Design and Selection of Novel C1s Inhibitors by In Silico and In Vitro Approaches

 ,
, 

Abstract
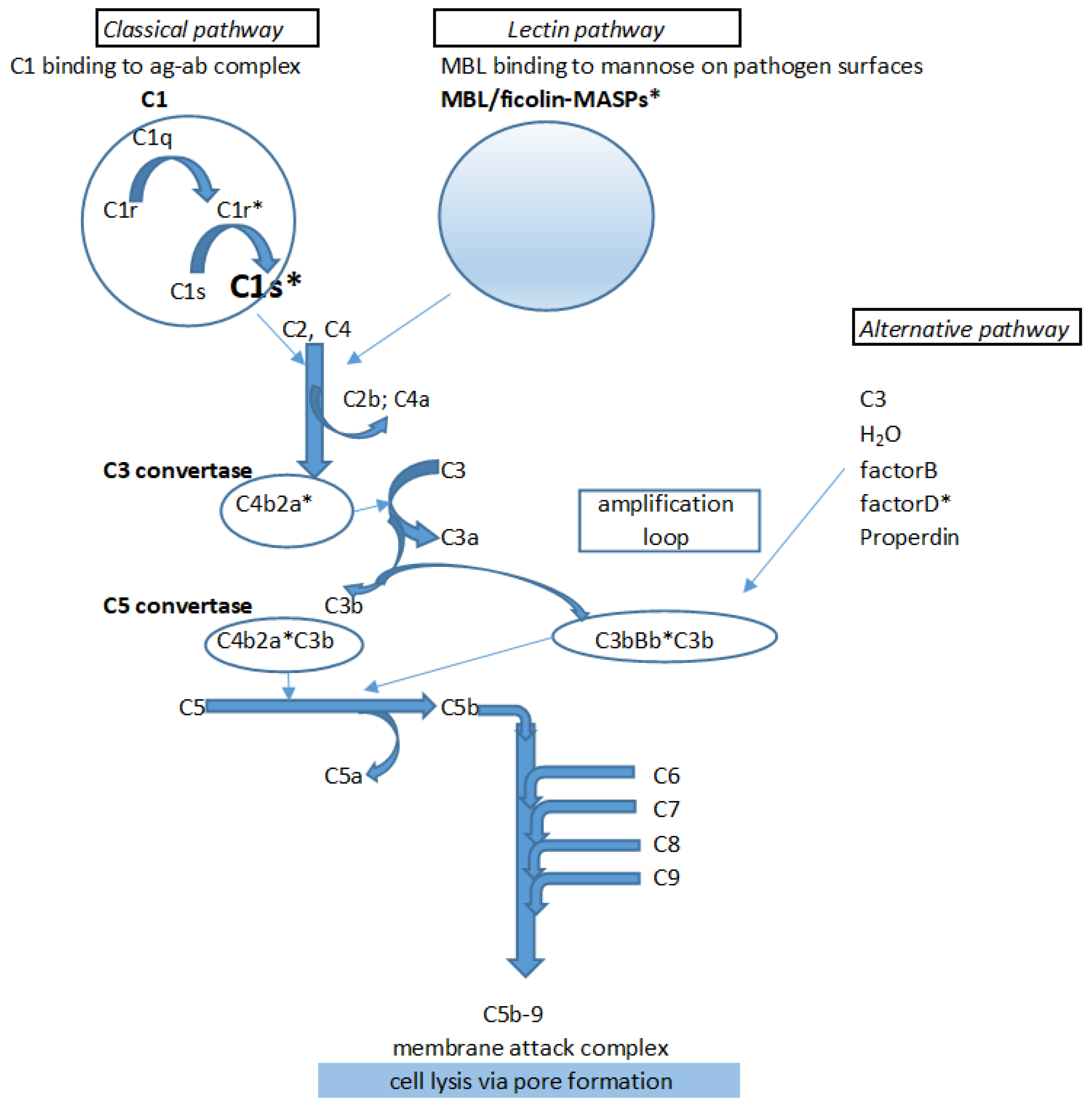
1. Introduction
2. Results
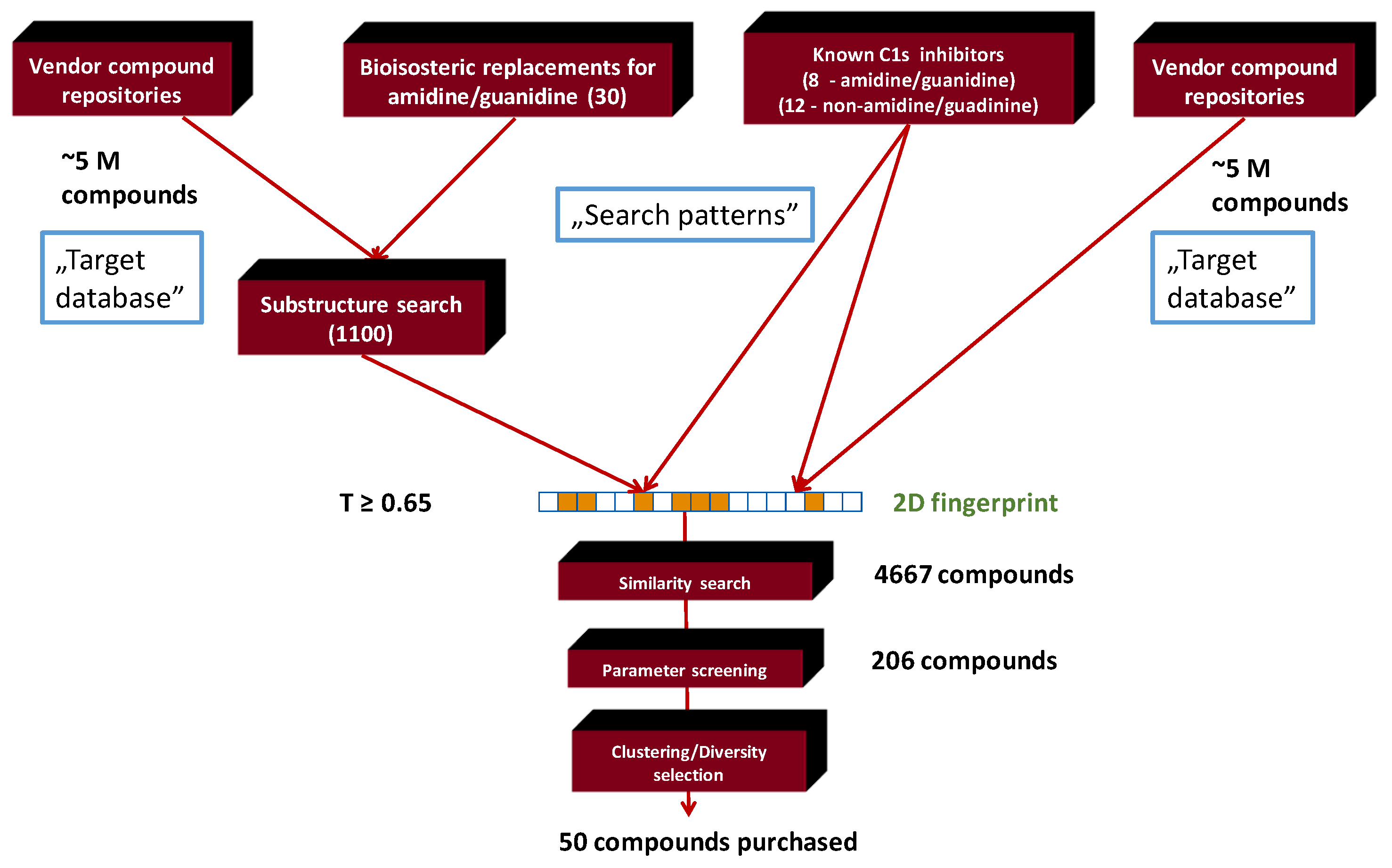
2.1. Generation of C1s Inhibitor Focused Library (by 2D Methods)
2.2. Generation of C1s Inhibitor Focused Library (Pharmacophore Search Methods)
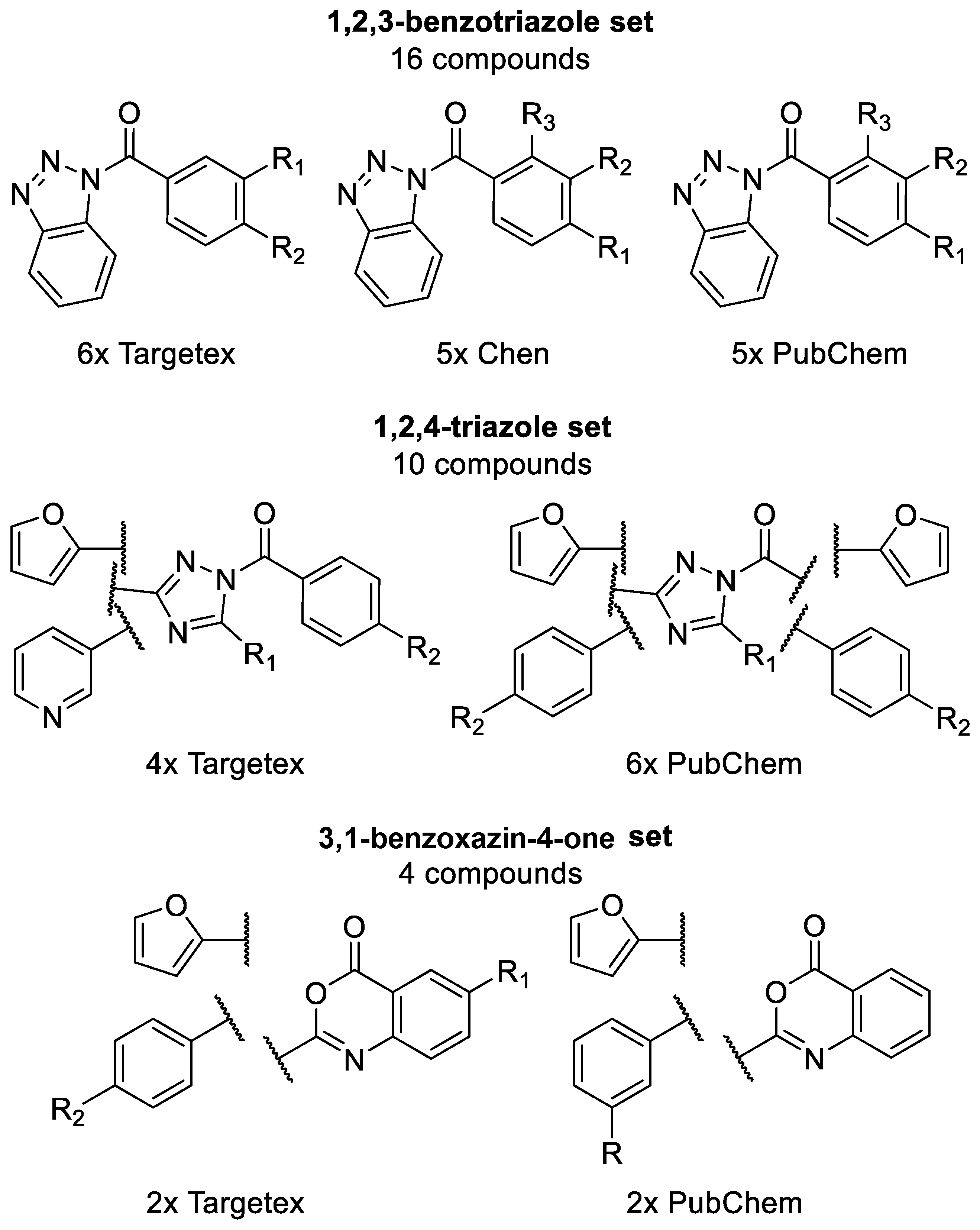
2.3. Generation of C1s Inhibitor Hit-Validation Small Library
2.4. In Vitro Screening Results
2.4.1. C1s Inhibition
2.4.2. Factor Xa Inhibition
3. Discussion
3.1. Structure–Activity Relationship of the Chemotypes
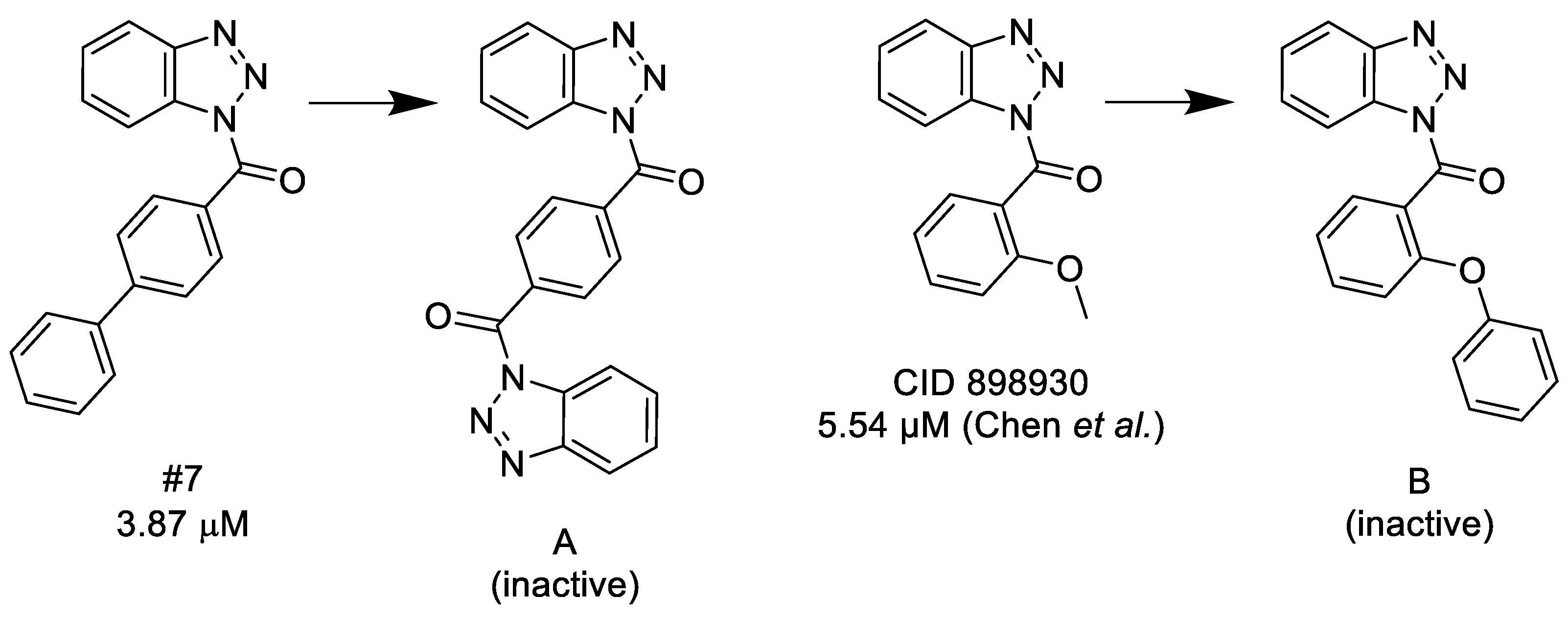
3.1.1. 1,2,3-Benzotriazoles
- (1)
- A substituted benzoyl group in the N-1 position is required for the activity. Unsubstituted benzoyl groups or replacement with an N-1 furoyl group is detrimental to the activity.
- (2)
- The position and nature of the functional groups on the benzoyl group have a significant impact on the activity. The para position is much favored, followed by the ortho position. Substituents in the meta position generally diminish or occasionally fully eliminate the activity. However, meta/para disubstitution somewhat counterbalances this negative effect.
- (3)
- Electron donating groups (amide, ether, thioether) at the para position are particularly beneficial. Alkyl groups are weaker in the para position, but interestingly bulky alkyl groups (t-butyl) look better. Interestingly, while a meta/para difluoro-substituted compound is relatively good inhibitor, analogous dichloro substitution and para-bromo substitution led to inactive compounds. Electron acceptor groups in the para position (such as nitro group) eliminate the activity.
