Synthesis of Novel Nicotinic Ligands with Multimodal Action: Targeting Acetylcholine α4β2, Dopamine and Serotonin Transporters

 , , , ,
, , , ,  and
and
Abstract
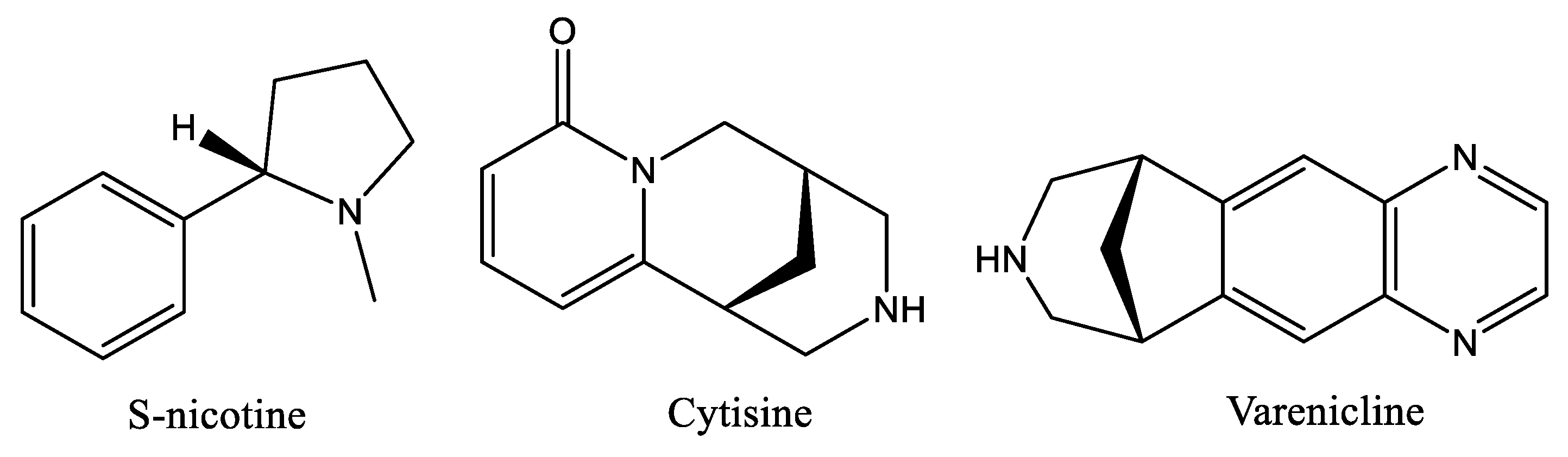
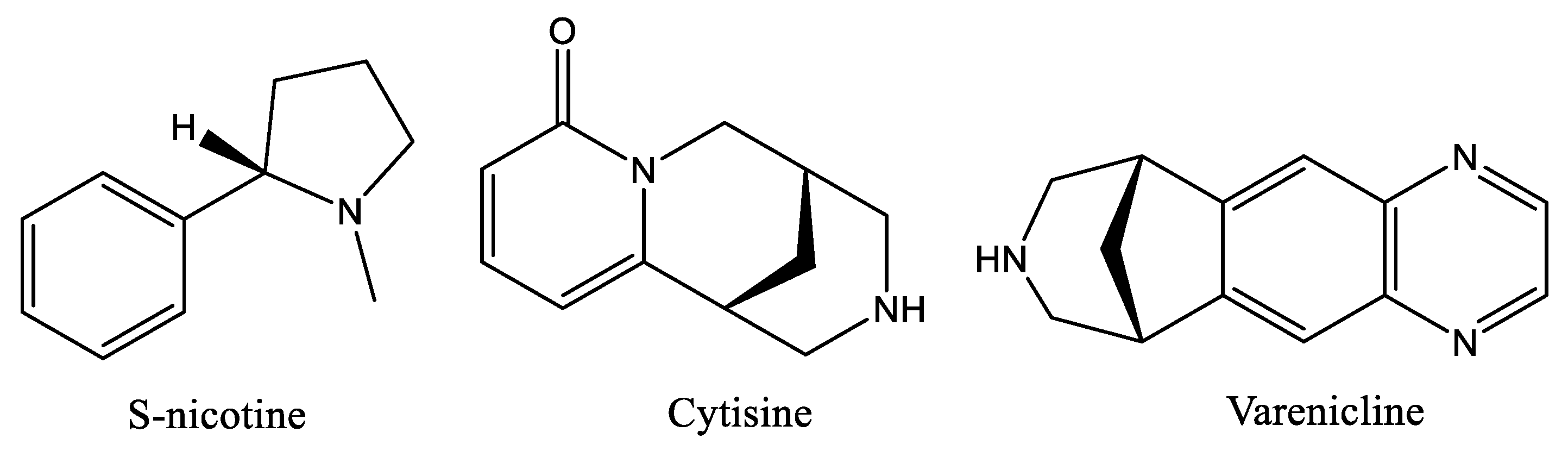
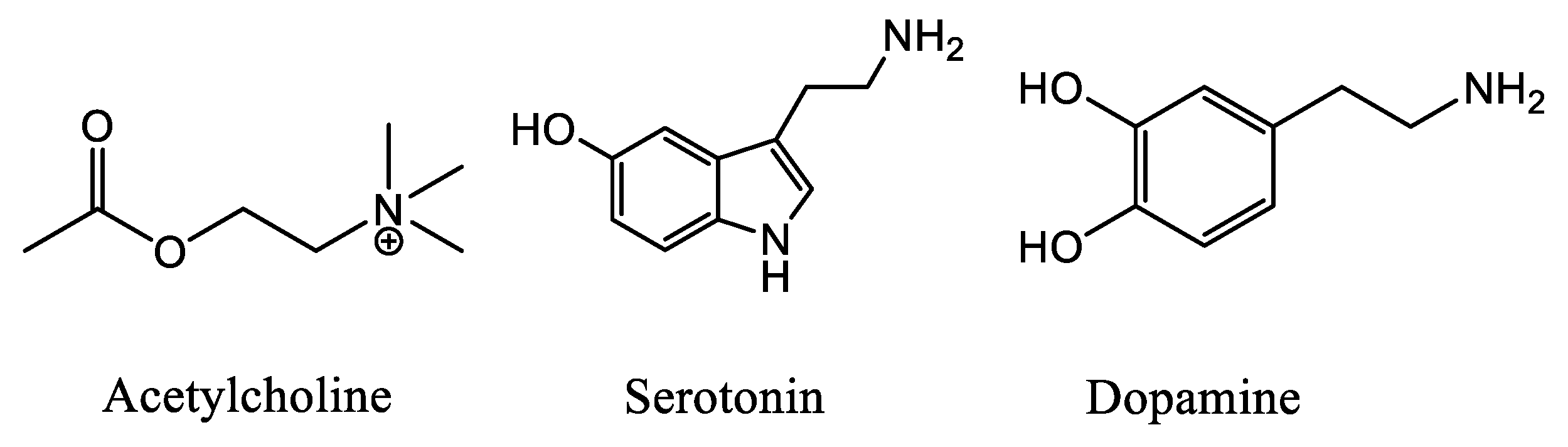
:1. Introduction
2. Results and Discussion
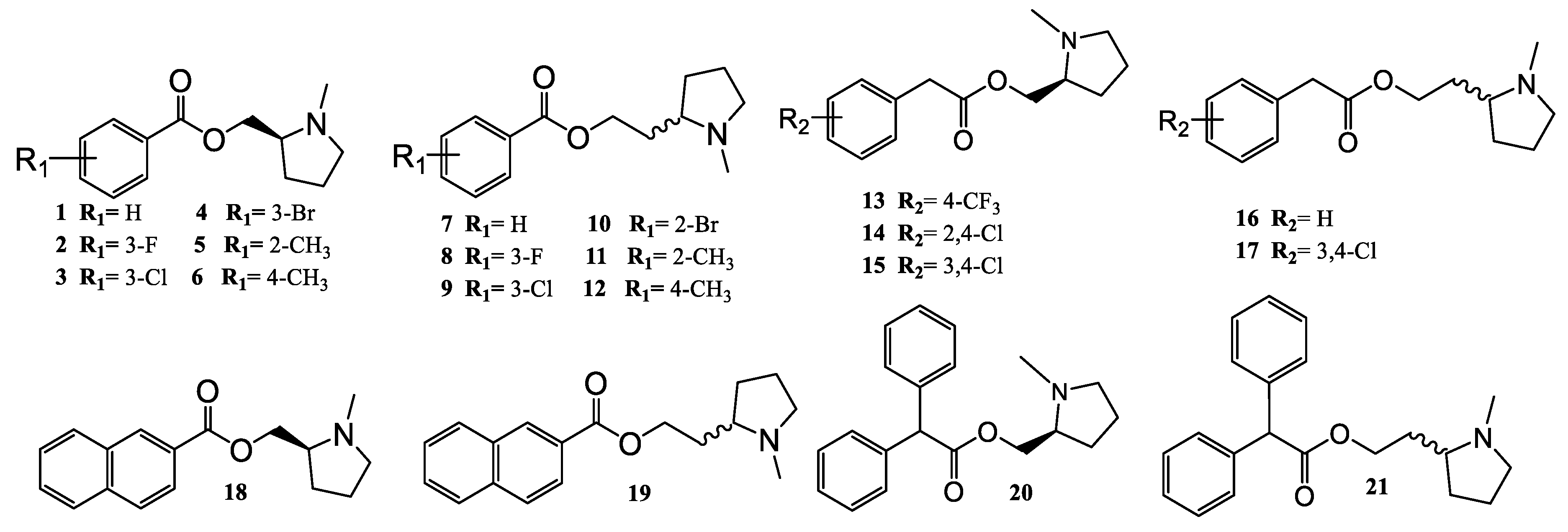
2.1. Chemistry
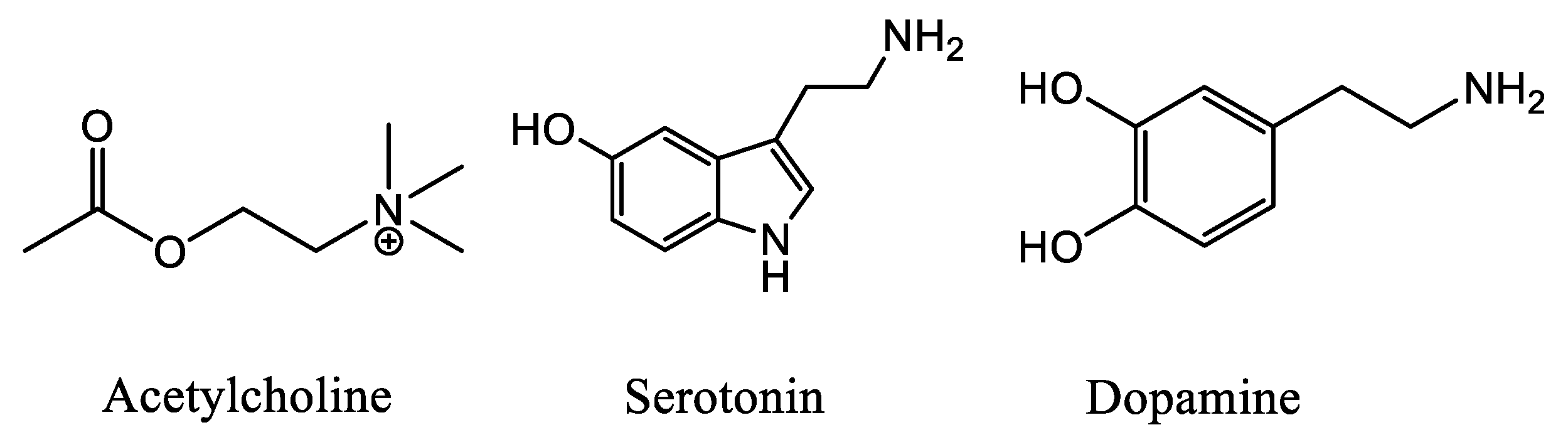
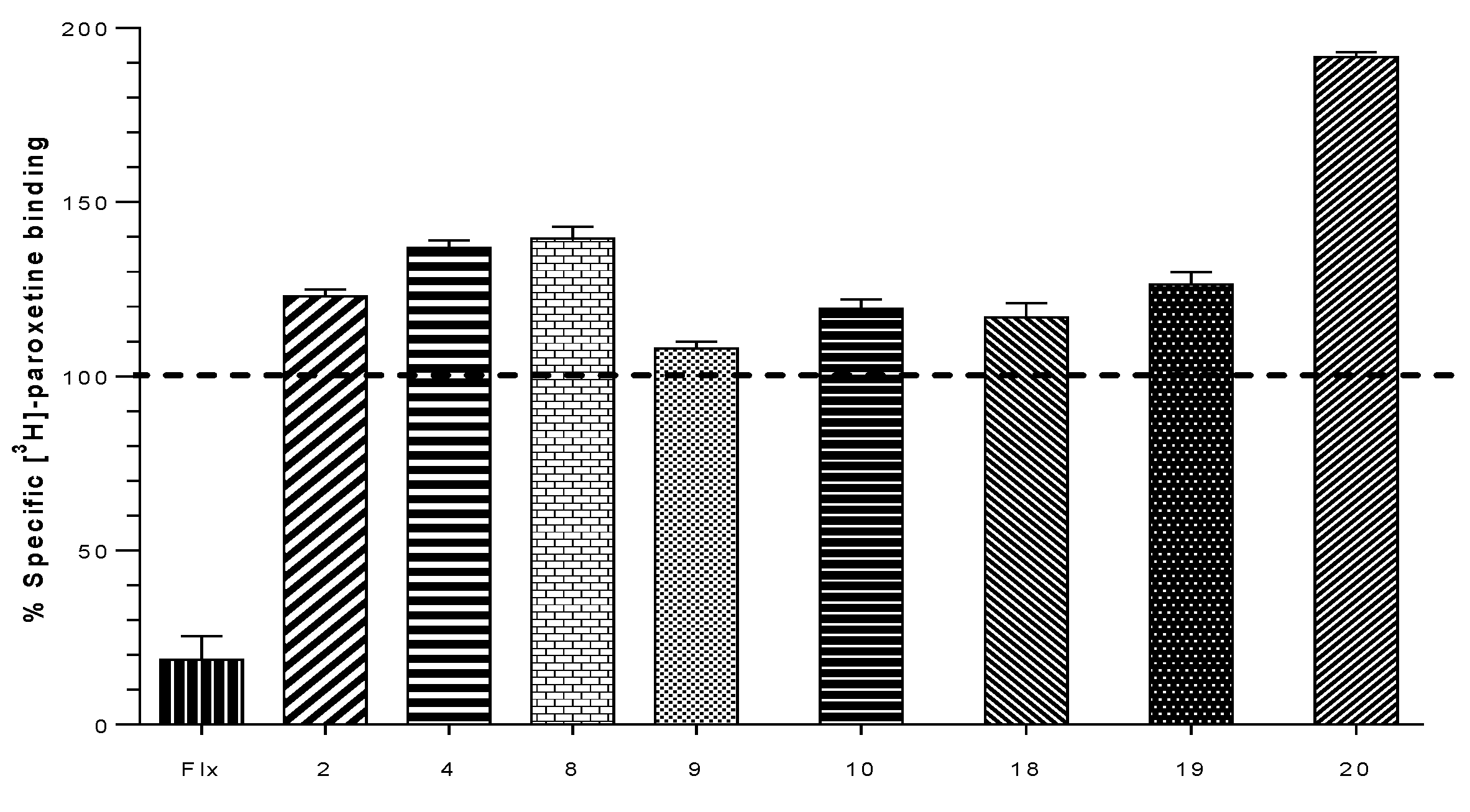
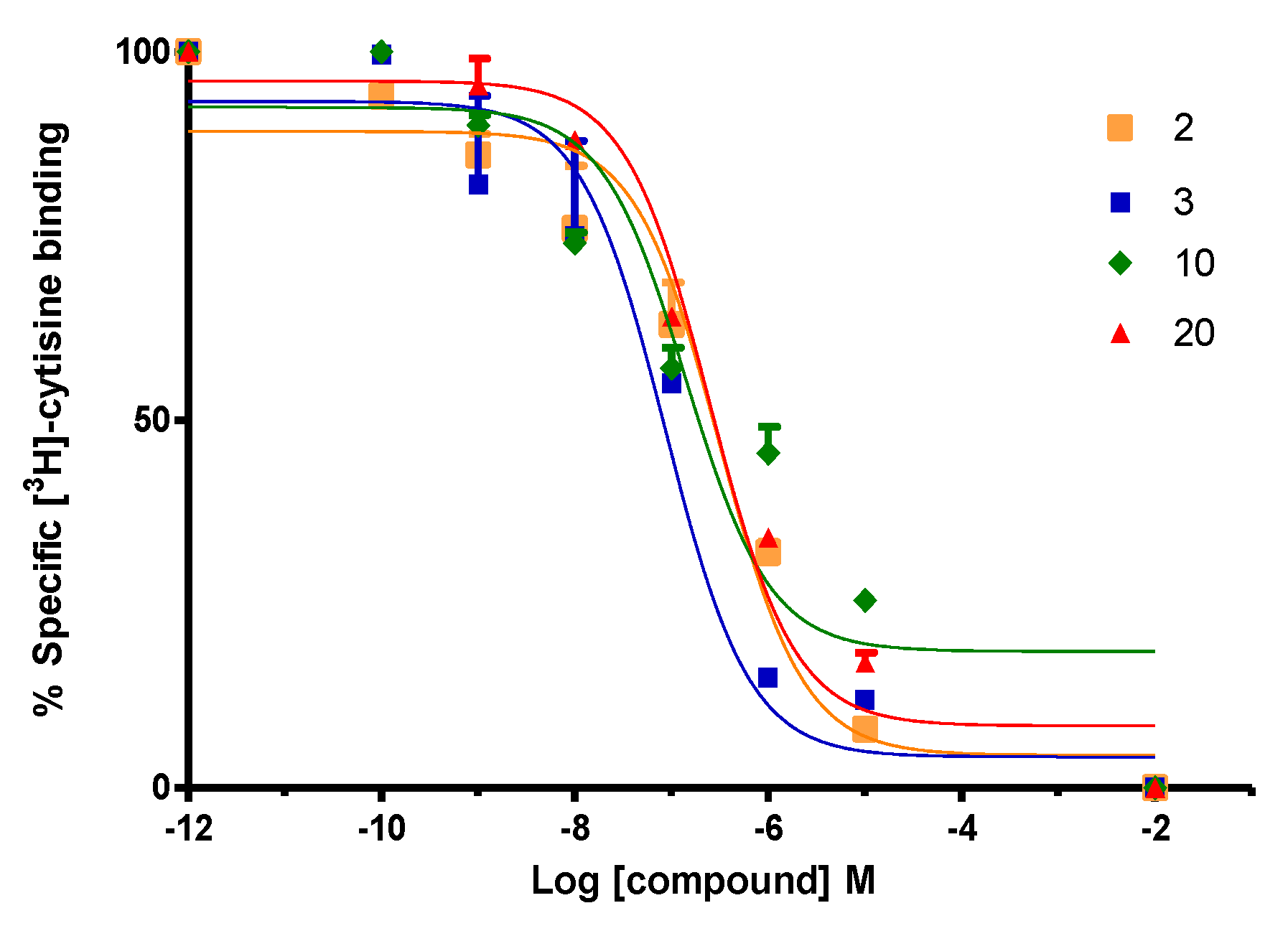
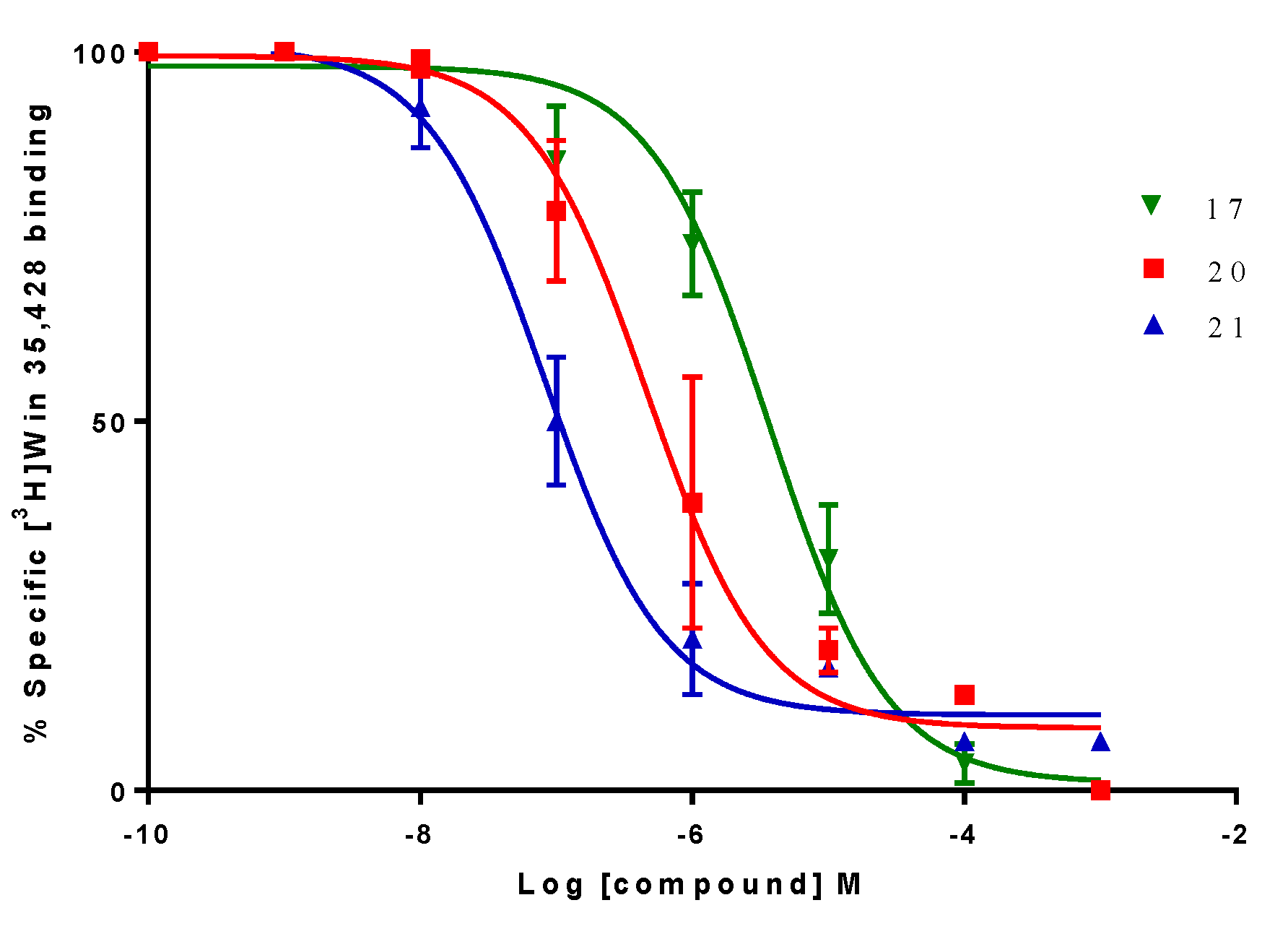
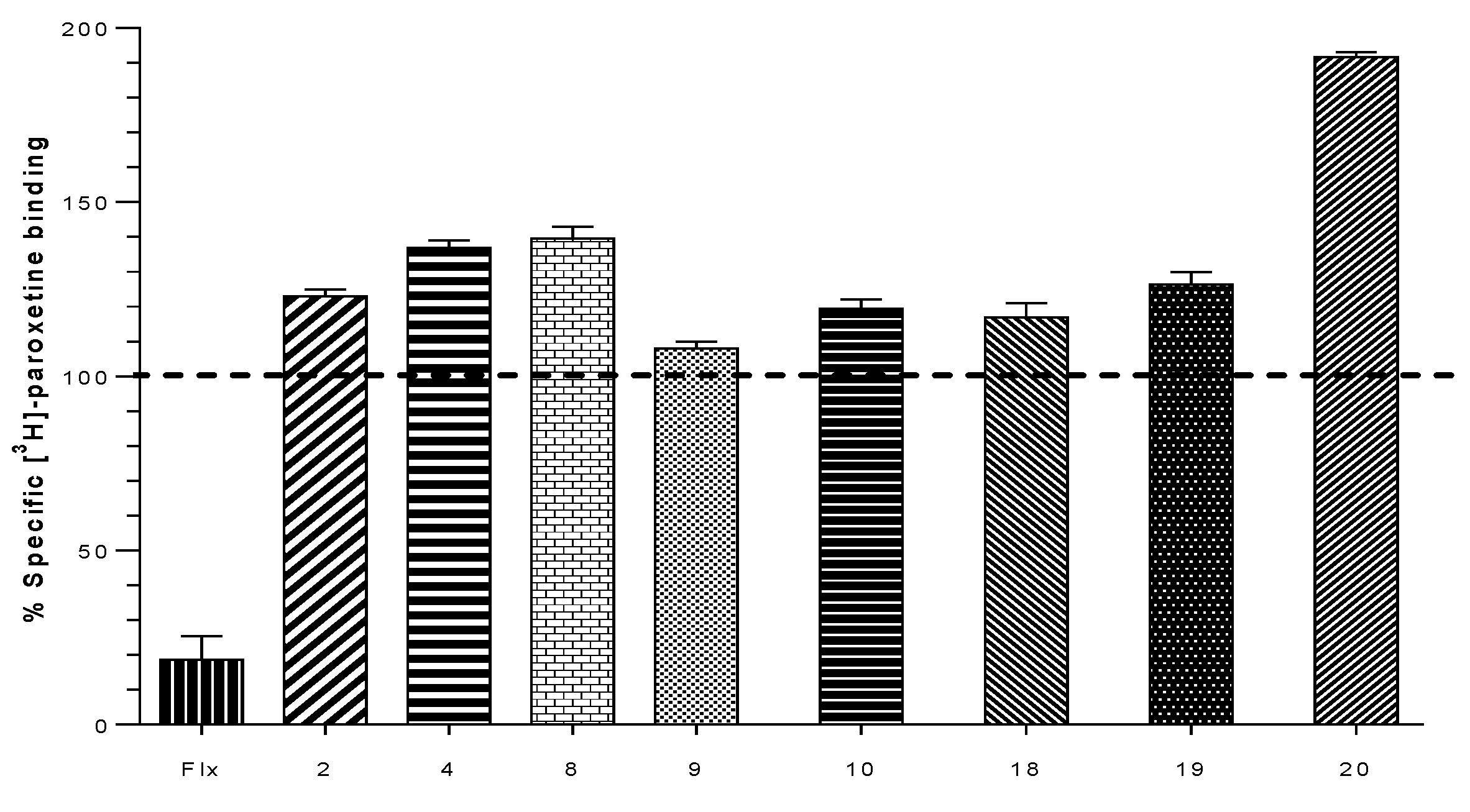
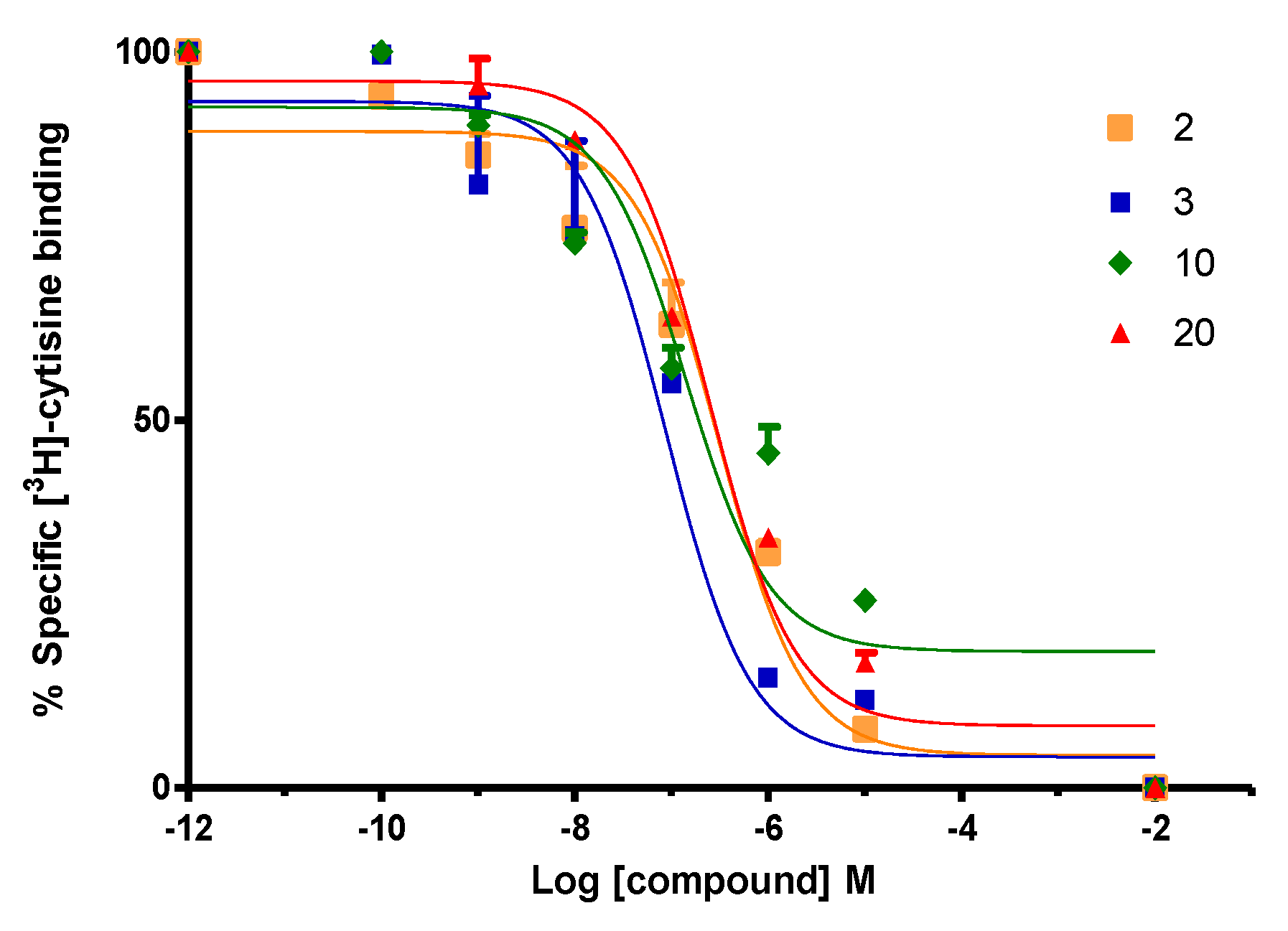
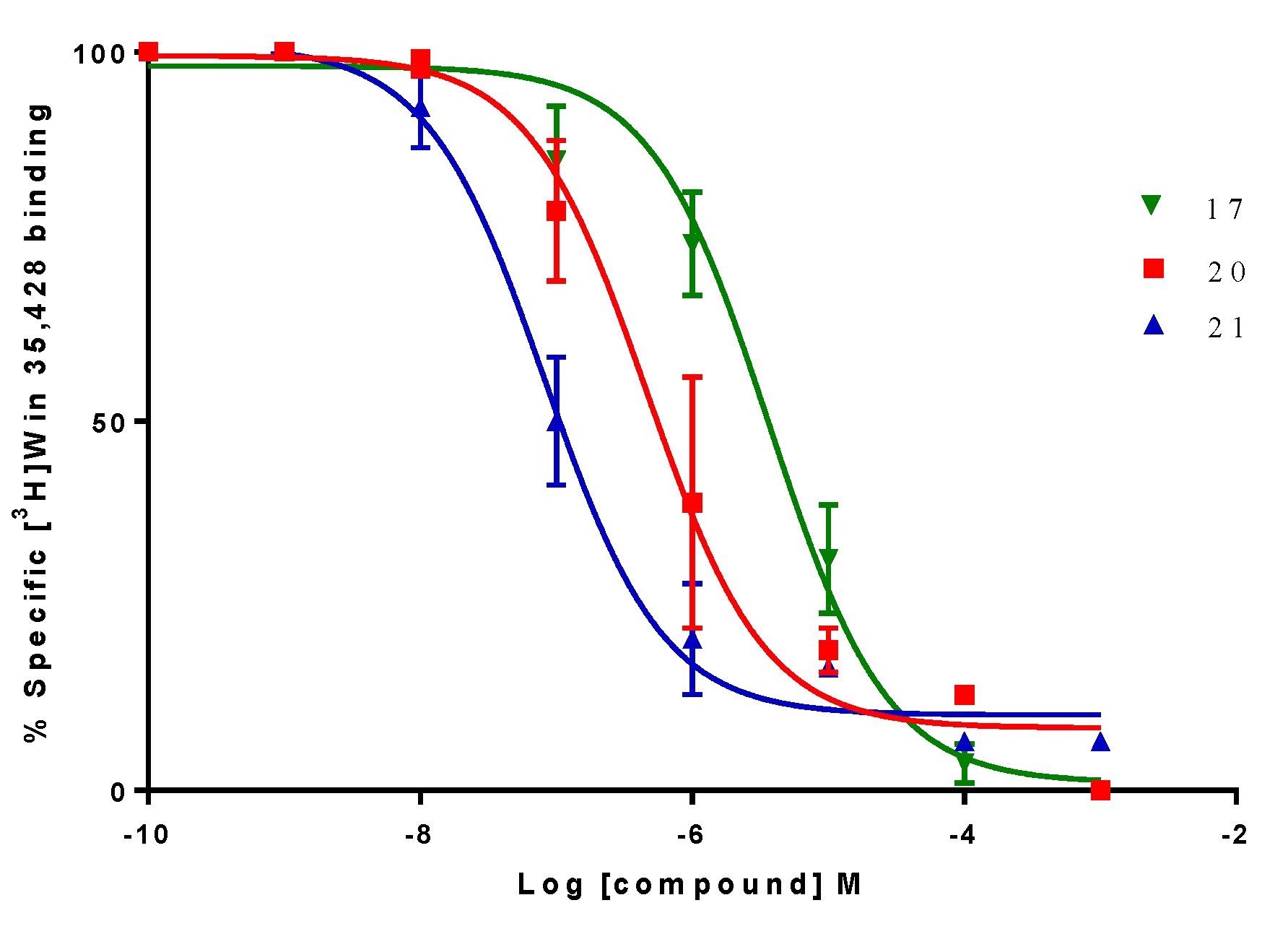
2.2. Biological Evaluation: Binding Affinities on h-DAT, h-SERT and α4β2 nAChR
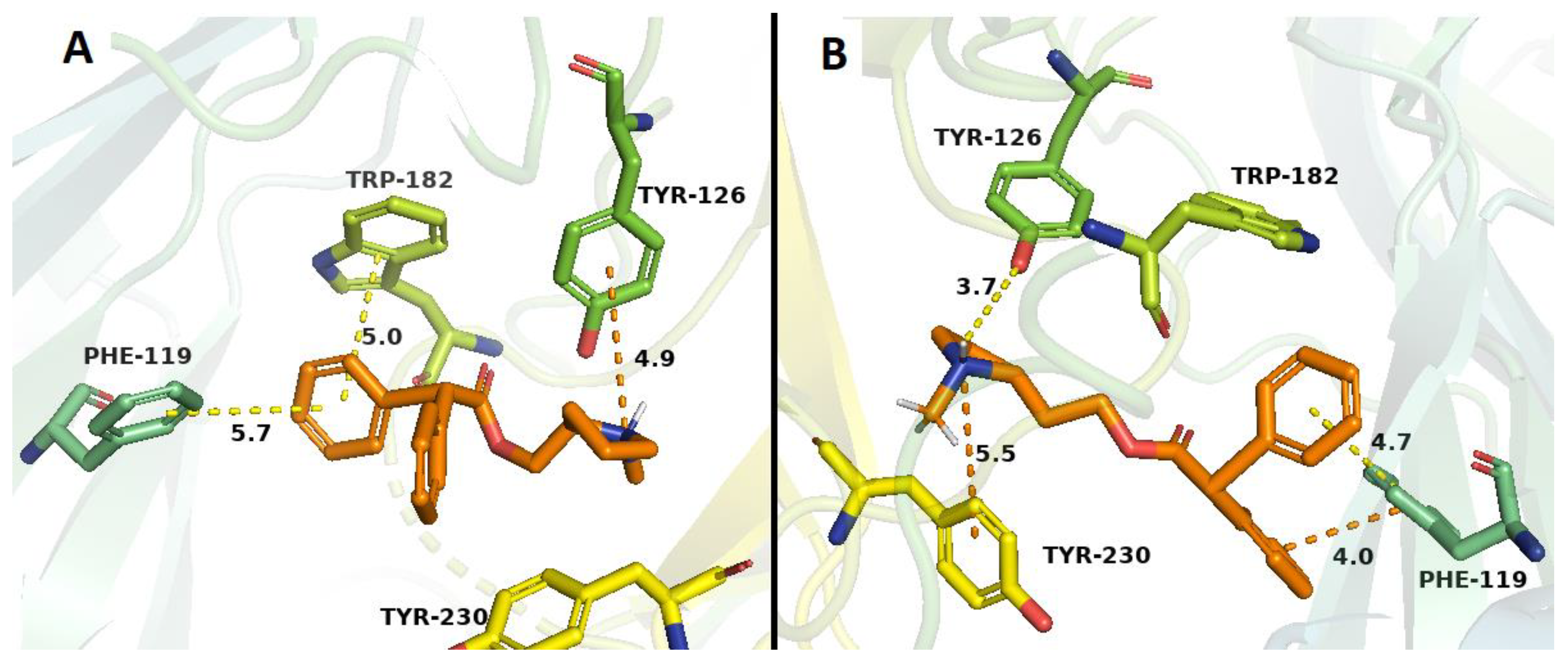
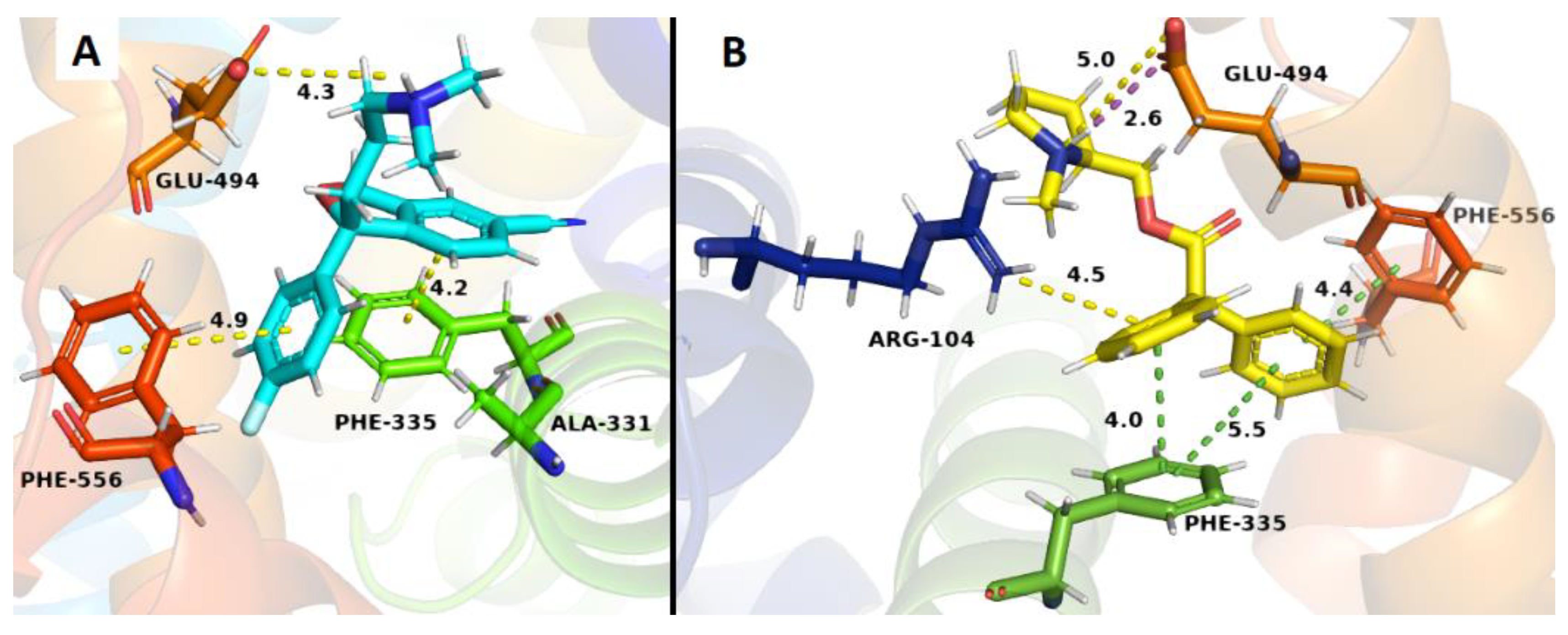
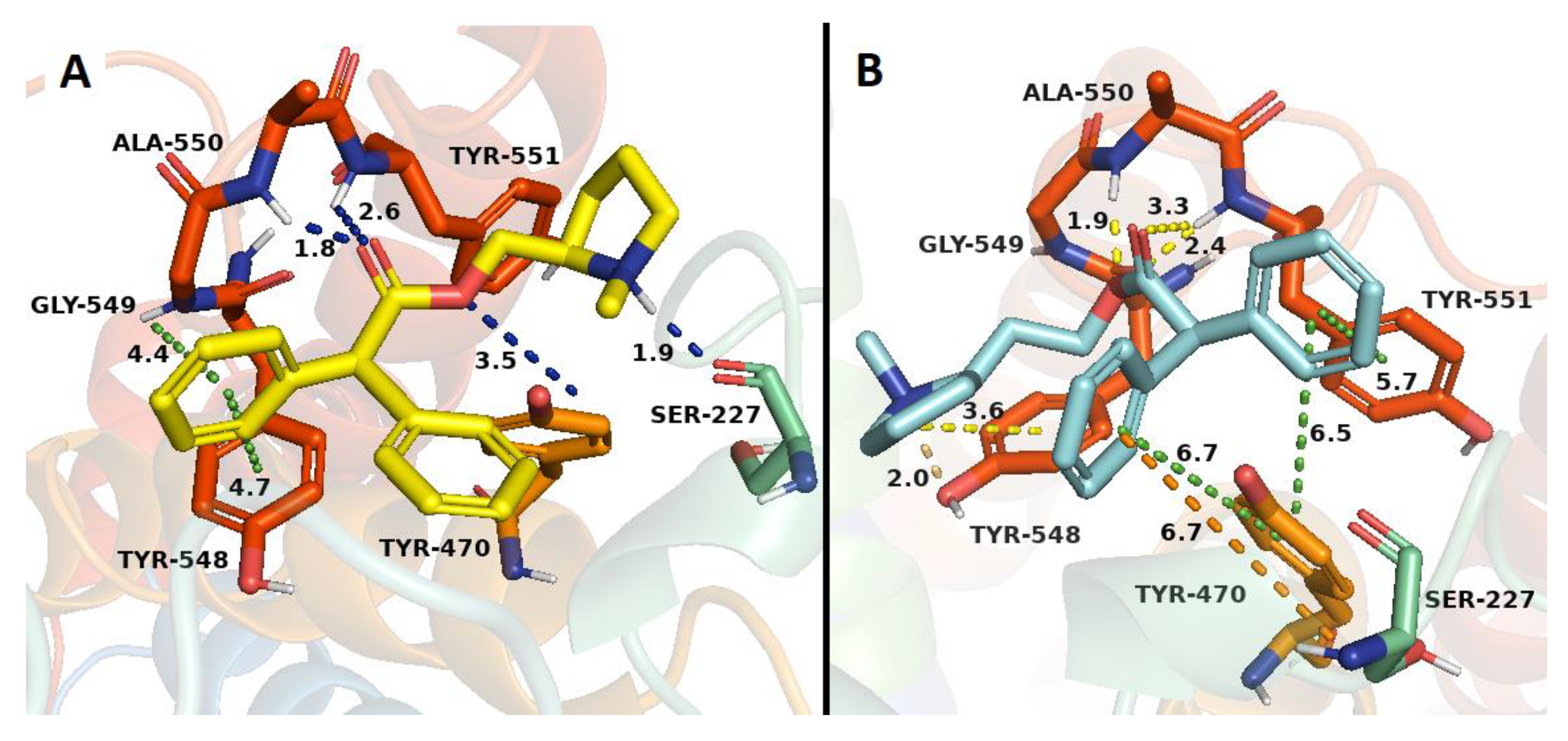
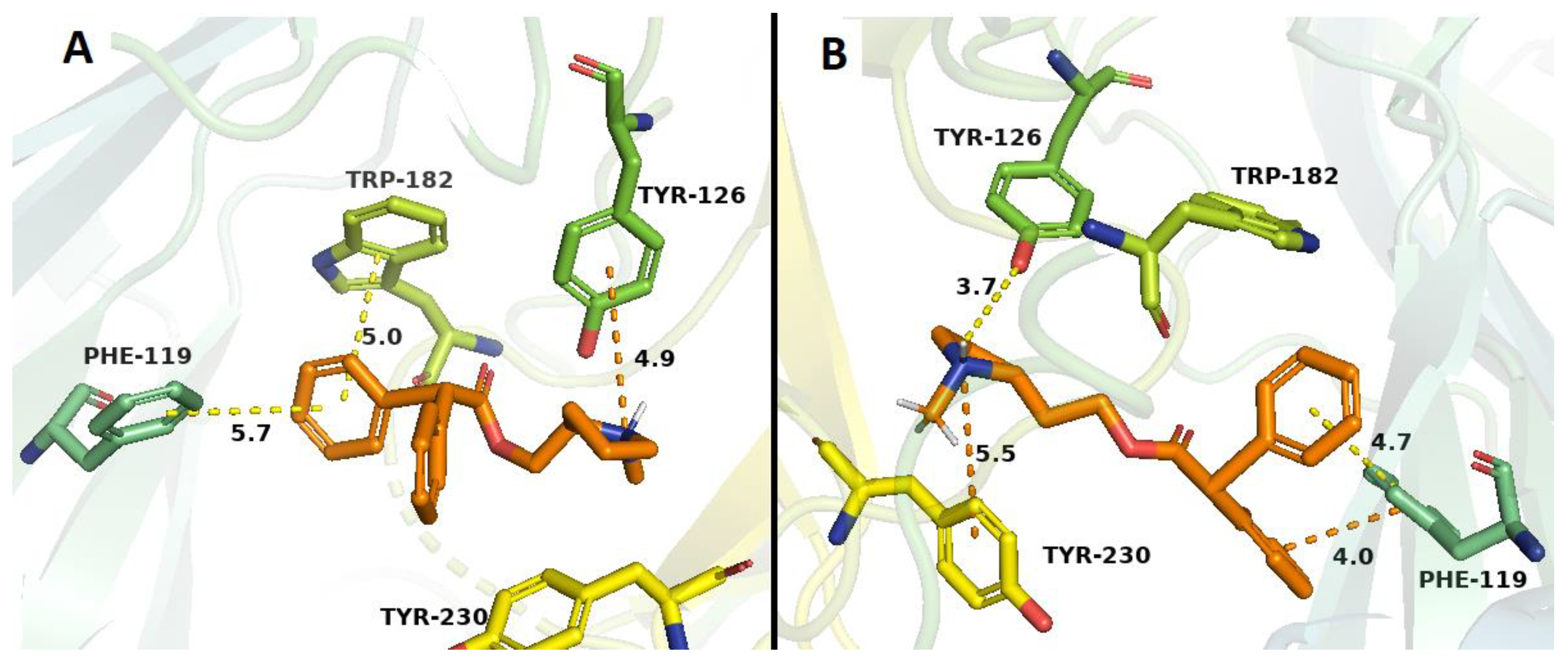
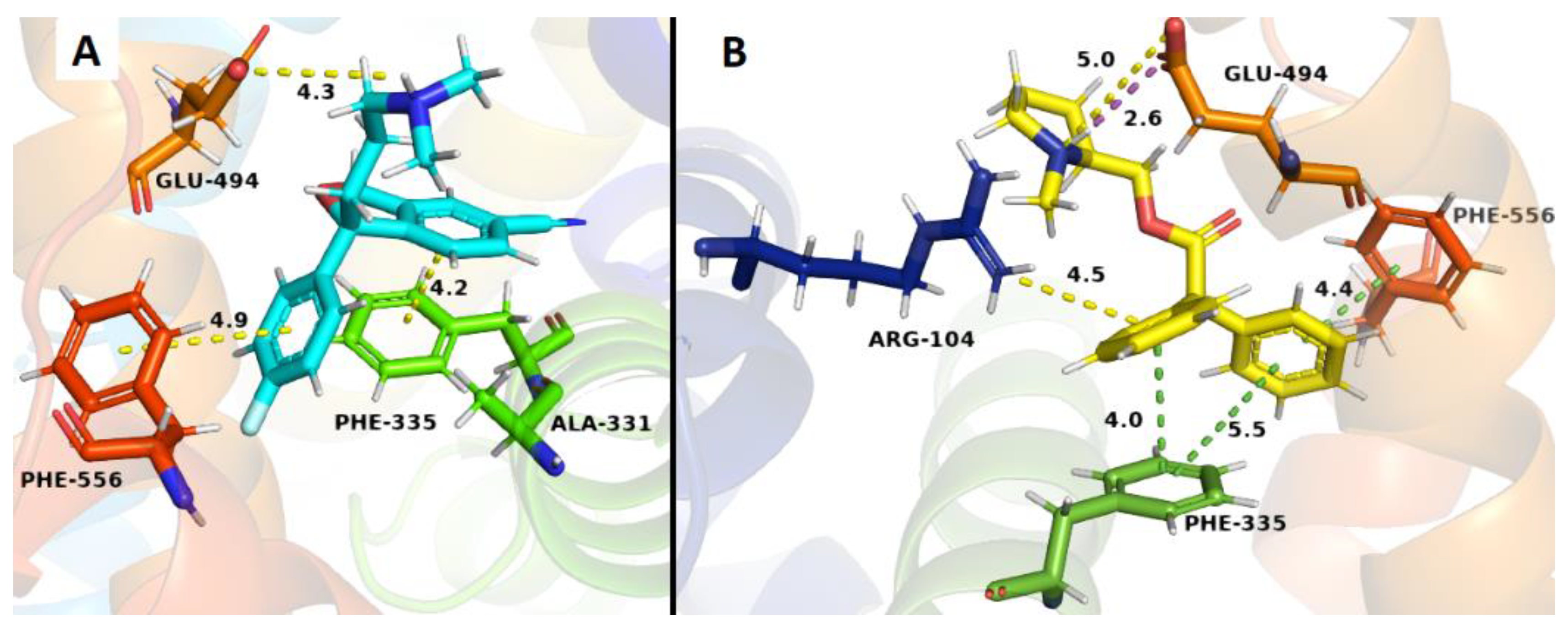
2.3. Docking Studies
3. Materials and Methods
3.1. Binding Protocol of [3H]-Cytisine on nAChR Synaptosomes
3.2. Protocol Binding of [3H]-Paroxetine on h-SERT Cells
3.3. Protocol Binding of [3H]-WIN 35,428 on h-DAT Cells
3.4. Docking Analysis
4. Synthetic Procedures
4.1. General Procedures for The synthesis of Benzoyl Chloride and 2-Phenylacetyl Chloride Derivatives (1–12)
4.2. General Procedures for the Synthesis of (S)-(1-methyl-2-pyrrolidinyl)methyl Benzoate Derivatives (1–6) and (S)-(1-methyl-2-pyrrolidinyl)methyl 2-naphtoate (18)
4.3. General Procedures for the Synthesis of (S)-(1-methyl-2-pyrrolidinyl)methyl 2-phenylacetate Derivatives (13–15) and (S)-(1-methyl-2-pyrrolidinyl)methyl 2,2-diphenylacetate (20)
4.4. General Procedures for the Synthesis of (±)-2-(1-methyl-2-pyrrolidinyl)ethyl Benzoate Derivates (7–12) and (±)-2-(1-methyl-2-pyrrolidinyl)ethyl 2-naphtoate (19)
4.5. General Procedures for the Synthesis of (±)-2-(1-methyl-2-pyrrolidinyl)ethyl 2-phenylacetate Derivatives (16–17) and (±)-2-(1-methyl-2-pyrrolidinyl)ethyl 2,2-diphenylacetate (21)
4.6. (S)-(1-Methyl-2-pyrrolidinyl)methyl Benzoate (1)
