Can we make Chitosan by Enzymatic Deacetylation of Chitin?

 , ,
, ,
Abstract
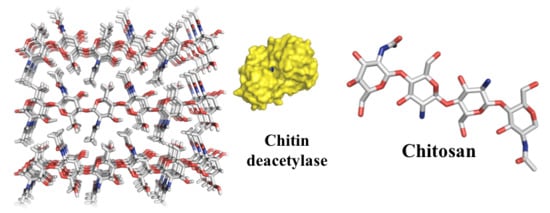
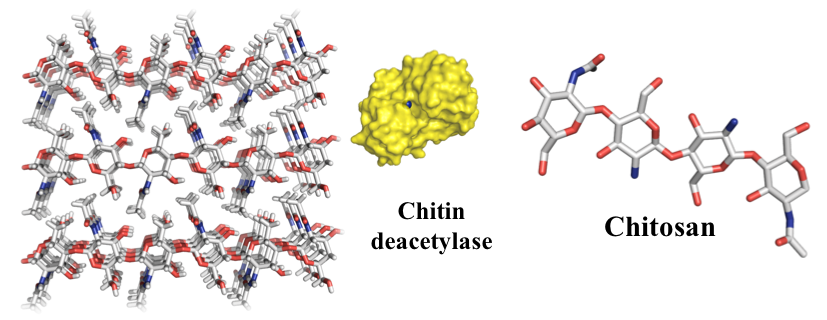{kind=link}
{kind=link}
{kind=link}
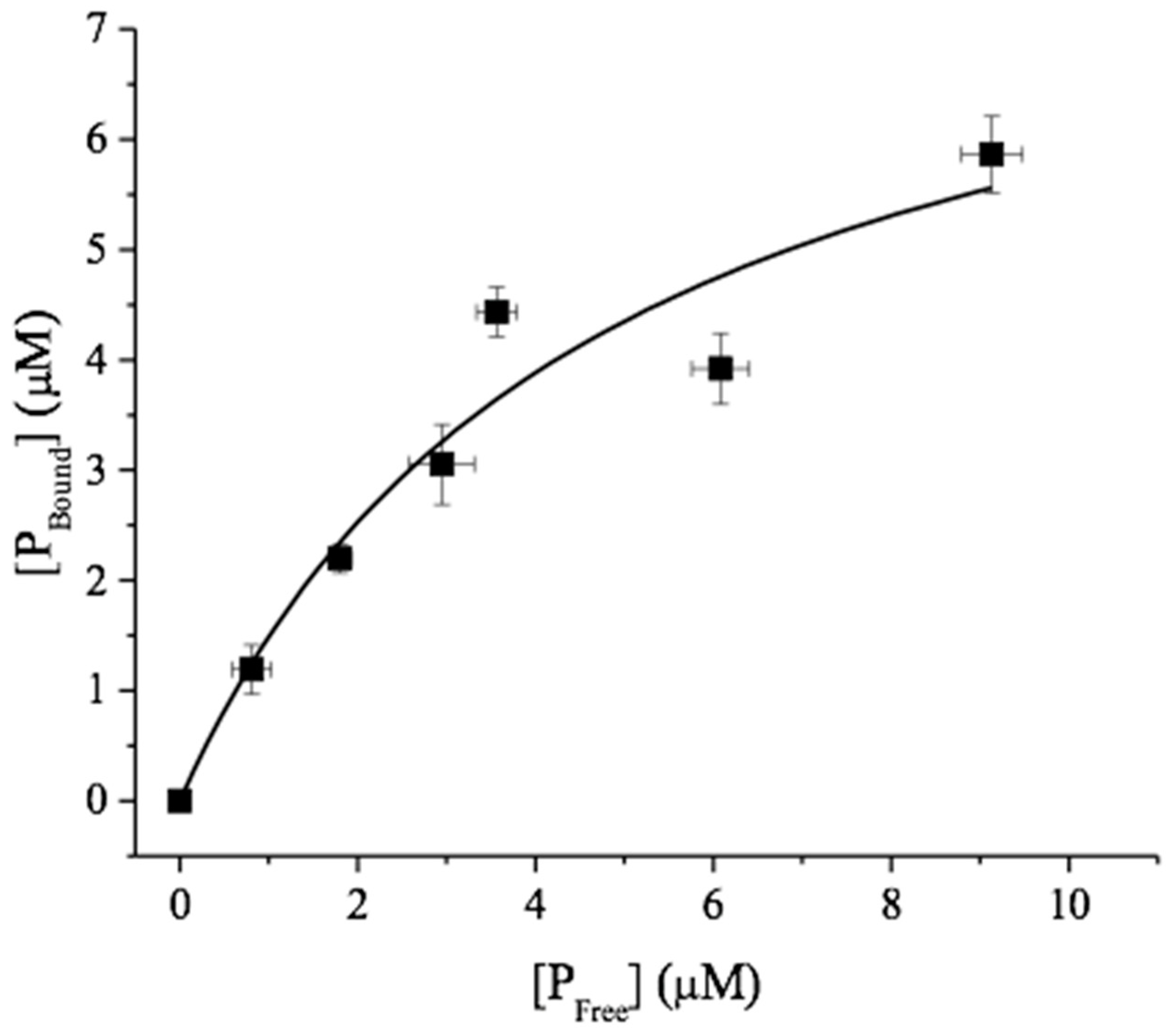{kind=link}
1. Introduction
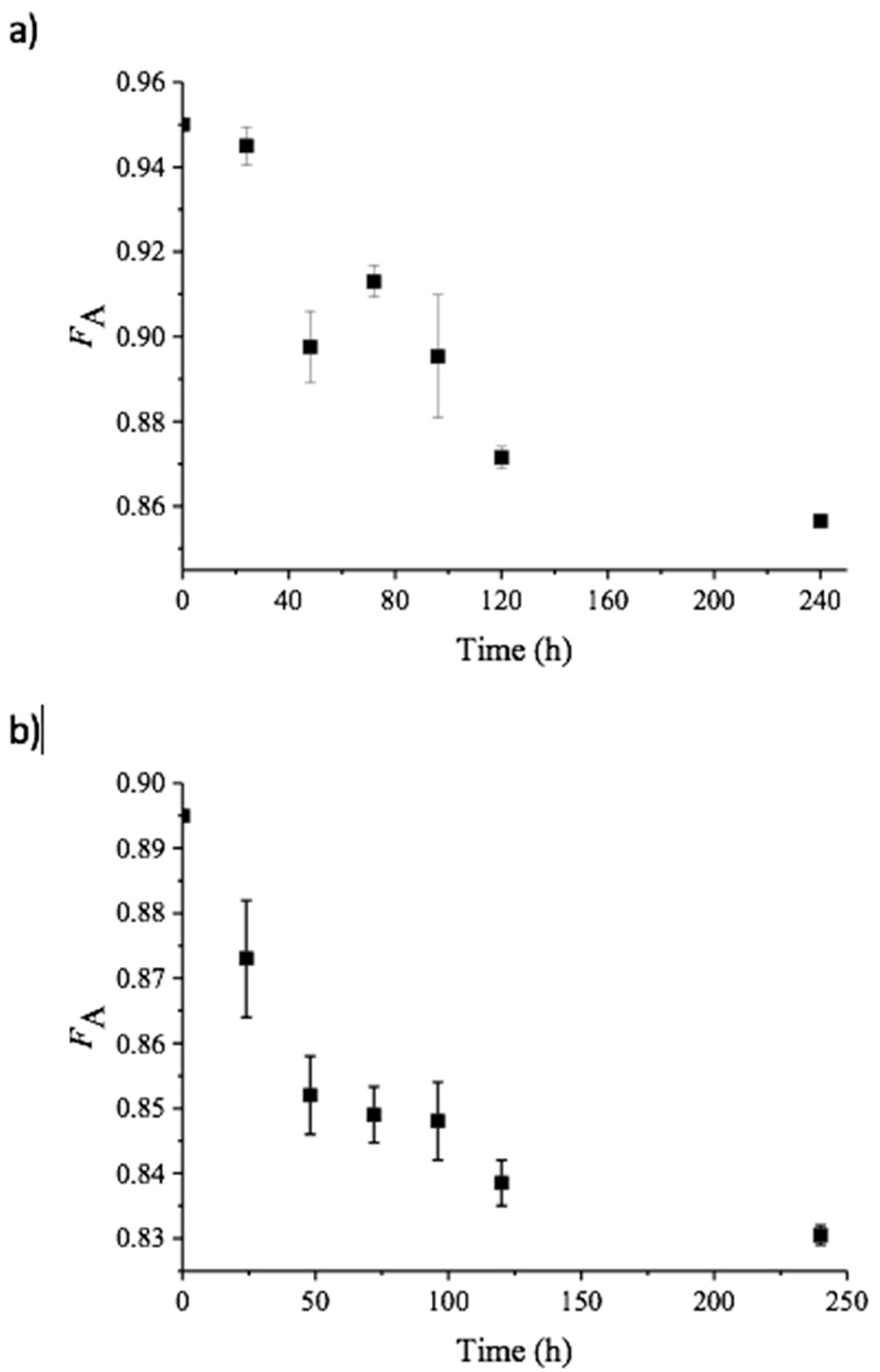
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Production of AnCDA9, SpPgdA, and VcCDA
3.3. Enzymatic Deacetylation of Chitin Nanofibers
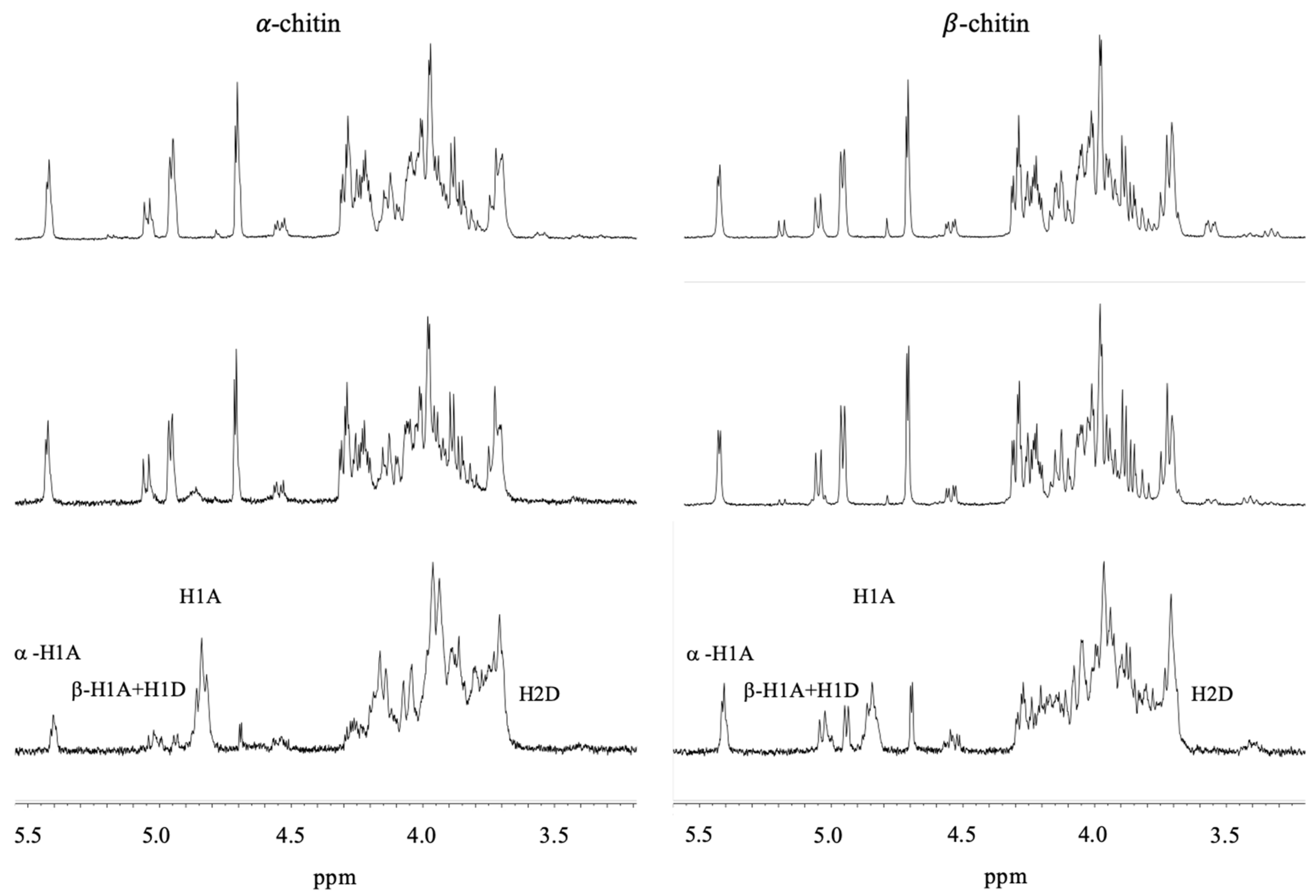
3.4. 1H NMR of α- and β-Chitin Nanofibers
3.5. Enzyme Activity Test
3.6. Binding Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aam, B.B.; Heggset, E.B.; Norberg, A.L.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G.H. Production of chitooligosaccharides and their potential applications in medicine. Mar. Drugs 2010, 8, 1482–1517. [Google Scholar] [CrossRef] [PubMed]
- Minke, R.; Blackwell, J. The structure of α-chitin. J. Mol. Biol. 1978, 120, 167–181. [Google Scholar] [CrossRef]
- Jang, M.-K.; Kong, B.-G.; Jeong, Y.-I.; Lee, C.H.; Nah, J.-W. Physicochemical characterization of α-chitin, β-chitin, and γ-chitin separated from natural resources. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 3423–3432. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Chitins and chitosans as immunoadjuvants and non-allergenic drug carriers. Mar. Drugs 2010, 8, 292–312. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.J. The effect of chitin size, shape, source and purification method on immune recognition. Molecules 2014, 19, 4433–4451. [Google Scholar] [CrossRef]
- Vårum, K.M.; Ottoy, M.H.; Smidsrød, O. Acid hydrolysis of chitosans. Carbohyd. Polym. 2001, 46, 89–98. [Google Scholar] [CrossRef]