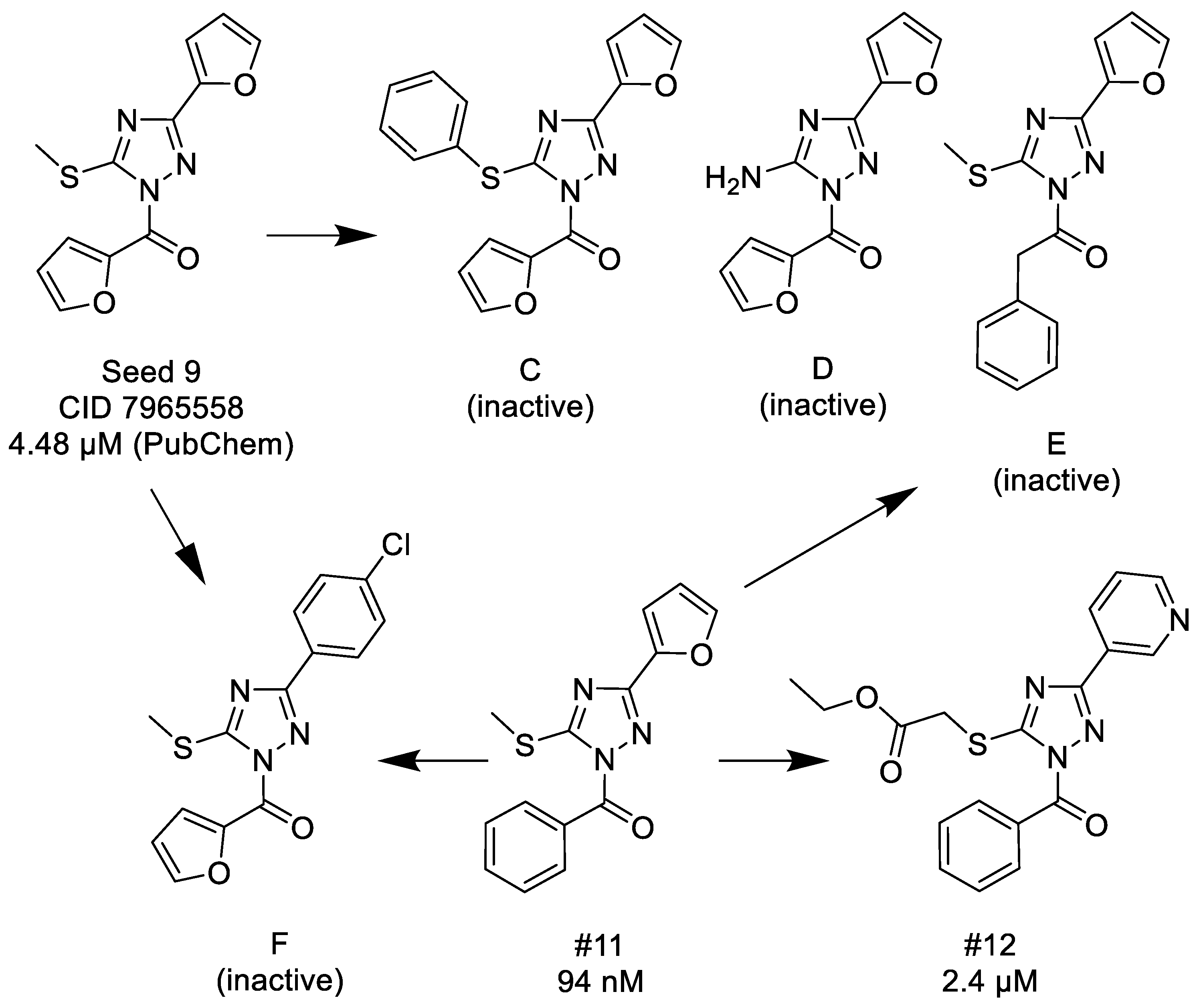
3.1.2. 1,2,4-Triazoles
- (1)
- Unsubstituted aroyl (furan-carbonyl, thiophene-carbonyl and benzoyl) groups in the N-1 position are beneficial. Ortho and para substitutions of the N-1 benzoyl group (such as methyl, methoxy, and fluoro) have also positive contributions. Replacing the N-1 aroyl groups to propionyl, benzyl-carbonyl leads to the loss of the activity.
- (2)
- Replacing the methyl-thio group to primary/secondary amines or bulky aralkyl groups results in inactive compounds.
- (3)
- Changing the C-3 furyl group to thiophenyl (PubChem, CID 4257399; IC50 = 880 nM) or 3-pyridyl group (#12; IC50 = 2.4 μM) retains the activity, while replacing with para-chloro-phenyl leads to inactivation.
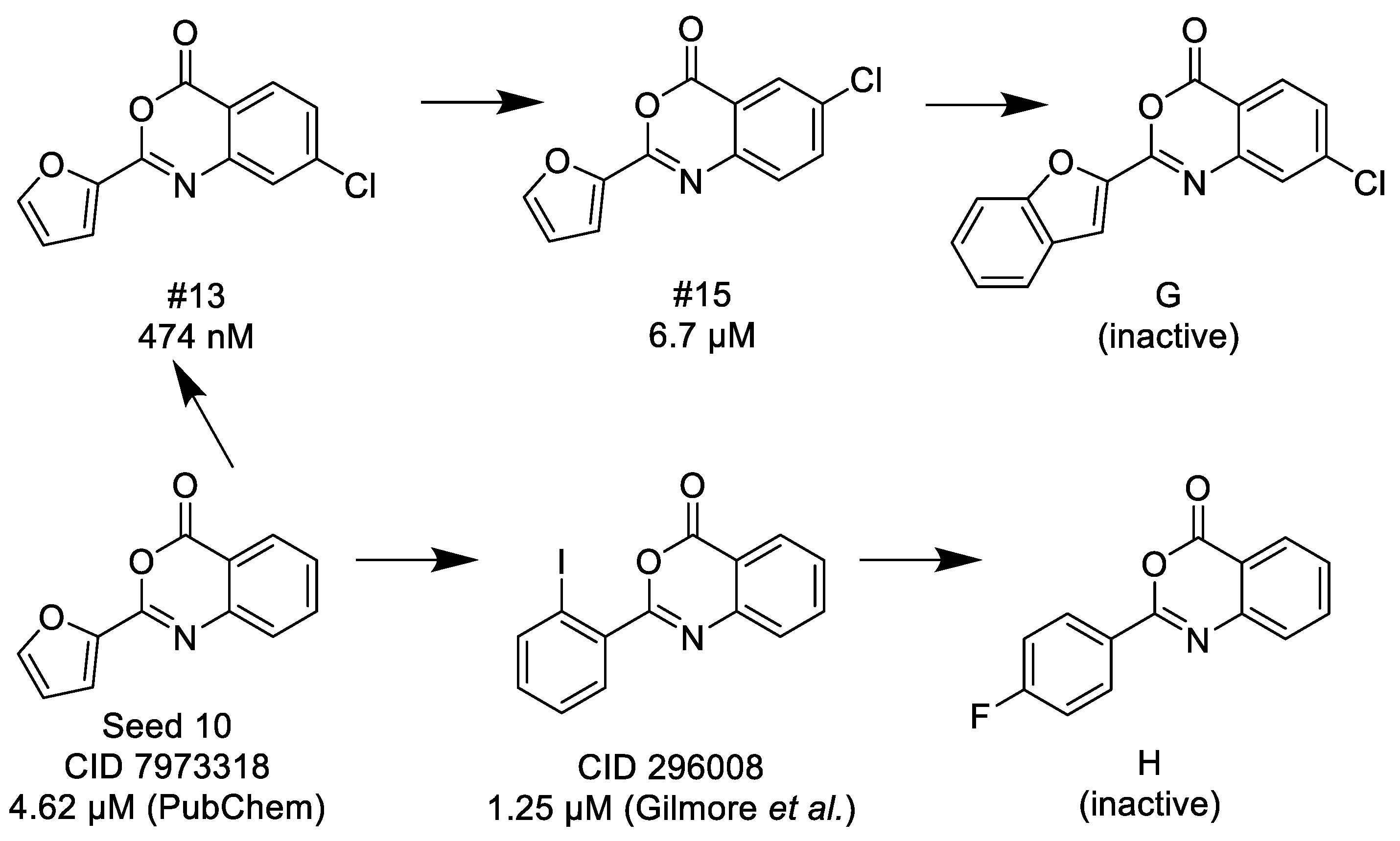
3.1.3. 3,1-Benzoxazin-4-ones
- (1)
- Halogen (chloro) in the C-7 position is much favored over the C-6 position as well as to the unsubstituted 2-aryl-3,1-benzoxazin-4-ones.
- (2)
- Replacing the furyl group to 2′-iodo-phenyl at the C-2 position improves the activity, while 4′-fluorophenyl group in the same position eliminates the activity.
- (3)
- If the furyl group at the C-2 position replaced with a bulky 2-benzofuryl group the compound loses the activity.
3.1.4. 1,3-Benzoxazin-4-ones
- (1)
- Since thiophene and furan are interchangeable in many bioactive compounds, therefore, the trend of the decreasing activity between #21 and #22 is comparable to the same direction between #13 and #15, thus, halogen (chloro) in the C-7 position is much favored over the C-6 position.
