc 7806. J. Antibiot. 1993, 46, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Deyle, K.; Kong, X.-D.; Heinis, C. Phage selection of cyclic peptides for application in research and drug development. Accounts Chem. Res. 2017, 50, 1866–1874. [Google Scholar] [CrossRef]
- Böcker, J.K.; Friedel, K.; Matern, J.C.; Bachmann, A.L.; Mootz, H.D. Generation of a genetically encoded, photoactivatable intein for the controlled production of cyclic peptides. Angew. Chem. Int. Ed. 2015, 54, 2116–2120. [Google Scholar] [CrossRef]
- Litovchick, A.; Szostak, J.W. Selection of cyclic peptide aptamers to HCV IRES RNA using mRNA display. Proc. Natl. Acad. Sci. USA 2008, 105, 15293–15298. [Google Scholar] [CrossRef]
- Lam, K.; Lebl, M. Synthesis and screening of a ‘one-bead-one-compound’ combinatorial peptide library. Methods Mol. Cell. Biol. 1996, 6, 46–56. [Google Scholar]
- Zhang, Z.; Lin, Z.; Zhou, Z.; Shen, H.C.; Yan, S.F.; Mayweg, A.V.; Xu, Z.; Qin, N.; Wong, J.C.; Zhang, Z.; et al. Structure-based design and synthesis of potent cyclic peptides inhibiting the YAP–TEAD protein–protein interaction. ACS Med. Chem. Lett. 2014, 5, 993–998. [Google Scholar] [CrossRef]
- Soto-Estrada, G.; Altamirano, L.M.; García-García, J.J.; Moreno, I.O.; Silberman, M. Trends in frequency of type 2 diabetes in Mexico and its relationship to dietary patterns and contextual factors. Gac. Sanit. 2018, 32, 283–290. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Dendi, V.S.R.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 2017, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, D.; Mooney, C.A.; Dose, A.; Sandford, G.; Coxon, C.R.; Cobb, S.L. The application of perfluoroheteroaromatic reagents in the preparation of modified peptide systems. Org. Biomol. Chem. 2017, 15, 4086–4095. [Google Scholar] [CrossRef] [PubMed]
- Kalhotra, P.; Chittepu, V.C.S.R.; Osorio-Revilla, G.; Gallardo-Velázquez, T. Structure–activity relationship and molecular docking of natural product library reveal Chrysin as a novel dipeptidyl peptidase-4 (DPP-4) inhibitor: An integrated in silico and in vitro study. Molecules 2018, 23, 1368. [Google Scholar] [CrossRef]
- Wang, F.; Yu, G.; Zhang, Y.; Zhang, B.; Fan, J. Dipeptidyl peptidase IV inhibitory peptides derived from oat (avena sativa L.), buckwheat (fagopyrum esculentum), and highland barley (hordeum vulgare trifurcatum (L.) trofim) proteins. J. Agric. Food Chem. 2015, 63, 9543–9549. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; Fitzgerald, R.J. Dipeptidyl peptidase IV inhibitory properties of a whey protein hydrolysate: Influence of fractionation, stability to simulated gastrointestinal digestion and food–drug interaction. Int. Dairy J. 2013, 32, 33–39. [Google Scholar] [CrossRef]
- Velarde-Salcedo, A.J.; Barrera-Pacheco, A.; Lara-González, S.; Montero-Morán, G.M.; Díaz-Gois, A.; De Mejia, E.G.; De La Rosa, A.P.B. In vitro inhibition of dipeptidyl peptidase IV by peptides derived from the hydrolysis of amaranth (Amaranthus hypochondriacus L.) proteins. Food Chem. 2013, 136, 758–764. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; Fitzgerald, R.J. Susceptibility of milk protein-derived peptides to dipeptidyl peptidase IV (DPP-IV) hydrolysis. Food Chem. 2014, 145, 845–852. [Google Scholar] [CrossRef]
- Jao, C.-L.; Hung, C.-C.; Tung, Y.-S.; Lin, P.-Y.; Chen, M.-C.; Hsu, K.-C. The development of bioactive peptides from dietary proteins as a dipeptidyl peptidase IV inhibitor for the management of type 2 diabetes. Biomed. 2015, 5, 14. [Google Scholar] [CrossRef]
- Klement, J.; Ott, V.; Rapp, K.; Brede, S.; Piccinini, F.; Cobelli, C.; Lehnert, H.; Hallschmid, M. Oxytocin improves β-cell responsivity and glucose tolerance in healthy men. Diabetes 2017, 66, 264–271. [Google Scholar] [CrossRef]
