
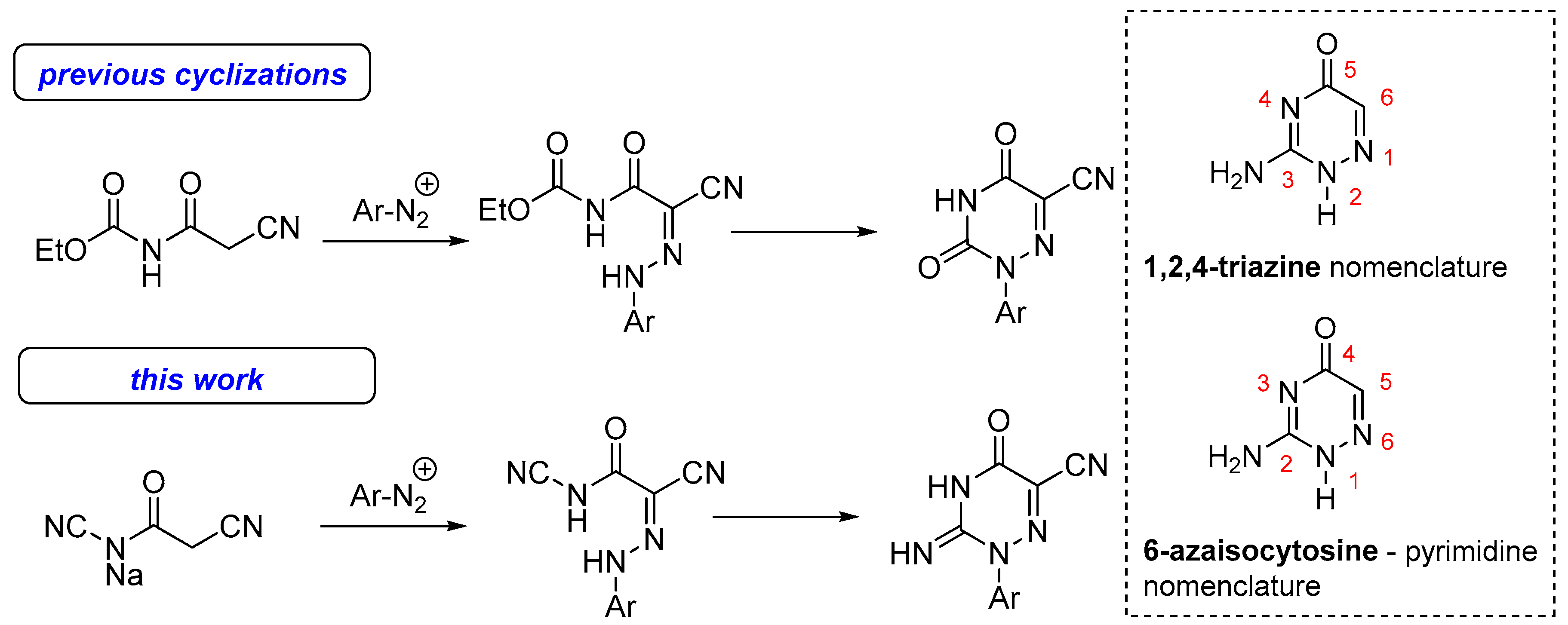
General Synthesis of 1-Aryl-6-azaisocytosines and Their Utilization for the Preparation of Related Condensed 1,2,4-Triazines


Abstract
:
1. Introduction
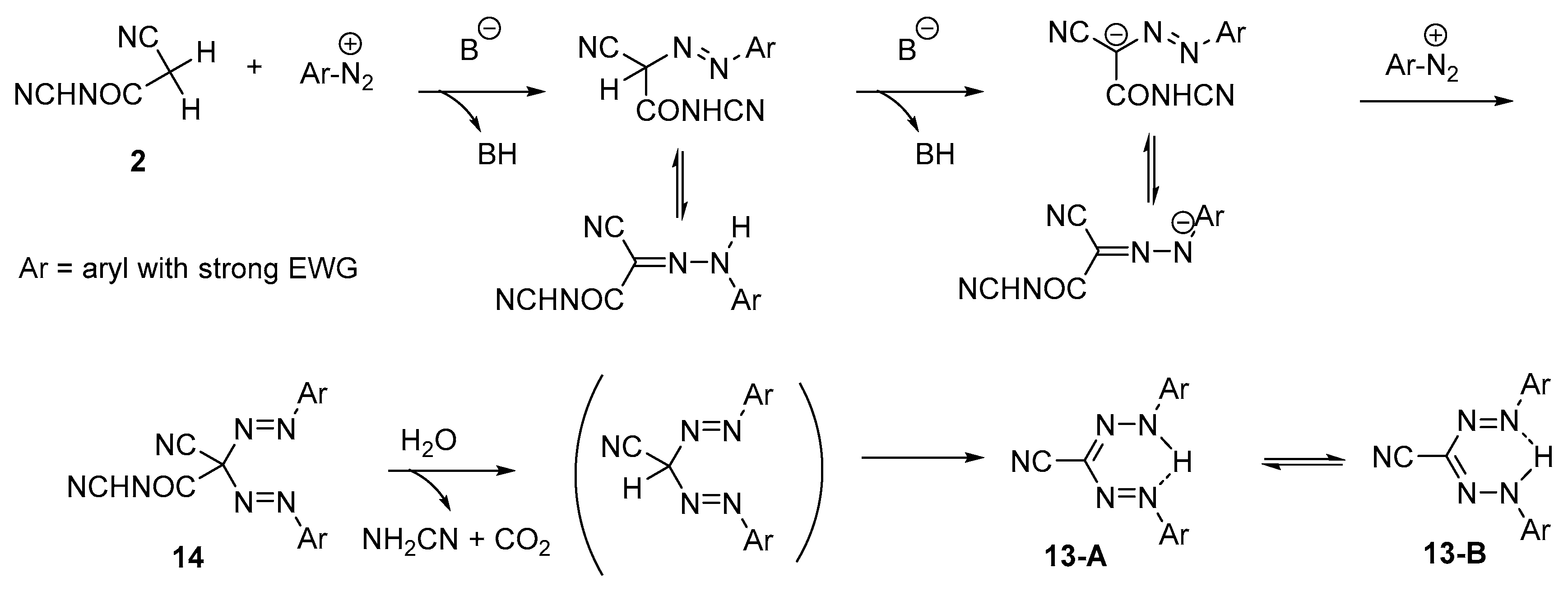
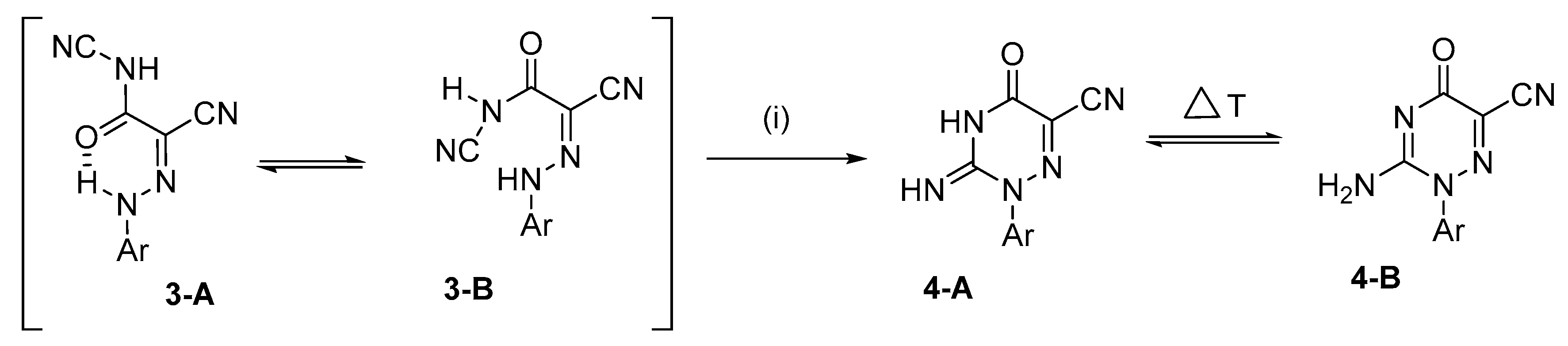
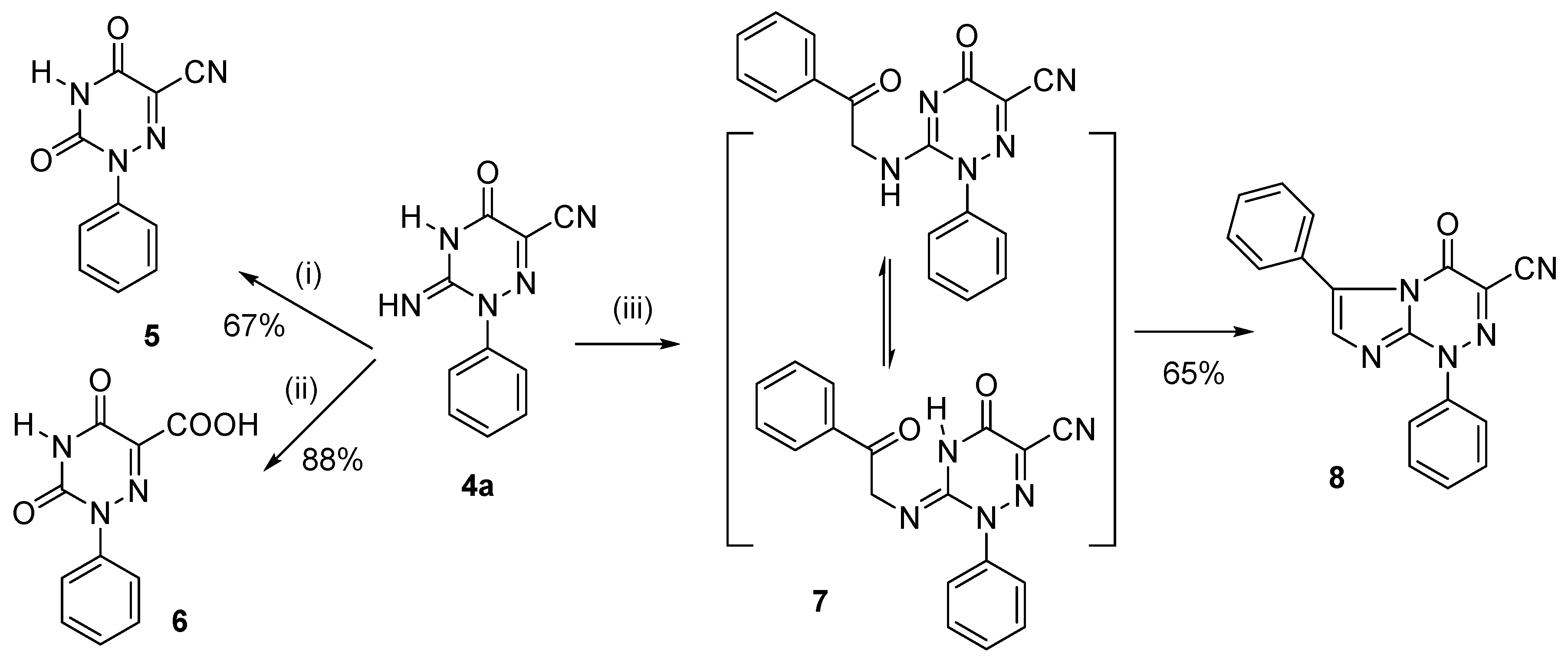
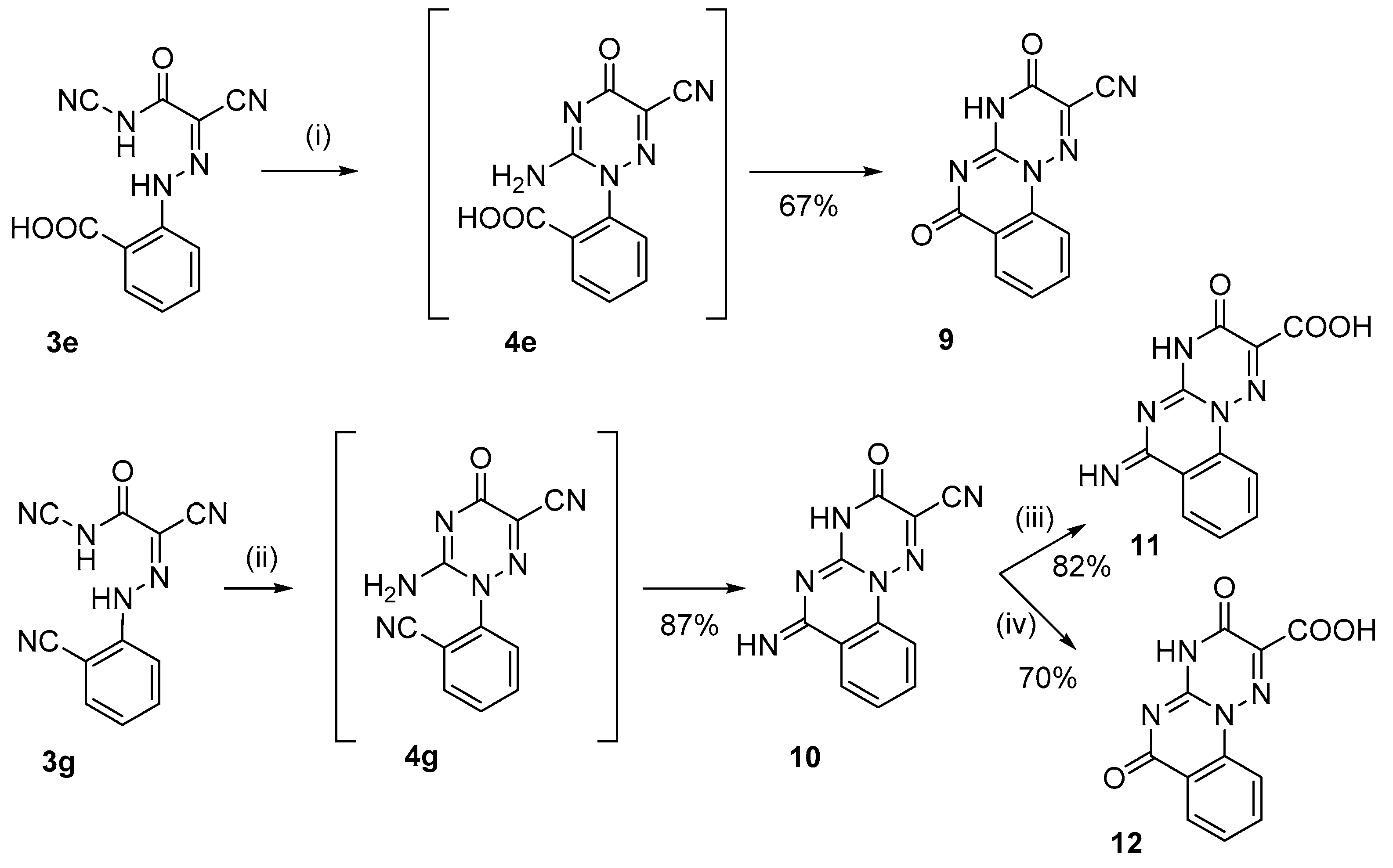
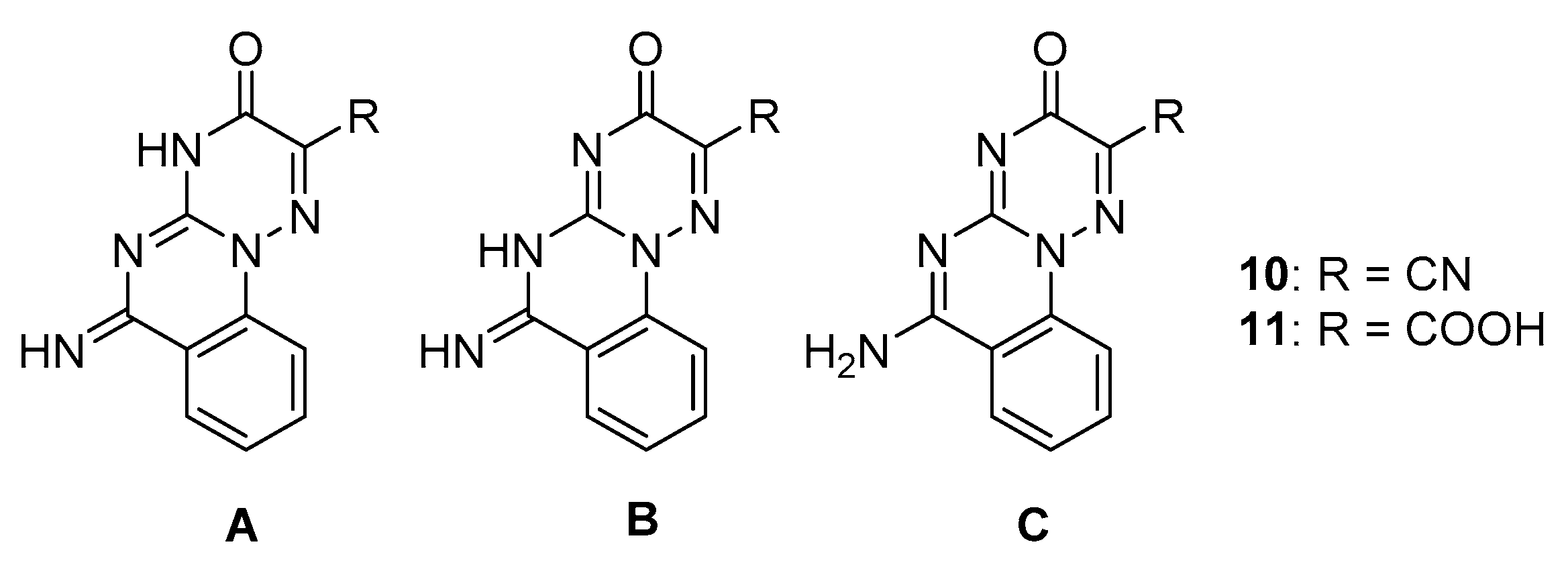
2. Results and Discussion
3. Materials and Methods
3.1. General Informations
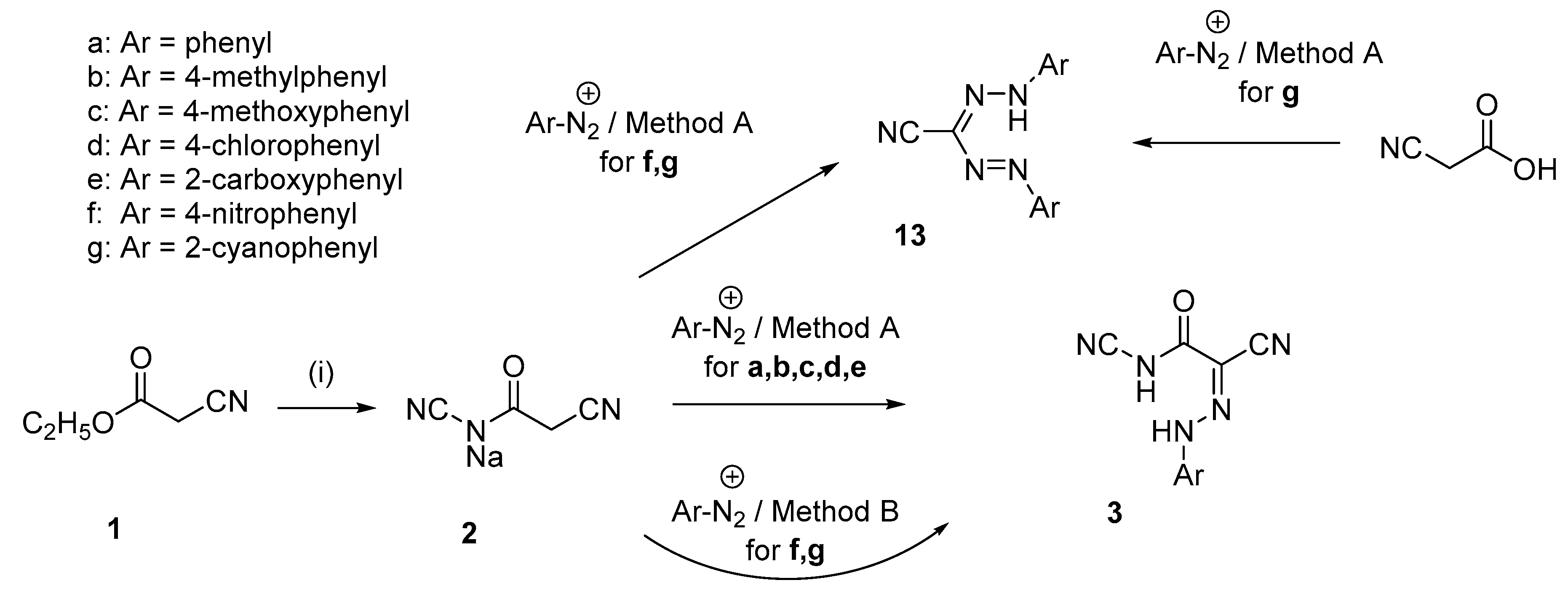
3.2. Synthesis of Compounds 2–13
3.2.1. General Procedure for the Preparations of 2-Arylhydrazono-2-cyanoacetylcyanamides (3)
Method A
Method B
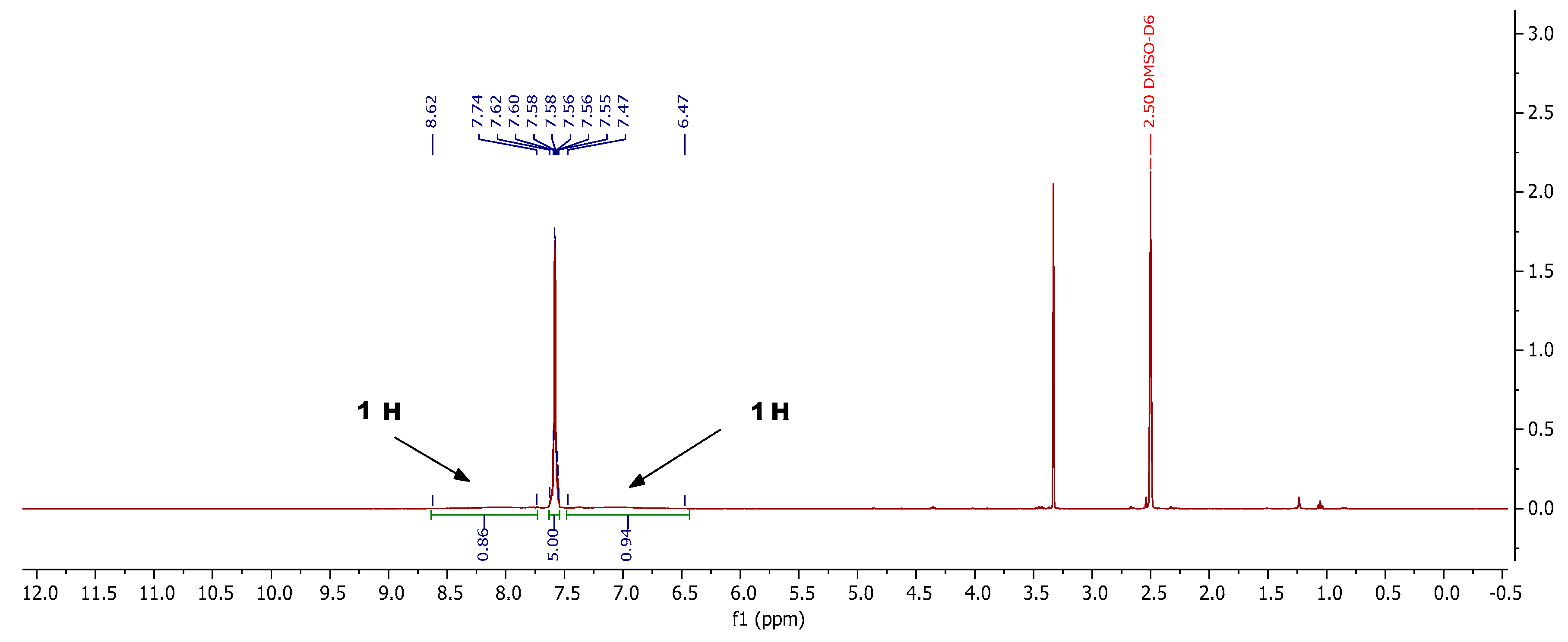
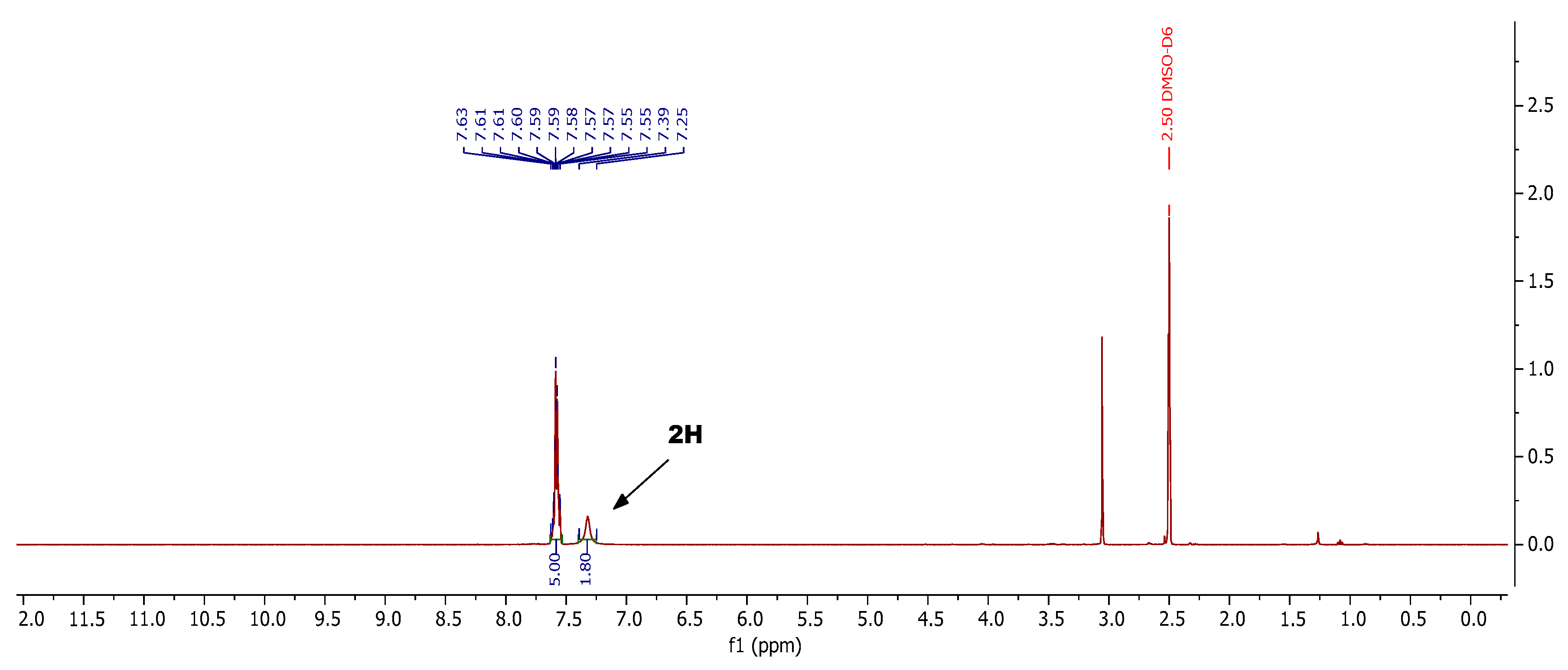
3.2.2. General Procedure for the Preparation of 2-Aryl-3-imino-5-oxo-2,3,4,5-tetrahydro-1,2,4- triazine-6-carbonitriles 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arshad, M.; Khan, T.A.; Khan, M.A. 1,2,4-Triazine Derivatives: Synthesis and Biological Applications. Int. J. Pharm. Sci. Res. 2015, 5, 149–162. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spanò, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. An overview on the recent developments of 1,2,4-triazine derivatives as anticancer compounds. Eur. J. Med. Chem. 2017, 142, 328–375. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sirohi, T.S.; Singh, H.; Yadav, R.; Roy, R.K.; Chaudhary, A.; Pandeya, S.N. 1,2,4-Triazine Analogs as Novel Class of Therapeutic Agents. Mini-Reviews Med. Chem. 2014, 14, 168–207. [Google Scholar] [CrossRef] [PubMed]
- Farràs, J.; del Mar Lleó, M.; Vilarrasa, J.; Castillón, S.; Matheu, M.; Solans, X.; Font-Bardia, M. New bicyclic nucleosides related to 6-azaisocytidine. Tetrahedron Lett. 1996, 37, 901–904. [Google Scholar] [CrossRef]
- Mahfouz, R.Z.; Jankowska, A.; Ebrahem, Q.; Gu, X.; Visconte, V.; Tabarroki, A.; Terse, P.; Covey, J.; Chan, K.; Ling, Y.; et al. Increased CDA Expression/Activity in Males Contributes to Decreased Cytidine Analog Half-Life and Likely Contributes to Worse Outcomes with 5-Azacytidine or Decitabine Therapy. Clin. Cancer Res. 2013, 19, 938–948. [Google Scholar] [CrossRef]