- (2)
- Introducing 4′-chlorophenyl group or alkyl groups at the C-2 position instead of furyl or thiophenyl groups leads to inactivation.
- (3)
- Finally, if N-3 is replaced with carbon forming the corresponding chromen-4-one, it results in a complete loss of the activity.
3.1.5. Thieno[2,3-d][1,3]oxazin-4-ones
- (1)
- Similarly to 3,1-benzoxazin-4-ones replacing the furyl group with 2′-halogeno (bromo)-phenyl at the C-2 position retains the activity, while unsubstituted phenyl, 4′-tolyl, 4′-halogeno (fluoro, chloro) substituted phenyl groups in the same position significantly reduce or completely eliminate the activity.
- (2)
- Introducing a bulky (thiophenyl) group into the thiophenyl part of the fused ring leads to complete inactivation.
3.1.6. Structural Features of the C1s Selectivity over Factor Xa
4. Materials and Methods
4.1. Molecular Biology. Protein Expression of C1s
4.2. Assay Development
4.3. Factor Xa (FXa) Assay Development
4.4. Chemoinformatics Methods
4.4.1. 2D Similarity Selection
4.4.2. 3D Modelling
X-Ray Structures
Ligand Preparation
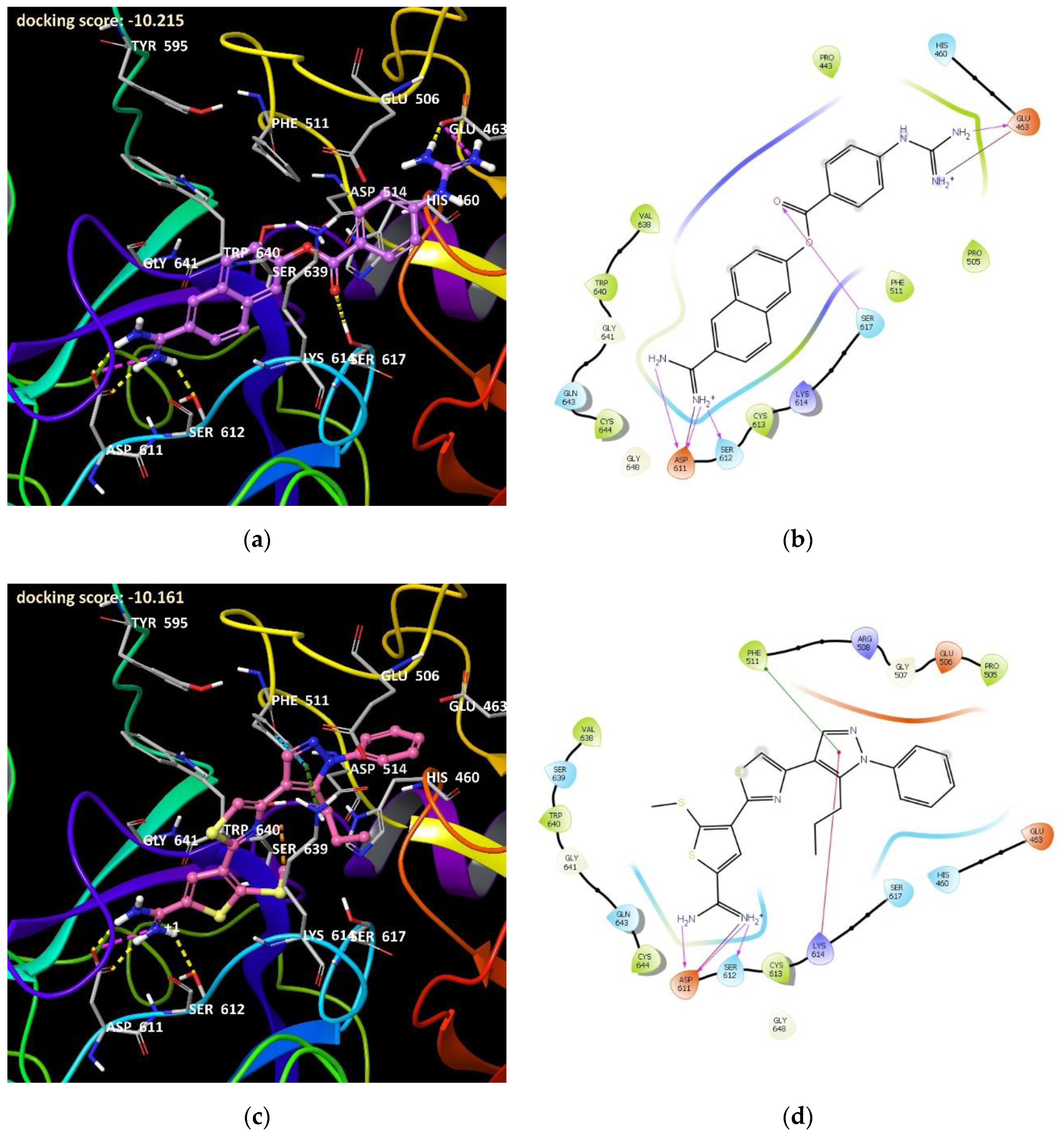
Protein Preparation and Docking
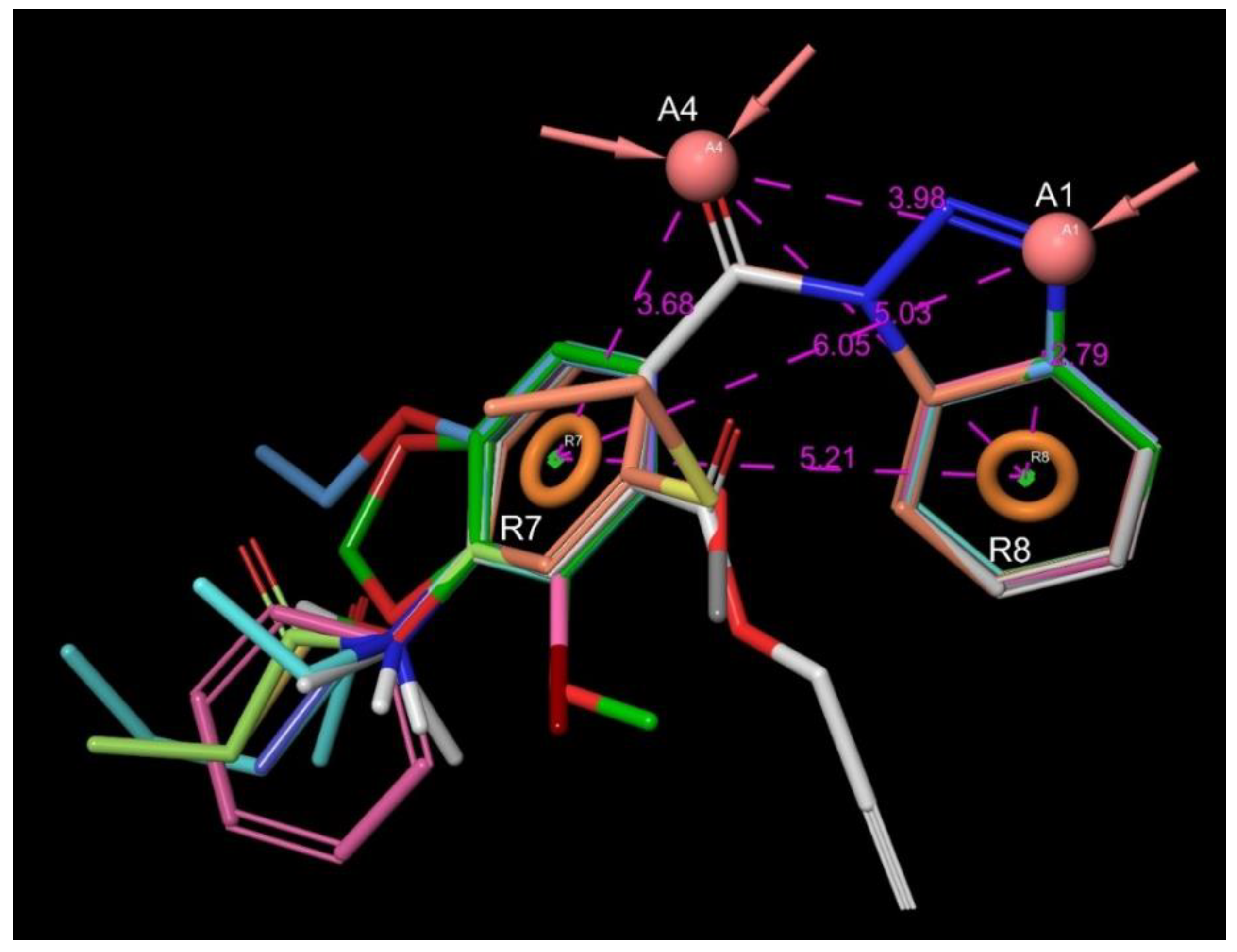
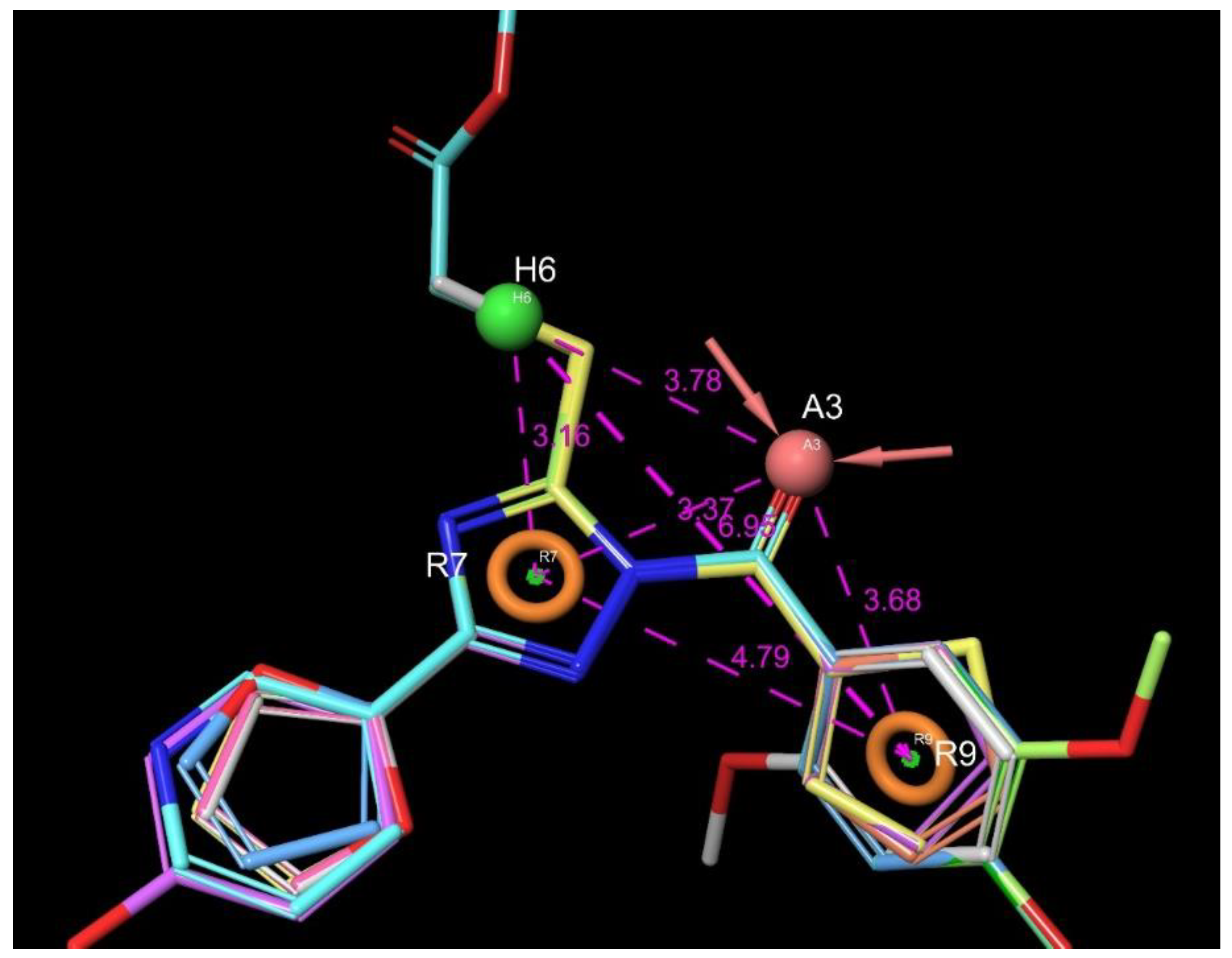
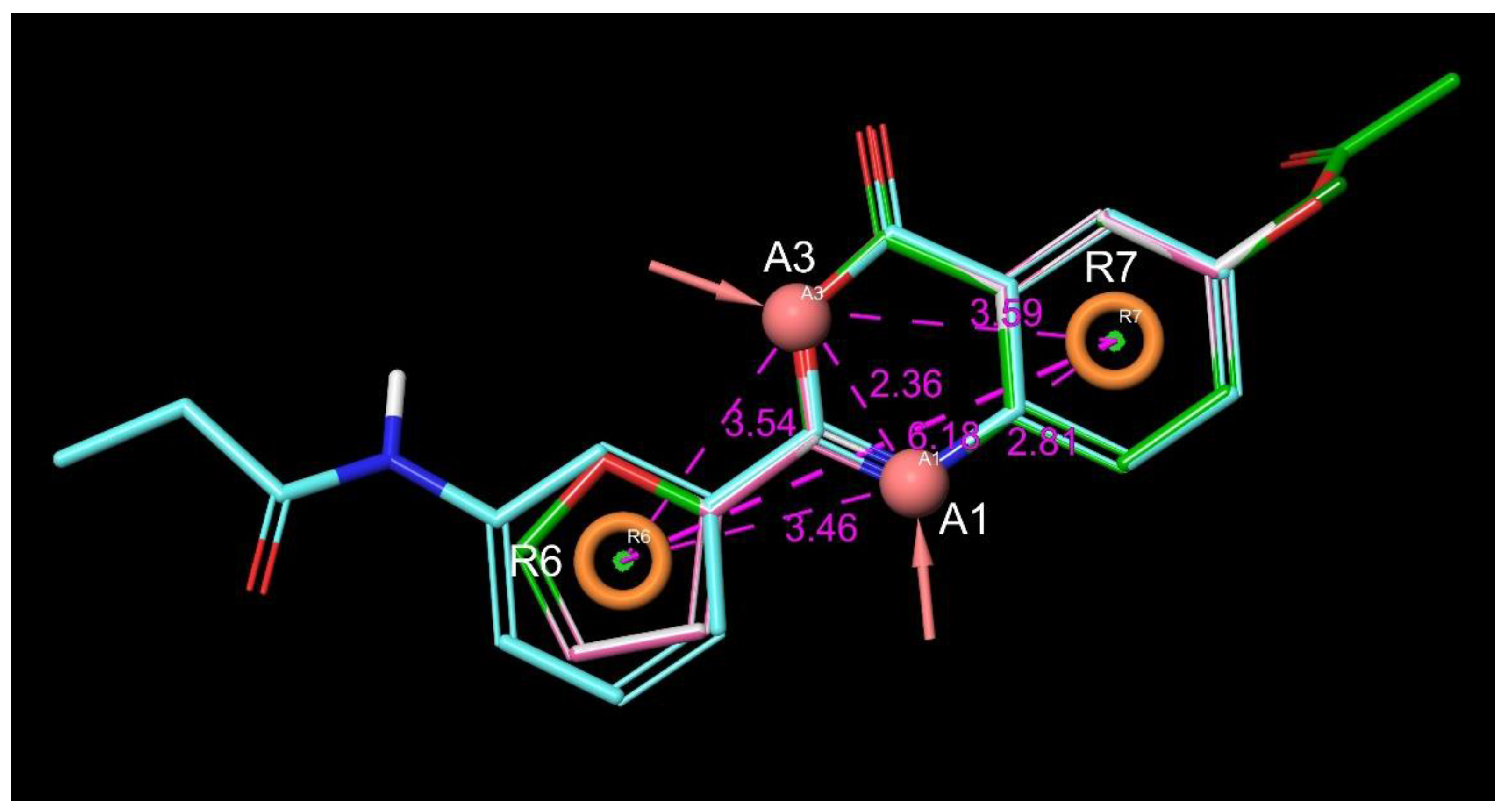
4.4.3. Pharmacophore Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dobo, J.; Kocsis, A.; Gal, P. Be on target: Strategies of targeting alternative and lectin pathway components in complement-mediated diseases. Front. Immunol. 2018, 9, 1851. [Google Scholar] [CrossRef] [PubMed]
- Gal, P.; Dobo, J.; Beinrohr, L.; Pal, G.; Zavodszky, P. Inhibition of the serine proteases of the complement system. Adv. Exp. Med. Biol. 2013, 735, 23–40. [Google Scholar] [PubMed]
- Sharp, J.A.; Whitley, P.H.; Cunnion, K.M.; Krishna, N.K. Peptide inhibitor of complement c1, a novel suppressor of classical pathway activation: Mechanistic studies and clinical potential. Front. Immunol. 2014, 5, 406. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Ino, Y.; Ozeki, M.; Oda, M.; Sato, T.; Koshiyama, Y.; Suzuki, S.; Fujita, M. Pharmacological studies of FUT-175, nafamstat mesilate. I. Inhibition of protease activity in in vitro and in vivo experiments. Jpn. J. Pharm. 1984, 35, 203–227. [Google Scholar] [CrossRef]
- Subasinghe, N.L.; Ali, A.; Illig, C.R.; Rudolph, M.J.; Klein, S.; Khalil, E.; Soll, R.M.; Bone, R.F.; Spurlino, J.C.; DesJarlais, R.L.; et al. A novel series of potent and selective small molecule inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2004, 14, 3043–3047. [Google Scholar] [CrossRef]
- Subasinghe, N.L.; Travins, J.M.; Ali, F.; Huang, H.; Ballentine, S.K.; Marugan, J.J.; Khalil, E.; Hufnagel, H.R.; Bone, R.F.; DesJarlais, R.L.; et al. A novel series of arylsulfonylthiophene-2-carboxamidine inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2006, 16, 2200–2204. [Google Scholar] [CrossRef]
- Travins, J.M.; Ali, F.; Huang, H.; Ballentine, S.K.; Khalil, E.; Hufnagel, H.R.; Pan, W.X.; Gushue, J.; Leonard, K.; Bone, R.F.; et al. Biphenylsulfonyl-thiophene-carboxamidine inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2008, 18, 1603–1606. [Google Scholar] [CrossRef]
- Subasinghe, N.L.; Khalil, E.; Travins, J.M.; Ali, F.; Ballentine, S.K.; Hufnagel, H.R.; Pan, W.; Leonard, K.; Bone, R.F.; Soll, R.M.; et al. Design and synthesis of polyethylene glycol-modified biphenylsulfonyl-thiophene-carboxamidine inhibitors of the complement component C1s. Bioorg. Med. Chem. Lett. 2012, 22, 5303–5307. [Google Scholar] [CrossRef]
- Diamond, S.L. Complement. Factor C1s IC50 from Mixture Screen; University of Pennsylvania: Pennsylvania, PA, USA, 2007. [Google Scholar]
- Diamond, S.L. Complement. Factor C1s; University of Pennsylvania: Pennsylvania, PA, USA, 2006. [Google Scholar]
- Chen, J.J.; Schmucker, L.N.; Visco, D.P. Pharmaceutical machine learning: Virtual high-throughput screens identifying promising and economical small molecule inhibitors of complement factor C1s. Biomolecules 2018, 8, 24. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.S.; Bologa, C.; Young, S.M.; Balakin, K.V.; Prossnitz, E.R.; Savchuck, N.P.; Sklar, L.A.; Oprea, T.I. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharm. 2005, 68, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Virtual Screening: Principles, Challenges, and Practical Guidelines, 1st ed.; Wiley-VCH: Weinheim, Germany, 2011; p. 519. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Emolecules. Available online: https://www.emolecules.com/ (accessed on 30 January 2019).