4.7. (S)-(1-Methyl-2-pyrrolidinyl)methyl 3-fluorobenzoate (2)
4.8. (S)-(1-Methyl-2-pyrrolidinyl)methyl 3-chlorobenzoate (3)
4.9. (S)-(1-Methyl-2-pyrrolidinyl)methyl 3-bromobenzoate (4)
4.10. (S)-(1-Methyl-2-pyrrolidinyl)methyl 2-methylbenzoate (5)
4.11. (S)-(1-Methyl-2-pyrrolidinyl)methyl 4-methylbenzoate (6)
4.12. (±)-2-(1-Methyl-2-pyrrolidinyl)ethyl benzoate (7)
4.13. (±)-2-(1-Methyl-2-pyrrolidinyl)ethyl 3-fluorobenzoate (8)
4.14. (±)-2-(1-Methyl-2-pyrrolidinyl)ethyl 3-chlorobenzoate (9)
4.15. (±)-2-(1-Methyl-2-pyrrolidinyl)ethyl 3-bromobenzoate (10)
4.16. (±)-2-(1-Methyl-2-pyrrolidinyl)ethyl 2-methylbenzoate (11)
4.17. (±)-2-(1-Methyl-2-pyrrolidinyl)ethyl 4-methylbenzoate (12)
4.18. (S)-(1-Methyl-2-pyrrolidinyl)methyl 2-(4-(trifluoromethyl)phenyl)acetate (13)
4.19. (S)-(1-Methyl-2-pyrrolidinyl)methyl 2-(2,4-dichlorophenyl)acetate (14)
4.20. (S)-(1-Methyl-2-pyrrolidinyl)methyl 2-(3,4-dichlorophenyl)acetate (15)
4.21. (±)-2-(-1-Methyl-2-pyrrolidinyl)ethyl 2-phenylacetate (16)
4.22. (±)-2-(-1-Methyl-2-pyrrolidinyl)ethyl 2-(3,4-dichlorophenyl)acetate (17)
4.23. (S)-(1-Methyl-2-pyrrolidinyl)methyl 2-naphthoate (18)
4.24. (±)-2-(1-Methyl-2-pyrrolidinyl)ethyl 2-naphthoate (19)
4.25. (S)-(1-Methyl-2-pyrrolidinyl)methyl 2,2-diphenylacetate (20)
4.26. (±)-2-(-1-Methyl-2-pyrrolidinyl)ethyl 2,2-diphenylacetate (21)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pistillo, F.; Clementi, F.; Zoli, M.; Gotti, C. Nicotinic, glutamatergic and dopaminergic synaptic transmission and plasticity in the mesocorticolimbic system: Focus on nicotine effects. Prog. Neurobiol. 2015, 124, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Rucktooa, P.; Haseler, C.A.; van Elk, R.; Smit, A.B.; Gallagher, T.; Sixma, T.K. Structural Characterization of Binding Mode of Smoking Cessation Drugs to Nicotinic Acetylcholine Receptors through Study of Ligand Complexes with Acetylcholine-binding Protein. J. Biol. Chem. 2012, 287, 23283–23293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iturriaga-Vasquez, P.; Carbone, A.; Garcia-Beltran, O.; Livingstone, P.D.; Biggin, P.C.; Cassels, B.K.; Wonnacott, S.; Zapata-Torres, G.; Bermudez, I. Molecular Determinants for Competitive Inhibition of 4 2 Nicotinic Acetylcholine Receptors. Mol. Pharmacol. 