- Tzeng, C.-C.; Hwang, L.-C.; Wang, C.-J.; Lee, G.-H.; Wang, Y. Synthesis and Structure Assignment of 1-[(2-Hydroxyethoxy)methyl]- and 1-[(1,3-Dihydroxy-2-propoxy)methyl]-6-azaisocytosine. Heterocycles 1995, 41, 293. [Google Scholar] [CrossRef]
- Piťha, J.; Fiedler, P.; Gut, J. Nucleic acids components and their analogues. LXXXII. The fine structure of 6-azaisocytosine and its derivatives. Collect. Czechoslov. Chem. Commun. 1966, 31, 1864–1871. [Google Scholar] [CrossRef]
- Sztanke, M.; Sztanke, K.; Rajtar, B.; Świątek, Ł.; Boguszewska, A.; Polz-Dacewicz, M. The influence of some promising fused azaisocytosine-containing congeners on zebrafish (Danio rerio) embryos/larvae and their antihaemolytic, antitumour and antiviral activities. Eur. J. Pharm. Sci. 2019, 132, 34–43. [Google Scholar] [CrossRef]
- Stock, M.L.; Elazab, S.T.; Hsu, W.H. Review of triazine antiprotozoal drugs used in veterinary medicine. J. Vet. Pharmacol. Ther. 2018, 41, 184–194. [Google Scholar] [CrossRef]