- ASINEX Screening Libraries. Available online: http://www.asinex.com/libraries-html/ (accessed on 27 March 2018).
- ChemBridge Screening Libraries. Available online: https://www.chembridge.com/screening_libraries/ (accessed on 14 April 2018).
- ChemDiv Compound Libraries and Screening Compounds. Available online: http://www.chemdiv.com/services-menu/screening-libraries/ (accessed on 12 March 2018).
- Enamine Building Blocks Screening Collection. Available online: https://enamine.net/hit-finding/compound-collections/screening-collection (accessed on 9 March 2018).
- InterBioScreen Synthetic Compounds. Available online: https://www.ibscreen.com/ (accessed on 10 March 2018).
- Life Chemicals Screening Libraries. Available online: https://lifechemicals.com/screening-libraries/hts-compound-collection (accessed on 13 April 2018).
- SPECS Compound Library. Available online: https://www.specs.net/ (accessed on 13 April 2018).
- UkrOrgSyntez Ltd. (UORSY) Screening Compounds. Available online: https://uorsy.com/screening-compounds/ (accessed on 7 January 2018).
- Gaboriaud, C.; Rossi, V.; Bally, I.; Arlaud, G.J.; Fontecilla-Camps, J.C. Crystal structure of the catalytic domain of human complement C1s: A serine protease with a handle. EMBO J. 2000, 19, 1755–1765. [Google Scholar] [CrossRef]
- Page, M.J.; Macgillivray, R.T.; Di Cera, E. Determinants of specificity in coagulation proteases. J. Thromb. Haemost. 2005, 3, 2401–2408. [Google Scholar] [CrossRef]
- Kerr, F.K.; O’Brien, G.; Quinsey, N.S.; Whisstock, J.C.; Boyd, S.; de la Banda, M.G.; Kaiserman, D.; Matthews, A.Y.; Bird, P.I.; Pike, R.N. Elucidation of the substrate specificity of the C1s protease of the classical complement pathway. J. Biol. Chem. 2005, 280, 39510–39514. [Google Scholar] [CrossRef]
- Hsu, H.J.; Tsai, K.C.; Sun, Y.K.; Chang, H.J.; Huang, Y.J.; Yu, H.M.; Lin, C.H.; Mao, S.S.; Yang, A.S. Factor Xa active site substrate specificity with substrate phage display and computational molecular modeling. J. Biol. Chem. 2008, 283, 12343–12353. [Google Scholar] [CrossRef]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Ujváry, H.; Hayward, J. Bioisosteres in Medicinal Chemistry, 1st ed.; Wiley-VCH: New York, NY, USA, 2012; p. 53. [Google Scholar]
- Morphy, R. The influence of target family and functional activity on the physicochemical properties of pre-clinical compounds. J. Med. Chem. 2006, 49, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Eckert, H.; Bajorath, J. Molecular similarity analysis in virtual screening: Foundations, limitations and novel approaches. Drug Discov Today 2007, 12, 225–233. [Google Scholar] [CrossRef]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef]
- Jasial, S.; Hu, Y.; Vogt, M.; Bajorath, J. Activity-relevant similarity values for fingerprints and implications for similarity searching. F1000Res 2016, 5. [Google Scholar]
- Tomori, T.; Hajdu, I.; Barna, L.; Lorincz, Z.; Cseh, S.; Dorman, G. Combining 2D and 3D in silico methods for rapid selection of potential PDE5 inhibitors from multimillion compounds’ repositories: Biological evaluation. Mol. Divers. 2012, 16, 59–72. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Martinez-Mayorga, K.; Meurice, N. Balancing novelty with confined chemical space in modern drug discovery. Expert Opin. Drug Discov. 2014, 9, 151–165. [Google Scholar] [CrossRef]
- Flachner, B.; Tömöri, T.; Hajdú, I.; Dobi, K.; Lőrincz, Z.; Cseh, S.; Dormán, G. Rapid in silico selection of an MCHR1 antagonists’ focused library from multi-million compounds’ repositories: Biological evaluation. Med. Chem. Res. 2013, 23, 1234–1247. [Google Scholar] [CrossRef]