- Vårum, K.M.; Anthonsen, M.W.; Grasdalen, H.; Smidsrød, O. 13c-nmr. Studies of the acetylation sequences in partially n-deacetylated chitins (chitosans). Carbohydr. Res. 1991, 217, 19–27. [Google Scholar] [CrossRef]
- Vårum, K.M.; Ottøy, M.H.; Smidsrød, O. Water-solubility of partially n-acetylated chitosans as a function of ph-effect of chemical-composition and depolymerization. Carbohydr. Polym. 1994, 25, 65–70. [Google Scholar] [CrossRef]
- Sannan, T.; Kurita, K.; Iwakura, Y. Studies on chitin: 2. Effect of deacetylation on solubility. Makromol. Chem. 1976, 177, 3589–3600. [Google Scholar] [CrossRef]
- Cai, J.; Yang, J.; Du, Y.; Fan, L.; Qiu, Y.; Li, J.; Kennedy, J.F. Enzymatic preparation of chitosan from the waste aspergillus niger mycelium of citric acid production plant. Carbohydr. Polym. 2006, 64, 151–157. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (cazy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Andrés, E.; Albesa-Jové, D.; Biarnés, X.; Moerschbacher, B.M.; Guerin, M.E.; Planas, A. Structural basis of chitin oligosaccharide deacetylation. Angew. Chem. Int. Ed. Engl. 2014, 53, 6882–6887. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Wang, L.X.; Wang, X.S.; Roseman, S. The chitin catabolic cascade in the marine bacterium vibrio cholerae: Characterization of a unique chitin oligosaccharide deacetylase. Glycobiology 2007, 17, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Gay, L.M.; Tuveng, T.R.; Agger, J.W.; Westereng, B.; Mathiesen, G.; Horn, S.J.; Vaaje-Kolstad, G.; van Aalten, D.M.F.; Eijsink, V.G.H. Structure and function of a broadspecificity chitin deacetylase from aspergillus nidulans fgsc a4. Sci. Rep. 2017, 7, 12. [Google Scholar]