2010, 78, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Faundez-Parraguez, M.; Farias-Rabelo, N.; Gonzalez-Gutierrez, J.P.; Etcheverry-Berrios, A.; Alzate-Morales, J.; Adasme-Carreño, F.; Varas, R.; Bermudez, I.; Iturriaga-Vasquez, P. Neonicotinic analogues: Selective antagonists for α4β2 nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2013, 21, 2687–2694. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Guiducci, S.; Tedesco, V.; Corbioli, S.; Zanetti, L.; Moretti, M.; Zanardi, A.; Rimondini, R.; Mugnaini, M.; Clementi, F.; et al. Nicotinic Acetylcholine Receptors in the Mesolimbic Pathway: Primary Role of Ventral Tegmental Area 6 2* Receptors in Mediating Systemic Nicotine Effects on Dopamine Release, Locomotion, and Reinforcement. J. Neurosci. 2010, 30, 5311–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascoli, V.; Terrier, J.; Hiver, A.; Lüscher, C. Sufficiency of Mesolimbic Dopamine Neuron Stimulation for the Progression to Addiction. Neuron 2015, 88, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Pierce, R.C.; Kumaresan, V. The mesolimbic dopamine system: The final common pathway for the reinforcing effect of drugs of abuse? Neurosci. Biobehav. Rev. 2006, 30, 215–238. [Google Scholar] [CrossRef]
- Salamone, J.D.; Pardo, M.; Yohn, S.E.; López-Cruz, L.; SanMiguel, N.; Correa, M. Mesolimbic Dopamine and the Regulation of Motivated Behavior. Curr. Top. Behav. Neurosci. 2015, 27, 231–257. [Google Scholar]
- Exley, R.; Maubourguet, N.; David, V.; Eddine, R.; Evrard, A.; Pons, S.; Marti, F.; Threlfell, S.; Cazala, P.; McIntosh, J.M.; et al. Distinct contributions of nicotinic acetylcholine receptor subunit 4 and subunit 6 to the reinforcing effects of nicotine. Proc. Natl. Acad. Sci. 2011, 108, 7577–7582. [Google Scholar] [CrossRef]