- Saddala, M.S.; Adi, P.J. Discovery of small molecules through pharmacophore modeling, docking and molecular dynamics simulation against Plasmodium vivax Vivapain-3 (VP-3). Heliyon 2018, 4, e00612. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Structure based design and molecular docking studies for phosphorylated tau inhibitors in Alzheimer’s disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, A.P.; Reddy, P.H. Pharmacophore-based models for therapeutic drugs against phosphorylated tau in Alzheimer’s disease. Drug Discov. Today 2019, 24, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2018, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, J.L.; Hays, S.J.; Caprathe, B.W.; Lee, C.; Emmerling, M.R.; Michael, W.; Jaen, J.C. Synthesis and evaluation of 2-aryl-4H-3,1-benzoxazin-4-ones as C1r serine protease inhibitors. Bioorg. Med. Chem. Lett. 1996, 6, 679–682. [Google Scholar] [CrossRef]
- Kardos, J.; Gal, P.; Szilagyi, L.; Thielens, N.M.; Szilagyi, K.; Lorincz, Z.; Kulcsar, P.; Graf, L.; Arlaud, G.J.; Zavodszky, P. The role of the individual domains in the structure and function of the catalytic region of a modular serine protease, C1r. J. Immunol. 2001, 167, 5202–5208. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Midorikawa, A.; Ino, Y.; Oda, M.; Nakamura, K.; Suzuki, S.; Kurumi, M. Inhibitory effects of newly synthesized active center-directed trypsin-like serine protease inhibitors on the complement system. Inflamm. Res. 2000, 49, 42–46. [Google Scholar] [CrossRef]
- Furugohri, T.; Isobe, K.; Honda, Y.; Kamisato-Matsumoto, C.; Sugiyama, N.; Nagahara, T.; Morishima, Y.; Shibano, T. DU-176b, a potent and orally active factor Xa inhibitor: In vitro and in vivo pharmacological profiles. J. Thromb. Haemost. 2008, 6, 1542–1549. [Google Scholar]
- Cortés-Cabrera, A.; Murcia, P.A.S.; Morreale, A.; Gago, F. In Silico Drug Discovery and Design: Theory, Methods, Challenges, and Applications, 1st ed.; Taylor & Francis: Boca Raton, FL, USA, 2015; p. 99. [Google Scholar]
- Koeppen, H.; Kriegl, J.; Lessel, U.; Tautermann, C.S.; Wellenzohn, B. Virtual Screening: Principles, Challenges, and Practical Guidelines, 1st ed.; Wiley-VCH: Weinheim, Germany, 2011; p. 61. [Google Scholar]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular similarity measures. Methods Mol. Biol. 2011, 672, 39–100. [Google Scholar]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Butina, D. Unsupervised data base clustering based on Daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem Inf Comp. Sci 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Willett, P. Similarity searching using 2D structural fingerprints. Methods Mol. Biol. 2011, 672, 133–158. [Google Scholar] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Polgar, T.; Keseru, G.M. Integration of virtual and high throughput screening in lead discovery settings. Comb. Chem High. Throughput Screen 2011, 14, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Polgar, T.; Baki, A.; Szendrei, G.I.; Keseru, G.M. Comparative virtual and experimental high-throughput screening for glycogen synthase kinase-3beta inhibitors. J. Med. Chem. 2005, 48, 7946–7959. [Google Scholar] [CrossRef] [PubMed]
- Stahura, F.L.; Bajorath, J. Virtual screening methods that complement HTS. Comb. Chem. High. T. Scr. 2004, 7, 259–269. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-like property concepts in pharmaceutical design. Curr. Pharm Des. 2009, 15, 2184–2194. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Willett, P.; Winterman, V. A comparison of some measures for the determination of intermolecular structural similarity measures of intermolecular structural similarity. Quant. Struct-Act. Rel. 1986, 5, 18–25. [Google Scholar] [CrossRef]
- Glen, R.C.; Adams, S.E. Similarity metrics and descriptor spaces-Which combinations to choose? Qsar Comb. Sci. 2006, 25, 1133–1142. [Google Scholar] [CrossRef]
- Dixon, S.L.; Koehler, R.T. The hidden component of size in two-dimensional fragment descriptors: Side effects on sampling in bioactive libraries. J. Med. Chem. 1999, 42, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Gregory, L.A.; Thielens, N.M.; Arlaud, G.J.; Fontecilla-Camps, J.C.; Gaboriaud, C. X-ray structure of the Ca2+-binding interaction domain of C1s. J. Biol. Chem. 2003, 278, 32157–32164. [Google Scholar] [CrossRef] [PubMed]
- Girija, U.V.; Gingras, A.R.; Marshall, J.E.; Panchal, R.; Sheikh, M.A.; Gal, P.; Schwaeble, W.J.; Mitchell, D.A.; Moody, P.C.E.; Wallis, R. Structural basis of the C1q/C1s interaction and its central role in assembly of the C1 complex of complement activation. Proc. Natl. Acad. Sci. USA 2013, 110, 13916–13920. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.J.; Wijeyewickrema, L.C.; Wilmann, P.G.; Gunzburg, M.J.; D’Andrea, L.; Irving, J.A.; Pang, S.S.; Duncan, R.C.; Wilce, J.A.; Whisstock, J.C.; et al. A molecular switch governs the interaction between the human complement protease C1s and its substrate, complement C4. J. Biol. Chem. 2013, 288, 15821–15829. [Google Scholar] [CrossRef]