- Diaferia, M.; Veronesi, F.; Morganti, G.; Nisoli, L.; Fioretti, D.P. Efficacy of Toltrazuril 5% Suspension (Baycox®, Bayer) and Diclazuril (Vecoxan®, Janssen-Cilag) in the Control of Eimeria spp. in Lambs. Parasitol. Res. 2013, 112, 163–168. [Google Scholar] [CrossRef]
- Kelly, M.J.; Pietranico-Cole, S.; Larigan, J.D.; Haynes, N.-E.; Reynolds, C.H.; Scott, N.; Vermeulen, J.; Dvorozniak, M.; Conde-Knape, K.; Huang, K.-S.; et al. Discovery of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dioxo-2,3,4,5-tetrahydro- [1,2,4]triazine-6-carbonitrile (MGL-3196), a Highly Selective Thyroid Hormone Receptor β Agonist in Clinical Trials for the Treatment of Dys. J. Med. Chem. 2014, 57, 3912–3923. [Google Scholar] [CrossRef] [PubMed]
- Slouka, J. 1-Aryl-6-azauracile: Die Synthese von 1-Phenyl-6-azauracil-5-carbonsäure und einiger ihrer Derivate. Monatshefte für Chemie 1963, 94, 258–262. [Google Scholar] [CrossRef]
- Neunhoeffer, H. Chemistry of 1,2,3-Triazines and 1,2,4-triazines, tetrazines and pentazines; J. Wiley and sons: New York, NY, USA, 1978. [Google Scholar]