- Jung, J.W.; Jeon, E.J.; Kim, J.G.; Yang, S.-Y.; Choi, J.C.; Shin, J.W.; Park, I.W.; Choi, B.W.; Kim, D.-K.; Kim, J.Y. Clinical experience of varenicline for smoking cessation. Clin. Respir. J. 2010, 4, 215–221. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Dravolina, O.A.; Bespalov, A.Y. Agonist and antagonist effects of cytisine in vivo. Neuropharmacology 2015, 95, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-K.; Eaton, J.B.; Fedolak, A.; Gunosewoyo, H.; Onajole, O.K.; Brunner, D.; Lukas, R.J.; Yu, L.-F.; Kozikowski, A.P. Synthesis and biological evaluation of novel hybrids of highly potent and selective α4β2-Nicotinic acetylcholine receptor (nAChR) partial agonists. Eur. J. Med. Chem. 2016, 124, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Coronado, N.; Walker, A.J.; Berk, M.; Dodd, S. Current and Emerging Pharmacotherapies for Cessation of Tobacco Smoking. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2018, 38, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Rollema, H.; Chambers, L.K.; Coe, J.W.; Glowa, J.; Hurst, R.S.; Lebel, L.A.; Lu, Y.; Mansbach, R.S.; Mather, R.J.; Rovetti, C.C.; et al. Pharmacological profile of the α4β2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology. 2007, 52, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Feduccia, A.A.; Simms, J.A.; Mill, D.; Yi, H.Y.; Bartlett, S.E. Varenicline decreases ethanol intake and increases dopamine release via neuronal nicotinic acetylcholine receptors in the nucleus accumbens. Br. J. Pharmacol. 2014, 171, 3420–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, S.G.; Legasse, A.J.; Uebelacker, L.A.; Brown, R.A.; Price, L.H.; Abrantes, A.M. Anxiety Sensitivity is Associated with Lower Enjoyment and an Anxiogenic Response to Physical Activity in Smokers. Cognit. Ther. Res. 2019, 43, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.R.; Hogarth, L.; Leventhal, A.M.; Cook, J.W.; Hitsman, B. Cigarette smoking and depression comorbidity: Systematic review and proposed theoretical model. Addiction 2017, 112, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Audrain-McGovern, J.; Leventhal, A.M.; Strong, D.R. The Role of Depression in the Uptake and Maintenance of Cigarette Smoking. Int. Rev. Neurobiol. 2015, 124, 209–243. [Google Scholar]