- Pang, S.S.; Wijeyewickrema, L.C.; Hor, L.; Tan, S.; Lameignere, E.; Conway, E.M.; Blom, A.M.; Mohlin, F.C.; Liu, X.Y.; Payne, R.J.; et al. The structural basis for complement inhibition by gigastasin, a protease inhibitor from the giant amazon leech. J. Immunol. 2017, 199, 3883–3891. [Google Scholar] [CrossRef]
- Almitairi, J.O.M.; Girija, U.V.; Furze, C.M.; Simpson-Gray, X.; Badakshi, F.; Marshall, J.E.; Schwaeble, W.J.; Mitchell, D.A.; Moody, P.C.E.; Wallis, R. Reply to Mortensen et al.: The zymogen form of complement component C1. Proc. Natl. Acad. Sci. USA 2018, 115, E3867–E3868. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2016, 45, D945–D954. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Truchon, J.-F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Modeling 2007, 47, 488–508. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | IDNUMBER | IC50 C1s; µM | LogP | TPSA | H Bond Acceptors | H Bond Donors | Rotatable Bonds | Lipinski Rule of 5 (4 of 4) | Chemoinformatics |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |  | BAS 00784557 | 0.083 | 2.6744 | 57.01 | 4 | 0 | 3 | true | 2D similarity |
| 2 |  | 2616-0473 | 0.091 | 1.713 | 76.88 | 4 | 1 | 2 | true | 2D similarity |
| 3 |  | 3502-2773 | 0.24 | 2.9458 | 57.01 | 4 | 0 | 3 | true | Pharmacophore |
| 4 |  | 8008-1157 | 1.30 | 4.0203 | 47.78 | 3 | 0 | 2 | true | 2D similarity |
| 5 |  | 4130-3864 (CHEMBL-143333)7 | 1.85 | 2.1599 | 66.24 | 5 | 0 | 3 | true | 2D similarity |
| 6 |  | 8012-0883 | 2.16 | 2.7607 | 47.78 | 3 | 0 | 1 | true | Pharmacophore |
| 7 |  | BAS 00785517 | 3.867 | 4.1225 | 47.78 | 3 | 0 | 2 | true | 2D similarity |
| 8 |  | 3455-3034 | 5.90 | 3.2969 | 51.02 | 4 | 0 | 4 | true | 2D similarity |
| Entry | Structure | IDNUMBER | IC50 C1s; µM | LogP | TPSA | H Bond Acceptors | H Bond Donors | Rotatable Bonds | Lipinski rule of 5 (4 of 4) | Chemoinformatics |
|---|---|---|---|---|---|---|---|---|---|---|
| 9 |  | BAS 07161957 (CHEMBL-490106) | 0.012 | 3.45 | 70.15 | 4 | 0 | 4 | true | 2D similarity |
| 10 |  | BAS 07161950 | 0.044 | 3.79 | 60.92 | 3 | 0 | 3 | true | 2D similarity |
| 11 |  | BAS 07161949 (CHEMBL-490107) | 0.094 | 3.65 | 60.92 | 3 | 0 | 3 | true | 2D similarity |
| 12 |  | ASN 01365717 | 2.4 | 2.97 | 86.97 | 5 | 0 | 7 | true | 2D similarity |
| Entry | Structure | IDNUMBER | IC50 C1s; µM | LogP | TPSA | H Bond Acceptors | H Bond Donors | Rotatable Bonds | Lipinski Rule of 5 (4 of 4) | Chemoinformatics |
|---|---|---|---|---|---|---|---|---|---|---|
| 13 |  | 3226-0357 | 0.474 | 3.02 | 51.80 | 2 | 0 | 1 | true | Pharmacophore |
| 14 |  | Z55930777 | 6.44 | 2.10 | 70.26 | 4 | 0 | 3 | true | Pharmacophore |
| 15 |  | BAS 01170083 | 6.7 | 3.02 | 51.80 | 2 | 0 | 1 | true | 2D similarity |
| 16 |  | 4334-1472 (CHEMBL-1449374 | 29 | 2.02 | 78.10 | 3 | 0 | 3 | true | 2D similarity |
| Entry | Structure | IDNUMBER | IC50 C1s; µM | LogP | TPSA | H Bond Acceptors | H Bond Donors | Rotatable Bonds | Lipinski Rule of 5 (4 of 4) | Chemoinformatics |
|---|---|---|---|---|---|---|---|---|---|---|
| 17 |  | STOCK1S-76323 (CHEMBL-1333976) | 0.549 | 3.52 | 51.80 | 2 | 0 | 1 | true | Pharmacophore |
| 18 |  | STOCK3S-12710 (CHEMBL-1501165) | 0.845 | 4.72 | 38.66 | 2 | 0 | 1 | true | Hit validation |
| 19 |  | STOCK2S-97295 (CHEMBL-1441606) | ~20 | 4.46 | 38.66 | 2 | 0 | 1 | true | Hit validation |
| 20 |  | STOCK2S-12571 | ~50 | 4.98 | 38.66 | 2 | 0 | 1 | true | Hit validation |
| Entry | Structure | IDNUMBER | IC50 C1s; µM | LogP | TPSA | H Bond Acceptors | H Bond Donors | Rotatable Bonds | Lipinski Rule of 5 (4 of 4) | Chemoinformatics |
|---|---|---|---|---|---|---|---|---|---|---|
| 21 |  | Z55992821 | 0.241 | 3.20 | 38.66 | 2 | 0 | 1 | true | Pharmacophore |
| 22 |  | Z55992803 | 3.6 | 2.35 | 51.80 | 2 | 0 | 1 | true | Hit validation |
| 23 |  | Z55992807 | 41 | 2.26 | 51.80 | 2 | 0 | 1 | true | Hit validation |
| Entry | Factor Xa 10 µM Remaining Activity % | Entry | Factor Xa 10 µM Remaining Activity % |
|---|---|---|---|
