
Enantioselective 5-exo-Fluorocyclization of Ene-Oximes

 and
and
Abstract
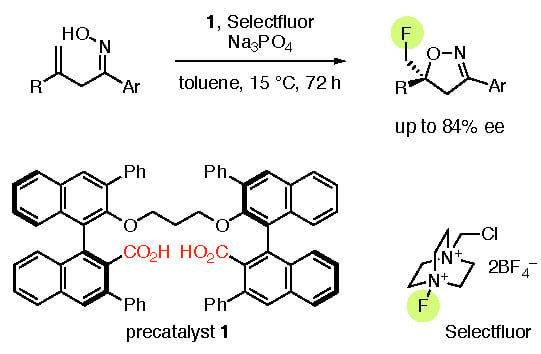
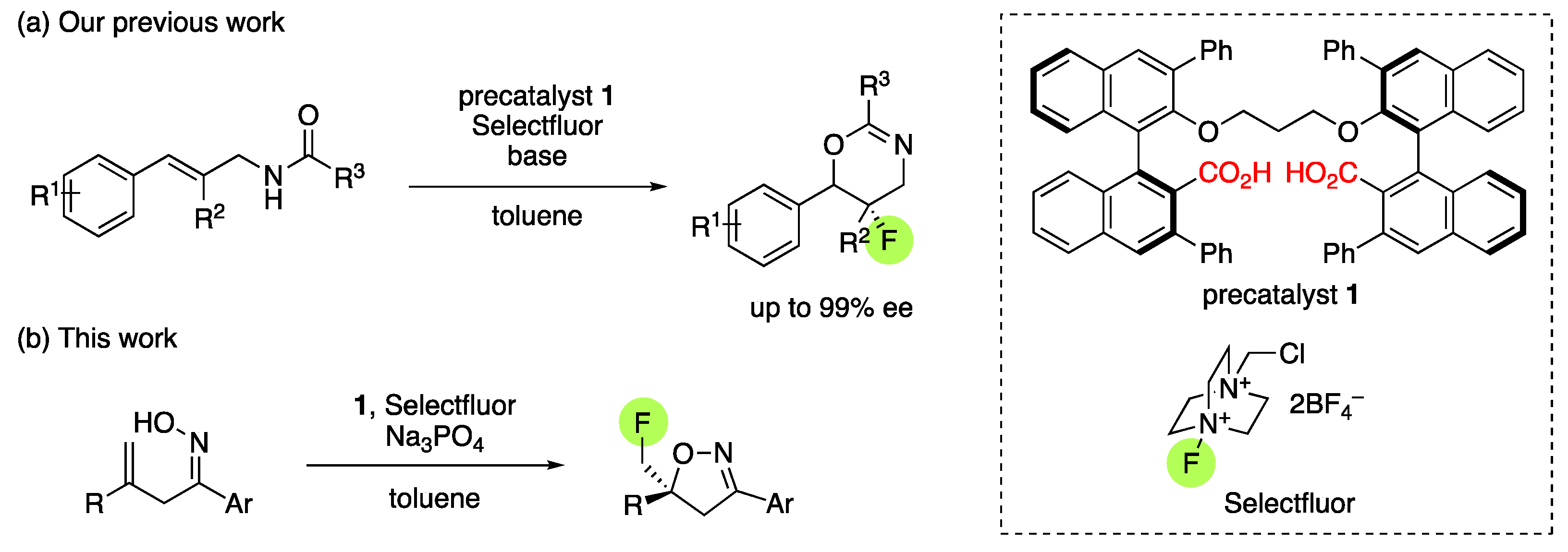
1. Introduction