- Zvolensky, M.J.; Shepherd, J.M.; Bakhshaie, J.; Garey, L.; Viana, A.G.; Peraza, N. Emotion dysregulation and cigarette dependence, perceptions of quitting, and problems during quit attempts among Spanish-speaking Latinx adult smokers. Addict. Behav. 2019, 96, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Picciotto, M.R. Nicotine receptors and depression: Revisiting and revising the cholinergic hypothesis. Trends Pharmacol. Sci. 2010, 31, 580–586. [Google Scholar] [CrossRef]
- Shytle, R.D.; Silver, A.A.; Lukas, R.J.; Newman, M.B.; Sheehan, D.V.; Sanberg, P.R. Nicotinic acetylcholine receptors as targets for antidepressants. Mol. Psychiatry. 2002, 7, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaweł, K.; Jenda, M.; Kotlińska, J.H. The role of acetylcholine in drug addiction. Curr. Issues Pharm. Med. Sci. 2012, 25, 212–217. [Google Scholar] [CrossRef]
- Lesuis, S.L.; Hoeijmakers, L.; Korosi, A.; de Rooij, S.R.; Swaab, D.F.; Kessels, H.W.; Lucassen, P.J.; Krugers, H.J. Vulnerability and resilience to Alzheimer’s disease: Early life conditions modulate neuropathology and determine cognitive reserve. Alzheimers. Res. Ther. 2018, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Coupé, P.; Manjón, J.V.; Lanuza, E.; Catheline, G. Lifespan Changes of the Human Brain In Alzheimer’s Disease. Sci. Rep. 2019, 9, 3998. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Grothe, M.J.; Ray, N.J.; Müller, M.L.T.M.; Teipel, S.J. Recent Advances in Cholinergic Imaging and Cognitive Decline—Revisiting the Cholinergic Hypothesis of Dementia. Curr. Geriatr. Reports. 2018, 7, 1–11. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Eldufani, J.; Blaise, G. The role of acetylcholinesterase inhibitors such as neostigmine and rivastigmine on chronic pain and cognitive function in aging: A review of recent clinical applications. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 175–183. [Google Scholar] [CrossRef]