- Frysova, I.; Mutina, V.; Slouka, J.; Hlavac, J. Chemistry of 1-Aryl-6-azauracil-5-carbonitriles. Acta Upol. Fac. Rer. Nat. Chem. 2004, 43, 15–58. [Google Scholar] [CrossRef]
- Slouka, J.; Peč, P. 1-Aryl-6-azauracile, 4. Mitt.: Die Synthese des 1-Phenyl-5-[5′-methyl-1′,2′,4′-oxdiazolyl-(3′)]-6-azauracils und einiger seiner Derivate. Monatshefte für Chemie 1965, 96, 1874–1877. [Google Scholar] [CrossRef]
- Wardakhan, W.W.; Ouf, S.A. Reaction of cyanoacetamide with benzoyl isothiocyanate: synthesis of pyridine, pyrido[2,1-a]triazine, pyrazolo[3,4-d]triazine and thiazole derivatives. Egypt. J. Chem. 2005, 48, 393–406. [Google Scholar]
- El-Bannany, A.A.A.; Ghozlan, S.A.S.; Ibraheim, L.I. Synthesis of some new 1,2,4-triazine, 1,2,3-triazole and, pyridazine derivatives. Pharmazie 1987, 42, 695–696. [Google Scholar]
- Hirayama, T.; Kamada, M.; Tsurumi, H.; Mimura, M. A novel synthesis of pyrimidines. I. Cyclization of N-cyano-cyanoaceto derivatives. Chem. Pharm. Bull. 1976, 24, 26–35. [Google Scholar] [CrossRef]
