
One-Pot Two-Step Synthesis of 2-Aryl benzimidazole N-oxides Using Microwave Heating as a Tool



Abstract
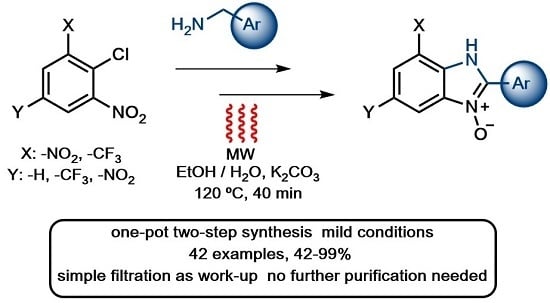
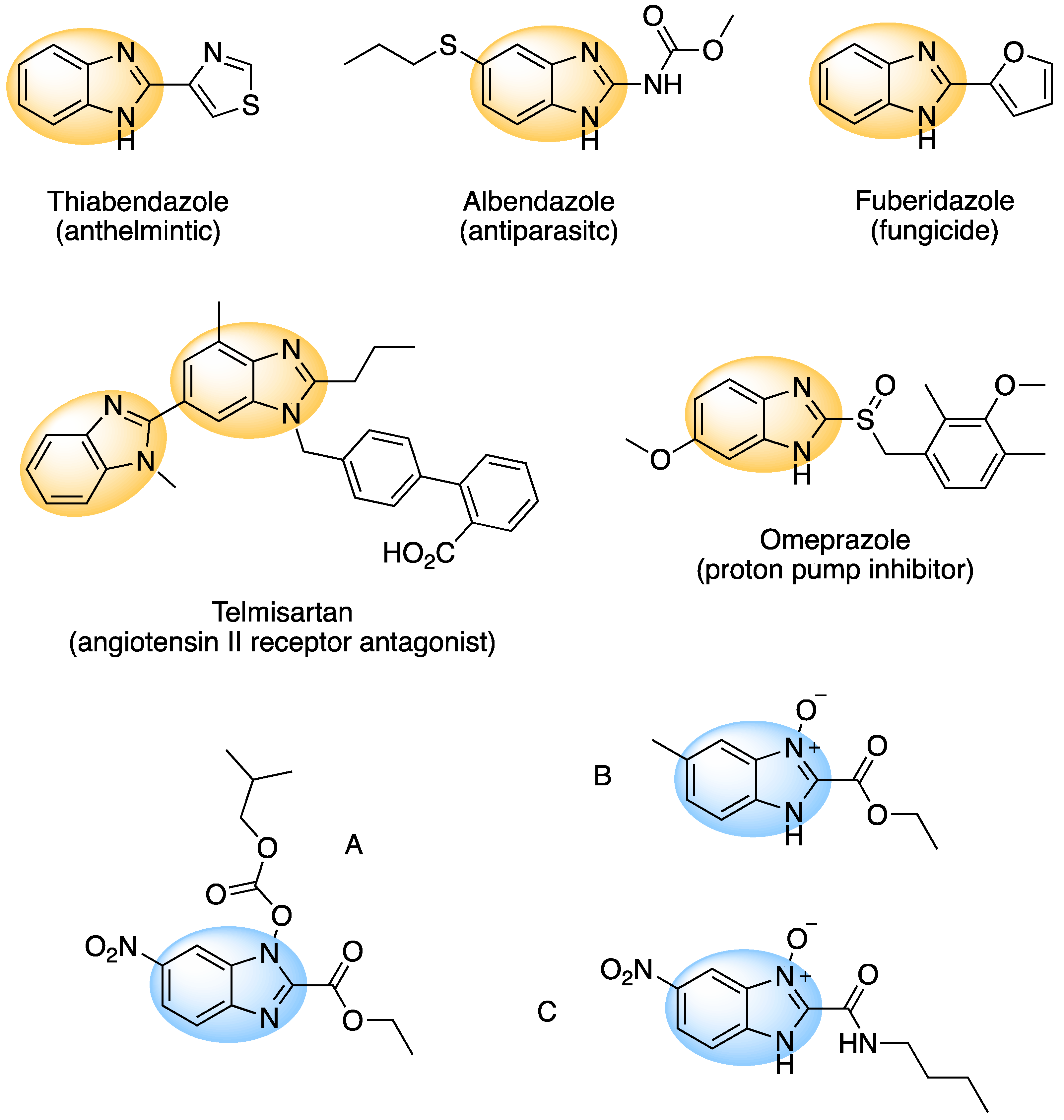
1. Introduction
2. Results and Discussion
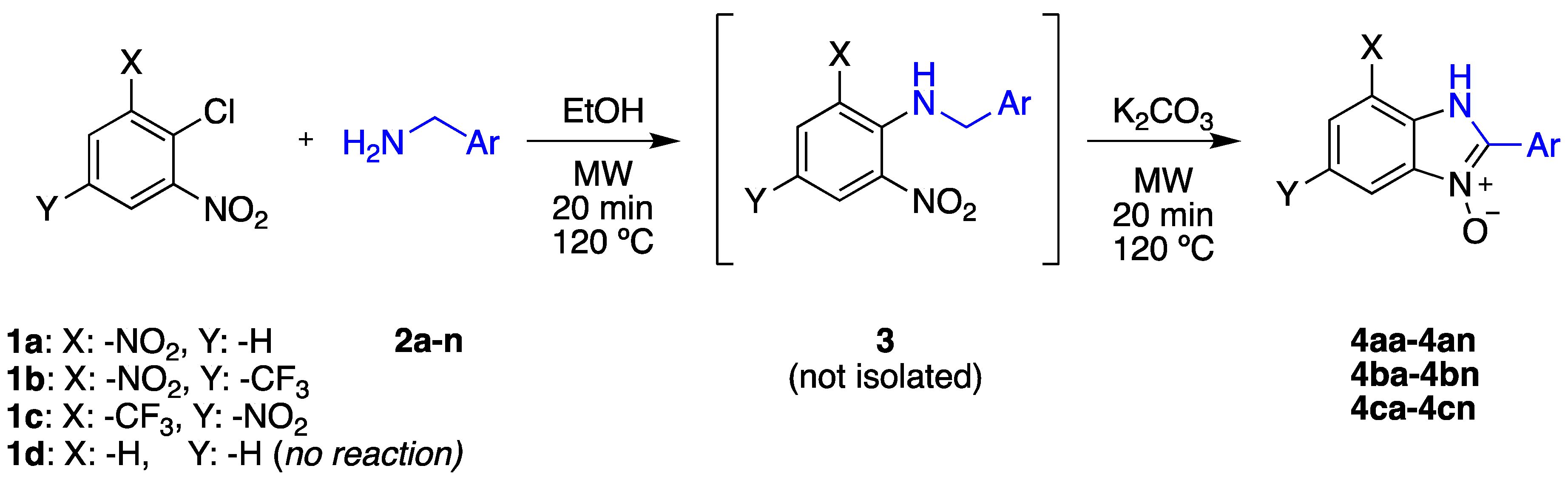
2.1. Synthesis
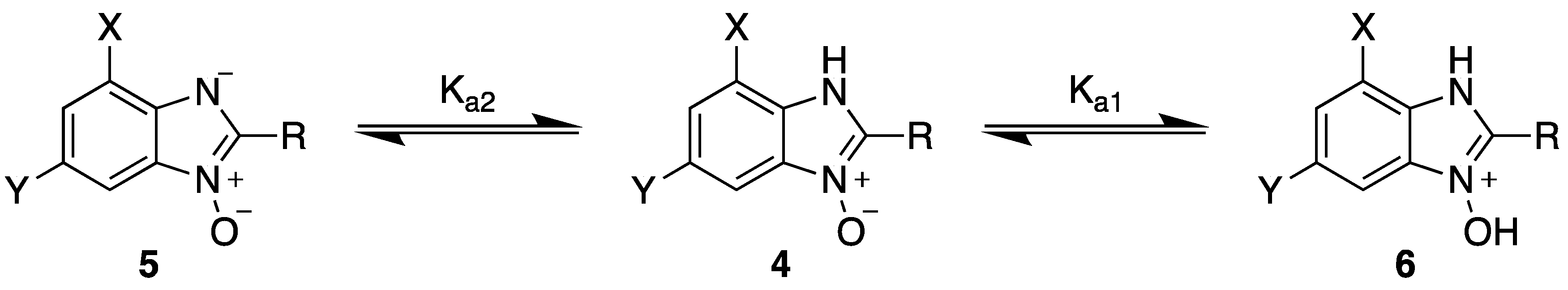
2.2. Product Isolation
2.3. Expanding the Substrate Scope, Applicability, and Scale of the Reaction
3. Materials and Methods
3.1. General
3.2. Chemicals
3.3. Representative Procedure for the Synthesis of Benzimidazole-N-oxides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeSimone, R.W.; Currie, K.S.; Mitchell, S.A.; Darrow, J.W.; Pippin, D.A. Privileged structures: Applications in drug discovery. Comb. Chem. High Throughput Screen. 2004, 7, 473–494. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Preston, P.N. Synthesis, reactions, and spectroscopic properties of benzimidazoles. Chem. Rev. 1974, 74, 279–314. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorganic Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef] [PubMed]
- Mamedov, V.A. Recent advances in the synthesis of benzimidazol(on)es: Via rearrangements of quinoxalin(on)es. RSC Adv. 2016, 6, 42132–42172. [Google Scholar] [CrossRef]
- Narasimhan, B.; Sharma, D.; Kumar, P. Benzimidazole: A medicinally important heterocyclic moiety. Med. Chem. Res. 2012, 21, 269–283. [Google Scholar] [CrossRef]
- Gardiner, J.M.; Loyns, C.R. Synthesis of novel 1-, 1,4- and 1,7-substituted 2-mercapto- and 2-methylmercapto- benzimidazoles: Acyclic analogues of the HIV-1 RT inhibitor, TIBO. Tetrahedron 1995, 51, 11515–11530. [Google Scholar] [CrossRef]
- Hayes, M.E.; Wallace, G.A.; Grongsaard, P.; Bischoff, A.; George, D.M.; Miao, W.; McPherson, M.J.; Stoffel, R.H.; Green, D.W.; Roth, G.P. Discovery of small molecule benzimidazole antagonists of the chemokine receptor CXCR3. Bioorganic Med. Chem. Lett. 2008, 18, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Scheers, J.; Johansson, P.; Szczeciński, P.; Wieczorek, W.; Armand, M.; Jacobsson, P. Benzimidazole and imidazole lithium salts for battery electrolytes. J. Power Sources 2010, 195, 6081–6087. [Google Scholar] [CrossRef]