- Van Dam, D.; Vermeiren, Y.D.; Dekker, A.; Dekker, A.J.W.; Naudé, P.; De Deyn, P.P. Neuropsychiatric Disturbances in Alzheimer’s Disease: What Have We Learned from Neuropathological Studies? Curr. Alzheimer Res. 2016, 13, 1145–1164. [Google Scholar] [CrossRef]
- Yohn, C.N.; Gergues, M.M.; Samuels, B.A. The role of 5-HT receptors in depression. Mol. Brain. 2017, 10, 28. [Google Scholar] [CrossRef]
- Fisher, J.R.; Wallace, C.E.; Tripoli, D.L.; Sheline, Y.I.; Cirrito, J.R. Redundant Gs-coupled serotonin receptors regulate amyloid-β metabolism in vivo. Mol. Neurodegener. 2016, 11, 45. [Google Scholar] [CrossRef]
- Wang, D.; Xiang, Y.K. β-Adrenergic Receptor, Amyloid β-Peptide, and Alzheimer’s Disease. Curr. Top. Membr. 2011, 67, 205–228. [Google Scholar] [PubMed]
- Cirrito, J.R.; Disabato, B.M.; Restivo, J.L.; Verges, D.K.; Goebel, W.D.; Sathyan, A.; Hayreh, D.; D’Angelo, G.; Benzinger, T.; Yoon, H.; et al. Serotonin signaling is associated with lower amyloid- levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. 2011, 108, 14968–14973. [Google Scholar] [CrossRef] [PubMed]
- Sanjari Moghaddam, H.; Zare-Shahabadi, A.; Rahmani, F.; Rezaei, N. Neurotransmission systems in Parkinson’s disease. Rev. Neurosci. 2017, 28, 509–536. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Sahu, T.; Ramanujam, P.L.; Banerjee, A.K.; Chakraborty, I.; Kumar R, A.; Arora, N. Neurochemicals, Behaviours and Psychiatric Perspectives of Neurological Diseases. Neuropsychiatry (London). 2018, 08, 395–424. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Niu, C.; Zhong, S.; Hu, H.; Chen, P.; Zhang, S.; Chen, G.; Deng, F.; Lai, S.; et al. Common and distinct abnormal frontal-limbic system structural and functional patterns in patients with major depression and bipolar disorder. NeuroImage Clin. 2018, 20, 42–50. [Google Scholar] [CrossRef]
- Politis, M.; Loane, C. Serotonergic Dysfunction in Parkinson’s Disease and Its Relevance to Disability. Sci. World J. 2011, 11, 1726–1734. [Google Scholar] [CrossRef]