- Blair, D.E.; Schüttelkopf, A.W.; MacRae, J.I.; van Aalten, D.M.F. Structure and metal-dependent mechanism of peptidoglycan deacetylase, a streptococcal virulence factor. Proc. Natl. Acad. Sci. USA 2005, 102, 15429–15434. [Google Scholar] [CrossRef] [PubMed]
- Einbu, A.; Vårum, K.M. Characterization of chitin and its hydrolysis to glcnac and glcn. Biomacromolecules 2008, 9, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Ifuku, S.; Nogi, M.; Abe, K.; Yoshioka, M.; Morimoto, M.; Saimoto, H.; Yano, H. Preparation of chitin nanofibers with a uniform width as α-chitin from crab shells. Biomacromolecules 2009, 10, 1584–1588. [Google Scholar] [CrossRef]
- Ifuku, S.; Saimoto, H. Chitin nanofibers: Preparations, modifications, and applications. Nanoscale 2012, 4, 3308–3318. [Google Scholar] [CrossRef]
- Savitsky, P.; Bray, J.; Cooper, C.D.O.; Marsden, B.D.; Mahajan, P.; Burgess-Brown, N.A.; Gileadi, O. High-throughput production of human proteins for crystallization: The sgc experience. J. Struct. Biol. 2010, 172, 3–13. [Google Scholar] [CrossRef]
- Aslanidis, C.; de Jong, P.J. Ligation-independent cloning of pcr products (lic-pcr). Nucleic Acids Res. 1990, 18, 6069–6074. [Google Scholar] [CrossRef]
- Cederkvist, F.H.; Parmer, M.P.; Vårum, K.M.; Eijsink, V.G.H.; Sørlie, M. Inhibition of a family 18 chitinase by chitooligosaccharides. Carbohyd. Polym. 2008, 74, 41–49. [Google Scholar] [CrossRef]
- Zakariassen, H.; Klemetsen, L.; Sakuda, S.; Vaaje-Kolstad, G.; Vårum, K.M.; Sørlie, M.; Eijsink, V.G.H. Effect of enzyme processivity on the efficacy of a competitive chitinase inhibitor. Carbohyd. Polym. 2010, 82, 779–785. [Google Scholar] [CrossRef]