- Niedzicki, L.; Oledzki, P.; Bitner, A.; Bukowska, M.; Szczecinski, P. Benzimidazole-derived anion for lithium-conducting electrolytes. J. Power Sources 2016, 306, 573–577. [Google Scholar] [CrossRef]
- Wang, F.M.; Pradanawati, S.A.; Yeh, N.H.; Chang, S.C.; Yang, Y.T.; Huang, S.H.; Lin, P.L.; Lee, J.F.; Sheu, H.S.; Lu, M.L.; et al. Robust Benzimidazole-Based Electrolyte Overcomes High-Voltage and High-Temperature Applications in 5 v Class Lithium Ion Batteries. Chem. Mater. 2017, 29, 5537–5549. [Google Scholar] [CrossRef]
- Li, D.; Wu, P.; Sun, N.; Lu, Y.-J.; Wong, W.-L.; Fang, Z.; Zhang, K. The Diversity of Heterocyclic N-oxide Molecules: Highlights on their Potential in Organic Synthesis, Catalysis and Drug Applications. Curr. Org. Chem. 2019, 23, 616–627. [Google Scholar] [CrossRef]
- Boiani, M.; Boiani, L.; Denicola, A.; Torres De Ortiz, S.; Serna, E.; Vera De Bilbao, N.; Sanabria, L.; Yaluff, G.; Nakayama, H.; Rojas De Arias, A.; et al. 2H-benzimidazole 1,3-dioxide derivatives: A new family of water-soluble anti-trypanosomatid agents. J. Med. Chem. 2006, 49, 3215–3224. [Google Scholar] [CrossRef]
- Chugunova, E.A.; Samsonov, V.A.; Gazizov, A.S.; Burilov, A.R.; Pudovik, M.A.; Sinyashin, O.G. 2H-Benzimidazole N-oxides: Synthesis, chemical properties, and biological activity. Russ. Chem. Bull. 2018, 67, 1955–1970. [Google Scholar] [CrossRef]
- Albini, A. Synthetic Utility of Amine N-Oxides. Synthesis (Stuttg). 1993, 1993, 263–277. [Google Scholar] [CrossRef]
- Boiani, M.; Gonzalez, M. Imidazole and Benzimidazole Derivatives as Chemotherapeutic Agents. Mini-Reviews Med. Chem. 2005, 5, 409–424. [Google Scholar] [CrossRef]