| 1 | 30.3 | 11 | 26.02 |
| 2 | 43.1 | 12 | 85.4 |
| 3 | 4.93 | 13 | 98.6 |
| 4 | 86.2 | 14 | 61.4 |
| 5 | 48.3 | 15 | 82 |
| 6 | 92.3 | 16 | 77.6 |
| 7 | 92.1 | 17 | 100 |
| 8 | 76.5 | 18 | 46.1 |
| 9 | 2.39 | 21 | 53.6 |
| 10 | 78.1 | 22 | 0.14 |
| Entry | IC50 (µM) | Glide Docking Score | Glide Emodel Score |
|---|---|---|---|
| 1 | 0.083 | −6.404 | −58.321 |
| 2 | 0.091 | −6.866 | −65.171 |
| 4 | 1.300 | −7.003 | −60.058 |
| 5 | 1.850 | −6.791 | −59.201 |
| 7 | 3.870 | −7.558 | −65.852 |
| 9 | 0.012 | −6.592 | −62.262 |
| 10 | 0.044 | −6.099 | −54.519 |
| 11 | 0.094 | −6.993 | −60.231 |
| Compound ID | IC50 (µM) | Glide Docking Score | Glide Emodel Scores |
|---|---|---|---|
| 17178137 | 11.0 | −5.538 | −53.585 |
| 4951143 | 19.1 | −5.595 | −57.808 |
| 2986934 | 0.34 | −6.892 | −67.866 |
| 710644 | 1.09 | −6.556 | −55.670 |
| 5146207 | >50 | −6.495 | −54.781 |
| 807111 | >50 | −5.909 | −58.543 |
| 1107361 | >50 | −6.621 | −67.010 |
| 827004 | 3.04 | −7.022 | −55.173 |
| 4957387 | 32.9 | −6.737 | −60.524 |
| 898930 | 5.54 | −6.733 | −57.680 |
| Pharmacophore Model | PhaseHypo-Score | Survival Score | Selectivity Score | BEDROC160.9 (from Validation) | ROC | EF1% |
|---|---|---|---|---|---|---|
| ARRR_1 | 1.319 | 5.317 | 1.219 | 0.67 | 0.63 | 2.78 |
| ARRR_2 | 1.318 | 5.305 | 1.211 | 0.74 | 0.51 | 2.78 |
| AARR_1 | 1.315 | 5.247 | 1.150 | 0.67 | 0.79 | 2.78 |
| AARR_2 | 1.314 | 5.236 | 1.139 | 0.67 | 0.80 | 2.78 |
| AARR_3 * | 1.314 | 5.231 | 1.141 | 0.92 | 0.81 | 5.56 |
| Pharmacophore Model | PhaseHypoScore | Survival Score | Selectivity Score | BEDROC160.9 (from Validation) | ROC Score | EF1% |
|---|---|---|---|---|---|---|
| AHRR_1 * | 1.283 | 5.175 | 1.358 | 0.52 | 0.40 | 2.78 |
| AAAHRR_1 | 1.259 | 5.833 | 2.034 | 0.29 | 0.09 | 2.78 |
| AAAHRR_2 | 1.258 | 5.828 | 2.026 | 0.14 | 0.11 | 0.00 |
| AAAHRR_3 | 1.258 | 5.826 | 2.029 | 0.11 | 0.11 | 0.00 |
| AHRRR_1 | 1.249 | 5.665 | 1.87 | 0.04 | 0.23 | 0.00 |
| Pharmacophore Model | PhaseHypoScore | Survival Score | Selectivity Score | BEDROC160.9 (from validation) | ROC | EF1% |
|---|---|---|---|---|---|---|
| AAARR_1 | 1.288 | 4.808 | 1.373 | 0.86 | 0.62 | 5.56 |
| ARRR_1 | 1.279 | 4.646 | 1.214 | 0.86 | 0.52 | 5.56 |
| AARR_1 | 1.274 | 4.574 | 1.142 | 0.86 | 0.76 | 5.56 |
| AARR_2 | 1.274 | 4.562 | 1.131 | 0.86 | 0.66 | 5.56 |
| AARR_3 * | 1.273 | 4.550 | 1.118 | 0.86 | 0.84 | 5.56 |
| Domains | Position | Length (aa) | X-Ray Structures | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1ELV * | 1NZI | 4J1Y * | 4LMF | 4LOR | 4LOS | 4LOT | 6F1C | 6F1H | 5UBM * | |||
| signal peptide | 1–15 | 15 | x | x | x | x | x | x | x | x | x | x |
| CUB 1 | 16–130 | 115 | − | + | − | + | + | − | − | + | + | − |
| EGF-like | 131–172 | 42 | − | + | − | + | + | − | − | + | + | − |
| CUB 2 | 175–290 | 116 | − | − | − | + | + | + | + | + | + | − |
| Sushi/CCP1 | 292–356 | 65 | − | − | + | − | - | + | + | − | − | + |
| Sushi/CCP2 | 357–423 | 67 | + | − | + | − | − | - | + | − | − | + |
| SP (peptidase) | 438–688 | 243 | + | − | + | − | − | − | − | − | − | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szilágyi, K.; Hajdú, I.; Flachner, B.; Lőrincz, Z.; Balczer, J.; Gál, P.; Závodszky, P.; Pirli, C.; Balogh, B.; Mándity, I.M.; et al. Design and Selection of Novel C1s Inhibitors by In Silico and In Vitro Approaches. Molecules 2019, 24, 3641. https://doi.org/10.3390/molecules24203641
Szilágyi K, Hajdú I, Flachner B, Lőrincz Z, Balczer J, Gál P, Závodszky P, Pirli C, Balogh B, Mándity IM, et al. Design and Selection of Novel C1s Inhibitors by In Silico and In Vitro Approaches. Molecules. 2019; 24(20):3641. https://doi.org/10.3390/molecules24203641
Chicago/Turabian StyleSzilágyi, Katalin, István Hajdú, Beáta Flachner, Zsolt Lőrincz, Júlia Balczer, Péter Gál, Péter Závodszky, Chiara Pirli, Balázs Balogh, István M. Mándity, and et al. 2019. "Design and Selection of Novel C1s Inhibitors by In Silico and In Vitro Approaches" Molecules 24, no. 20: 3641. https://doi.org/10.3390/molecules24203641
APA StyleSzilágyi, K., Hajdú, I., Flachner, B., Lőrincz, Z., Balczer, J., Gál, P., Závodszky, P., Pirli, C., Balogh, B., Mándity, I. M., Cseh, S., & Dormán, G. (2019). Design and Selection of Novel C1s Inhibitors by In Silico and In Vitro Approaches. Molecules, 24(20), 3641. https://doi.org/10.3390/molecules24203641