- Khattab, T.A.; Haggag, K.M.; Elnagdi, M.H.; Abdelrahman, A.A.; Abdelmoez Aly, S. Microwave-Assisted Synthesis of Arylazoaminopyrazoles as Disperse Dyes for Textile Printing. Zeitschrift fur Anorg. und Allg. Chemie 2016, 642, 766–772. [Google Scholar] [CrossRef]
- Slouka, J.; Bekárek, V. Synthesis and cyclization of some N-oxides of 2-pyridylhydrazones of mesoxalic acid derivatives. Collect. Czechoslov. Chem. Commun. 1988, 53, 626–632. [Google Scholar] [CrossRef]
- Slouka, J. 1-Aryl-6-azauracile, 3. Mitt.: Die Synthese von 1-Phenyl-6-azauracil und einiger seiner Derivate. Monatshefte für Chemie 1965, 96, 134–137. [Google Scholar] [CrossRef]
- Slouka, J.; Bekárek, V. 1-Aryl-6-azauracile. XVII. Synthese einiger 1,2,4-Triazino[2,3-a]chinazolin-Derivate. J. für Prakt. Chemie 1974, 316, 943–951. [Google Scholar] [CrossRef]
- Li, S.; Cooper, V.R.; Thonhauser, T.; Lundqvist, B.I.; Langreth, D.C. Stacking interactions and DNA intercalation. J. Phys. Chem. B 2009, 113, 11166–11172. [Google Scholar] [CrossRef] [PubMed]