- Aguirre, G.; Boiani, M.; Cerecetto, H.; Gerpe, A.; González, M.; Sainz, Y.F.; Denicola, A.; de Ocáriz, C.O.; Nogal, J.J.; Montero, D.; et al. Novel Antiprotozoal Products: Imidazole and BenzimidazoleN-Oxide Derivatives and Related Compounds. Arch. Pharm. (Weinheim) 2004, 337, 259–270. [Google Scholar] [CrossRef]
- Nikitina, P.A.; Perevalov, V.P. Methods of synthesis and physicochemical properties of 1-hydroxyimidazoles, imidazole 3-oxides, and their benzoannulated analogs. Chem. Heterocycl. Compd. 2017, 53, 123–149. [Google Scholar] [CrossRef]
- Blaszczak-Światkiewicz, K.; Mirowski, M.; Kaplinska, K.; Kruszynśki, R.; Trzesowska-Kruszyńska, A.; Mikiciuk-Olasik, E. New benzimidazole derivatives with potential cytotoxic activity—Study of their stability by RP-HPLC. Acta Biochim. Pol. 2012, 59, 279–288. [Google Scholar] [CrossRef]
- Buján de Vargas, E.I.; Cañas, A.I. From N-n-butyl-2,6-dinitroaniline to a Fused Heterocyclic N-oxide. Tetrahedron Lett. 1996, 37, 767–770. [Google Scholar] [CrossRef]
- Buján, E.I.; Salum, M.L. A simple synthesis of benzimidazole N-oxides from 2-nitroaniline derivatives—Scope and limitations. Can. J. Chem. 2004, 82, 1322–1327. [Google Scholar] [CrossRef]
- Buján, E.I.; Salum, M.L. FromN-(dinitrophenyl) amino acids to benzimidazole N-oxides. Synthesis, kinetics and mechanism. J. Phys. Org. Chem. 2006, 19, 187–195. [Google Scholar] [CrossRef]
- Prat, D.; Pardigon, O.; Flemming, H.W.; Letestu, S.; Ducandas, V.; Isnard, P.; Guntrum, E.; Senac, T.; Ruisseau, S.; Cruciani, P.; et al. Sanofi’s Solvent Selection Guide: A Step Toward More Sustainable Processes. Org. Process Res. Dev. 2013, 17, 1517–1525. [Google Scholar] [CrossRef]
- Prat, D.; Hayler, J.; Wells, A. A Survey of Solvent Selection Guides. Green Chem. 2014, 16, 4546–4551. [Google Scholar] [CrossRef]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C.R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef]