- de Kloet, S.F.; Mansvelder, H.D.; De Vries, T.J. Cholinergic modulation of dopamine pathways through nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 97, 425–438. [Google Scholar] [CrossRef]
- Soukup, O.; Winder, M.; Killi, U.K.; Wsol, V.; Jun, D.; Kuca, K.; Tobin, G. Acetylcholinesterase Inhibitors and Drugs Acting on Muscarinic Receptors- Potential Crosstalk of Cholinergic Mechanisms During Pharmacological Treatment. Curr. Neuropharmacol. 2017, 15, 637–653. [Google Scholar] [CrossRef] [Green Version]
- Posadas, I.; Lopez-Hernandez, B.; Cena, V. Nicotinic Receptors in Neurodegeneration. Curr. Neuropharmacol. 2013, 11, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Mortensen, O.V. Overview of Monoamine Transporters. Curr. Protoc. Pharmacol. 2017, 79. [Google Scholar] [CrossRef]
- Mortensen, O.V.; Kortagere, S. Designing modulators of monoamine transporters using virtual screening techniques. Front. Pharmacol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Iturriaga-Vásquez, P.; Alzate-Morales, J.; Bermudez, I.; Varas, R.; Reyes-Parada, M. Multiple binding sites in the nicotinic acetylcholine receptors: An opportunity for polypharmacolgy. Pharmacol. Res. 2015, 101, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Parada, M.; Iturriaga-Vasquez, P. The development of novel polypharmacological agents targeting the multiple binding sites of nicotinic acetylcholine receptors. Expert Opin. Drug Discov. 2016, 11, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, F.; Schneider, H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided. Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % of Binding [3H]-Paroxetine h-SERT | Ki (µM) h-DAT | Ki (µM) α4β2 nAChR |
|---|---|---|---|
| Bupropion | N.E | 0.370 | 10.0 |
| 1 | 86.0 ± 0.6 | N.E | 0.023 ± 0.006 |
| 2 | 123.0 ± 1.2 | N.E | 0.094 ± 0.002 |
| 3 | 87.5 ± 1.5 | N.E | 0.009 ± 0.001 |
| 4 | 137.0 ± 1.2 | N.E | 0.132 ± 0.038 |
| 7 | 100.0 ± 0.5 | N.E | 1.788 ± 0.378 |
| 8 | 139.5 ± 2.0 | N.E | N.E |
| 9 | 108.0 ± 1.2 | N.E | 3.461 ± 0.360 |
| 10 | 119.3 ± 2.7 | N.E | 0.042 ± 0.004 |