- Almaqwashi, A.A.; Paramanathan, T.; Rouzina, I.; Williams, M.C. Mechanisms of small molecule-DNA interactions probed by single-molecule force spectroscopy. Nucleic Acids Res. 2016, 44, 3971–3988. [Google Scholar] [CrossRef]
- Almaqwashi, A.A.; Andersson, J.; Lincoln, P.; Rouzina, I.; Westerlund, F.; Williams, M.C. DNA intercalation optimized by two-step molecular lock mechanism. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Biebricher, A.S.; Heller, I.; Roijmans, R.F.H.; Hoekstra, T.P.; Peterman, E.J.G.; Wuite, G.J.L. The impact of DNA intercalators on DNA and DNA-processing enzymes elucidated through force-dependent binding kinetics. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diazonium Salt Ar-N2+ | Arylhydrazone 3 [Isolated Yield %] | |
|---|---|---|
| Method A | Method B | |
| Ar: phenyl | 3a [80%] | 3a [5%] * |
| Ar: 4-methylphenyl | 3b [87%] | 3b [0%] |
| Ar: 4-methoxyphenyl | 3c [83%] | 3c [0%] |
| Ar: 4-chlorophenyl | 3d [79%] | 3d [5%] * |
| Ar: 2-carboxyphenyl | 3e [72%] | 3e [50%] |
| Ar: 4-nitrophenyl | 3f [15%] * | 3f [93%] |