- Alfonsi, K.; Colberg, J.; Dunn, P.J.; Fevig, T.; Jennings, S.; Johnson, T.A.; Kleine, H.P.; Knight, C.; Nagy, M.A.; Perry, D.A.; et al. Green chemistry tools to influence a medicinal chemistry and research chemistry based organisation. Green Chem. 2008, 10, 31–36. [Google Scholar] [CrossRef]
- Alder, C.M.; Hayler, J.D.; Henderson, R.K.; Redman, A.M.; Shukla, L.; Shuster, L.E.; Sneddon, H.F. Updating and further expanding GSK’s solvent sustainability guide. Green Chem. 2016, 18, 3879–3890. [Google Scholar] [CrossRef]
- Diorazio, L.J.; Hose, D.R.J.; Adlington, N.K. Toward a More Holistic Framework for Solvent Selection. Org. Process Res. Dev. 2016, 20, 760–773. [Google Scholar] [CrossRef]
- Politano, F.; Buján, E.I.; Leadbeater, N.E. Preparation of benzimidazole N-oxides by a two-step continuous flow process. Chem. Heterocycl. Compd. 2016, 52, 952–957. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Loupy, A.; de la Hoz, A. (Eds.) Microwaves in Organic Synthesis, 3rd ed.; Wiley: Weinheim, Germany, 2012. [Google Scholar]
- Leadbeater, N. Microwave Heating as a Tool for Sustainable Chemistry; CRC Press: Boca Raton, FL, USA, 2011; ISBN 9781439812709. [Google Scholar]
- Kappe, C.O.; Dallinger, D.; Murphree, S.S. Practical Microwave Synthesis for Organic Chemists; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; ISBN 9783527623907. [Google Scholar]
- Politano, F. Síntesis de N-óxidos de Bencimidazol Asistida por Microondas y en Flujo Continuo. Ph.D. Thesis, Universidad Nacional de Córdoba, Córdoba, Argentina, 2018. [Google Scholar]
- Szczeciński, P.; Bartusik, D. Cyclisation of N-alkyl-2,4-dinitro-6-trifluoromethylanilines. J. Chem. Res. 2002, 2002, 84–85. [Google Scholar] [CrossRef]
- Buján, E.I.; Cañas, A.I.; de Rossi, R.H. Amines as leaving groups in nucleophilic aromatic substitution reactions. Part 5.1 Substitution vs. N-oxide formation in the reaction of N-n-butyl-2,6-dinitroaniline with hydroxide ions. J. Chem. Soc. Perkin Trans. 2 2001, 1973–1977. [Google Scholar]
Sample Availability: Samples of the compounds reported here are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Isolated Yield (%) | |