| 16 | 92.0 ± 0.3 | 22.690 ± 7.099 | N.E |
| 17 | 78.0 ± 0.6 | 3.317 ± 0.923 | N.E |
| 18 | 117.0 ± 2.3 | 99.330 ± 1.411 | 0.120 ± 0.037 |
| 19 | 126.3 ± 1.9 | 44.240 ± 8.120 | N.E |
| 20 | 191.5 ± 0.9 | 1.208 ± 0.230 | 0.023 ± 0.006 |
| 21 | 86.5 ± 1.5 | 0.075 ± 0.009 | 0.113 ± 0.037 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Gutiérrez, J.P.; Pessoa-Mahana, H.A.; Iturriaga-Vásquez, P.E.; Reyes-Parada, M.I.; Guerra-Díaz, N.E.; Hodar-Salazar, M.; Viscarra, F.; Paillali, P.; Núñez-Vivanco, G.; Lorca-Carvajal, M.A.; et al. Synthesis of Novel Nicotinic Ligands with Multimodal Action: Targeting Acetylcholine α4β2, Dopamine and Serotonin Transporters. Molecules 2019, 24, 3808. https://doi.org/10.3390/molecules24203808
González-Gutiérrez JP, Pessoa-Mahana HA, Iturriaga-Vásquez PE, Reyes-Parada MI, Guerra-Díaz NE, Hodar-Salazar M, Viscarra F, Paillali P, Núñez-Vivanco G, Lorca-Carvajal MA, et al. Synthesis of Novel Nicotinic Ligands with Multimodal Action: Targeting Acetylcholine α4β2, Dopamine and Serotonin Transporters. Molecules. 2019; 24(20):3808. https://doi.org/10.3390/molecules24203808
Chicago/Turabian StyleGonzález-Gutiérrez, Juan Pablo, Hernán Armando Pessoa-Mahana, Patricio Ernesto Iturriaga-Vásquez, Miguel Iván Reyes-Parada, Nicolas Esteban Guerra-Díaz, Martin Hodar-Salazar, Franco Viscarra, Pablo Paillali, Gabriel Núñez-Vivanco, Marcos Antonio Lorca-Carvajal, and et al. 2019. "Synthesis of Novel Nicotinic Ligands with Multimodal Action: Targeting Acetylcholine α4β2, Dopamine and Serotonin Transporters" Molecules 24, no. 20: 3808. https://doi.org/10.3390/molecules24203808
APA StyleGonzález-Gutiérrez, J. P., Pessoa-Mahana, H. A., Iturriaga-Vásquez, P. E., Reyes-Parada, M. I., Guerra-Díaz, N. E., Hodar-Salazar, M., Viscarra, F., Paillali, P., Núñez-Vivanco, G., Lorca-Carvajal, M. A., Mella-Raipán, J., & Zúñiga, M. C. (2019). Synthesis of Novel Nicotinic Ligands with Multimodal Action: Targeting Acetylcholine α4β2, Dopamine and Serotonin Transporters. Molecules, 24(20), 3808. https://doi.org/10.3390/molecules24203808