| Ar: 2-cyanophenyl | 3g [10%] * | 3g [80%] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zálešák, F.; Slouka, J.; Stýskala, J. General Synthesis of 1-Aryl-6-azaisocytosines and Their Utilization for the Preparation of Related Condensed 1,2,4-Triazines. Molecules 2019, 24, 3558. https://doi.org/10.3390/molecules24193558
Zálešák F, Slouka J, Stýskala J. General Synthesis of 1-Aryl-6-azaisocytosines and Their Utilization for the Preparation of Related Condensed 1,2,4-Triazines. Molecules. 2019; 24(19):3558. https://doi.org/10.3390/molecules24193558
Chicago/Turabian StyleZálešák, František, Jan Slouka, and Jakub Stýskala. 2019. "General Synthesis of 1-Aryl-6-azaisocytosines and Their Utilization for the Preparation of Related Condensed 1,2,4-Triazines" Molecules 24, no. 19: 3558. https://doi.org/10.3390/molecules24193558
APA StyleZálešák, F., Slouka, J., & Stýskala, J. (2019). General Synthesis of 1-Aryl-6-azaisocytosines and Their Utilization for the Preparation of Related Condensed 1,2,4-Triazines. Molecules, 24(19), 3558. https://doi.org/10.3390/molecules24193558