- Synowiecki, J.; Al-Khateeb, N.A. Production, properties, and some new applications of chitin and its derivatives. Crit. Rev. Food Sci. 2003, 43, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Beckham, G.T.; Crowley, M.F. Examination of the α-chitin structure and decrystallization thermodynamics at the nanoscale. J. Phys. Chem. B 2011, 115, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Monreal, J.; Reese, E.T. Chitinase of Serratia marcescens. Can. J. Microbiol. 1969, 15, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Chylenski, P.; Bissaro, B.; Sørlie, M.; Røhr, Å.K.; Várnai, A.; Horn, S.J.; Eijsink, V.G.H. Lytic polysaccharide monooxygenases in enzymatic processing of lignocellulosic biomass. ACS Catal. 2019, 9, 4970–4991. [Google Scholar] [CrossRef]
- Beckham, G.T.; Ståhlberg, J.; Knott, B.C.; Himmel, M.E.; Crowley, M.F.; Sandgren, M.; Sørlie, M.; Payne, C.M. Towards a molecular-level theory of carbohydrate processivity in glycoside hydrolases. Curr. Opin. Struct. Biol. 2014, 27, 96–106. [Google Scholar] [CrossRef]
- Hamre, A.G.; Jana, S.; Holen, M.M.; Mathiesen, G.; Väljamäe, P.; Payne, C.M.; Sørlie, M. Thermodynamic relationships with processivity in Serratia marcescens family 18 chitinases. J. Phys. Chem. B 2015, 119, 9601–9613. [Google Scholar] [CrossRef]
- Norberg, A.L.; Karlsen, V.; Hoell, I.A.; Bakke, I.; Eijsink, V.G.H.; Sørlie, M. Determination of substrate binding energies in individual subsites of a family 18 chitinase. FEBS Lett. 2010, 584, 4581–4585. [Google Scholar] [CrossRef]