|---|---|---|
| Previously Reported Work-Up | New Work-Up | |
 | 31 | 86 |
| 4ai | ||
 | 14 | 84 |
| 4ac | ||
 | 31 | 85 |
| 4ah | ||
 | 20 | 79 |
| 4al | ||
| Amine |  |  |  |
|---|---|---|---|
 |  |  |  |
| 2a | 4aa (95%; 95% *) | 4ba (88%) | 4ca (88%) |
 |  |  |  |
| 2b | 4ab (88%) | 4bb (98%) | 4cb (90%) |
 |  |  |  |
| 2c | 4ac (76%) | 4bc (93%) | 4cc (83%) |
 |  |  |  |
| 2d | 4ad (99%) | 4bd (86%) | 4cd (76%) |
 |  |  |  |
| 2e | 4ae (99%) | 4be (90%) | 4ce (78%) |
 |  |  |  |
| 2f | 4af (98%) | 4bf (91%) | 4cf (84%) |
 |  |  |  |
| 2g | 4ag (51%) | 4bg (99%) | 4cg (92%) |
 |  |  |  |
| 2h | 4ah (77%) | 4bh (96%) | 4ch (67%) |
 |  |  |  |
| 2i | 4ai (86%) | 4bi (89%) | 4ci (99%) |
 |  |  |  |
| 2j | 4aj (88%) | 4bj (85%) | 4cj (80%) |
 |  |  |  |
| 2k | 4ak (74%) | 4bk (96%) | 4ck (94%) |
 |  |  |  |
| 2l | 4al (79%) | 4bl (98%) | 4cl (84%) |
 |  |  |  |
| 2m | 4am (61%) | 4bm (87%) | 4cm (84%) |
 |  |  |  |
| 2n | 4an (42%) | 4bn (52%) | 4cn (73%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Politano, F.; Gran-Magano, A.K.; Leadbeater, N.E. One-Pot Two-Step Synthesis of 2-Aryl benzimidazole N-oxides Using Microwave Heating as a Tool. Molecules 2019, 24, 3639. https://doi.org/10.3390/molecules24203639
Politano F, Gran-Magano AK, Leadbeater NE. One-Pot Two-Step Synthesis of 2-Aryl benzimidazole N-oxides Using Microwave Heating as a Tool. Molecules. 2019; 24(20):3639. https://doi.org/10.3390/molecules24203639
Chicago/Turabian StylePolitano, Fabrizio, Ana K. Gran-Magano, and Nicholas E. Leadbeater. 2019. "One-Pot Two-Step Synthesis of 2-Aryl benzimidazole N-oxides Using Microwave Heating as a Tool" Molecules 24, no. 20: 3639. https://doi.org/10.3390/molecules24203639
APA StylePolitano, F., Gran-Magano, A. K., & Leadbeater, N. E. (2019). One-Pot Two-Step Synthesis of 2-Aryl benzimidazole N-oxides Using Microwave Heating as a Tool. Molecules, 24(20), 3639. https://doi.org/10.3390/molecules24203639