- Elabd, S.; Sabry, I. Two birds with one stone: Possible dual-role of oxytocin in the treatment of diabetes and osteoporosis. Front. Endocrinol. 2015, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Leow, M.S.; Magkos, F. Oxytocin in metabolic homeostasis: Implications for obesity and diabetes management. Obes. Rev. 2019, 20, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Scerbo, M.J.; Gerdes, J.M. Bonding with β-cells—A role for oxytocin in glucose handling. Diabetes 2017, 66, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Zhu, T.; Tang, B.; Yu, S.; Hu, H.; Sun, W.; Pan, R.; Wang, J.; Wang, D.; Yang, L.; et al. Decreased circulating levels of oxytocin in obesity and newly diagnosed type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2014, 99, 4683–4689. [Google Scholar] [CrossRef] [PubMed]
- Snider, B.; Geiser, A.; Yu, X.-P.; Beebe, E.C.; Willency, J.A.; Qing, K.; Guo, L.; Lu, J.; Wang, X.; Yang, Q.; et al. Long-acting and selective oxytocin peptide analogs show antidiabetic and antiobesity effects in male mice. J. Endocr. Soc. 2019, 3, 1423–1444. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, C.; Chen, Q.; Chen, X.; Xu, Z.; Wu, J.; Cai, D. Treatment of obesity and diabetes using oxytocin or analogs in patients and mouse models. PLOS ONE 2013, 8, e61477. [Google Scholar] [CrossRef]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead finder: An approach to improve accuracy of protein−ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Zeifman, A.A.; Stroylov, V.S.; Chilov, G.G. TSAR, a new graph-theoretical approach to computational modeling of protein side-chain flexibility: Modeling of ionization properties of proteins. Proteins: Struct. Funct. Bioinform. 2011, 79, 2693–2710. [Google Scholar] [CrossRef]
- Truchon, J.-F.; Pettitt, B.M.; Labute, P. A cavity corrected 3D-RISM functional for accurate solvation free energies. J. Chem. Theory Comput. 2014, 10, 934–941. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company Name | Protease (Class) | Drug Name | Class |
|---|---|---|---|
| Novartis | Thrombin (Serine), Renin (aspartic) | Desirudin, Aliskiren | Peptidyl, Nonpeptidyl |
| Merck | DPP4 (Serine) | Sitagliptin | Nonpeptidyl |
| Bayer | Factor Xa | Rivaroxaban | Nonpeptidyl |
| Boehringer Ingelheim | Thrombin (Serine), HIV protease (aspartic) | Dabigatran, Tipranavir | Nonpeptidyl |
| GSK | Thrombin (Serine) | Argatroban | Nonpeptidyl |
| Bristol-Myers Squibb | ACE (Metallo) | Captopril | Peptidyl |
| Velcade | Proteosome (threonine) | Bortezomib | Peptidyl |
| No. | Clinical Use | Name of Peptides |
|---|---|---|
| 1 | Bacterial and fungal infections | Telavancin, Dalbavancin, Oritavancin, and Anidulafungin |
| 2 | Oncology | Lanreotide, Romidepsin, and Pasireotide |
| 3 | Gastrointestinal disorders | Linaclotide |
| 4 | Anemia, chronic kidney disease | Peginesatide |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chittepu, V.C.S.R.; Kalhotra, P.; Osorio-Gallardo, T.; Jiménez-Martínez, C.; Robles-de la Torre, R.R.; Gallardo-Velazquez, T.; Osorio-Revilla, G. New Molecular Insights into the Inhibition of Dipeptidyl Peptidase-4 by Natural Cyclic Peptide Oxytocin. Molecules 2019, 24, 3887. https://doi.org/10.3390/molecules24213887
Chittepu VCSR, Kalhotra P, Osorio-Gallardo T, Jiménez-Martínez C, Robles-de la Torre RR, Gallardo-Velazquez T, Osorio-Revilla G. New Molecular Insights into the Inhibition of Dipeptidyl Peptidase-4 by Natural Cyclic Peptide Oxytocin. Molecules. 2019; 24(21):3887. https://doi.org/10.3390/molecules24213887
Chicago/Turabian StyleChittepu, Veera C. S. R., Poonam Kalhotra, Tzayhri Osorio-Gallardo, Cristian Jiménez-Martínez, Raúl René Robles-de la Torre, Tzayhri Gallardo-Velazquez, and Guillermo Osorio-Revilla. 2019. "New Molecular Insights into the Inhibition of Dipeptidyl Peptidase-4 by Natural Cyclic Peptide Oxytocin" Molecules 24, no. 21: 3887. https://doi.org/10.3390/molecules24213887
APA StyleChittepu, V. C. S. R., Kalhotra, P., Osorio-Gallardo, T., Jiménez-Martínez, C., Robles-de la Torre, R. R., Gallardo-Velazquez, T., & Osorio-Revilla, G. (2019). New Molecular Insights into the Inhibition of Dipeptidyl Peptidase-4 by Natural Cyclic Peptide Oxytocin. Molecules, 24(21), 3887. https://doi.org/10.3390/molecules24213887