- Zolotnitsky, G.; Cogan, U.; Adir, N.; Solomon, V.; Shoham, G.; Shoham, Y. Mapping glycoside hydrolase substrate subsites by isothermal titration calorimetry. Proc. Natl. Acad. Sci. USA 2004, 101, 11275–11280. [Google Scholar] [CrossRef]
- Hedegård, E.D.; Ryde, U. Targeting the reactive intermediate in polysaccharide monooxygenases. J. Biol. Inorg. Chem. 2017, 22, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Kritchenkov, A.S.; Skorik, Y.A. Click reactions in chitosan chemistry. Russ. Chem. Bull. 2017, 66, 769–781. [Google Scholar] [CrossRef]
- Peng, P.; Cao, X.; Peng, F.; Bian, J.; Xu, F.; Sun, R. Binding cellulose and chitosan via click chemistry: Synthesis, characterization, and formation of some hollow tubes. Polym. Chem. 2012, 50, 5201–5210. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harmsen, R.A.G.; Tuveng, T.R.; Antonsen, S.G.; Eijsink, V.G.H.; Sørlie, M. Can we make Chitosan by Enzymatic Deacetylation of Chitin? Molecules 2019, 24, 3862. https://doi.org/10.3390/molecules24213862
Harmsen RAG, Tuveng TR, Antonsen SG, Eijsink VGH, Sørlie M. Can we make Chitosan by Enzymatic Deacetylation of Chitin? Molecules. 2019; 24(21):3862. https://doi.org/10.3390/molecules24213862
Chicago/Turabian StyleHarmsen, Rianne A. G., Tina R. Tuveng, Simen G. Antonsen, Vincent G.H. Eijsink, and Morten Sørlie. 2019. "Can we make Chitosan by Enzymatic Deacetylation of Chitin?" Molecules 24, no. 21: 3862. https://doi.org/10.3390/molecules24213862
APA StyleHarmsen, R. A. G., Tuveng, T. R., Antonsen, S. G., Eijsink, V. G. H., & Sørlie, M. (2019). Can we make Chitosan by Enzymatic Deacetylation of Chitin? Molecules, 24(21), 3862. https://doi.org/10.3390/molecules24213862