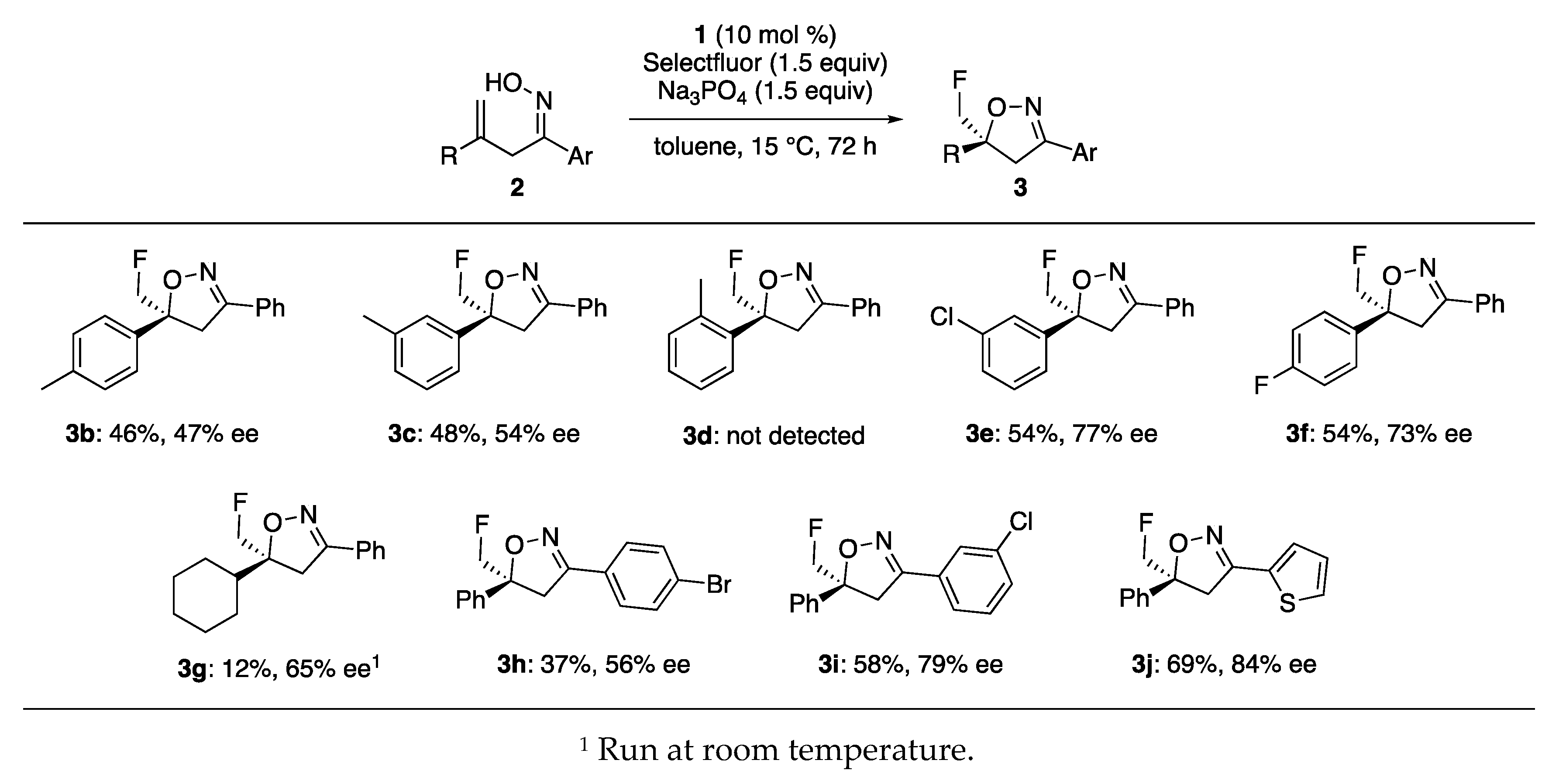
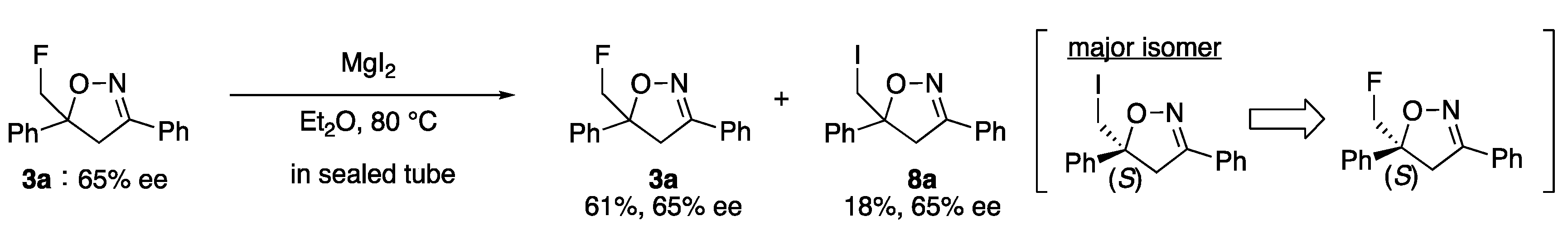
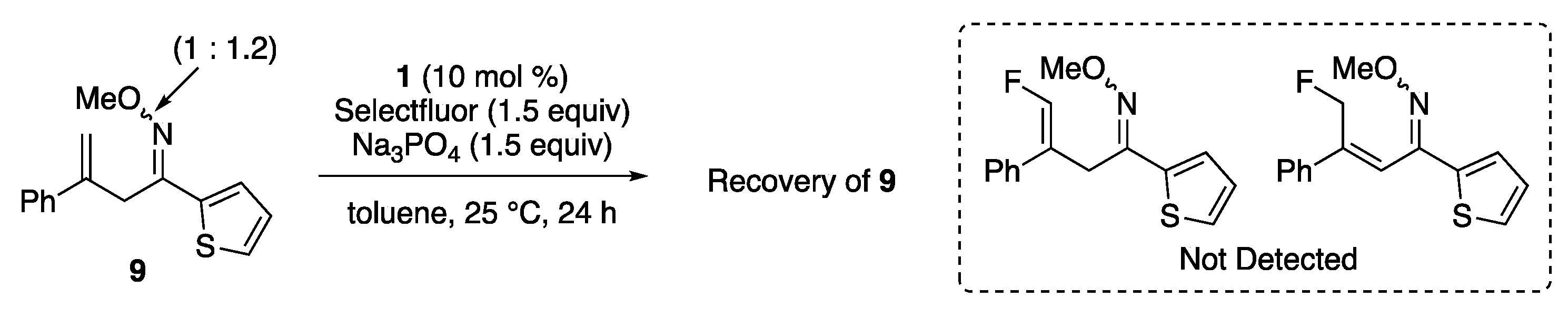
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Asymmetric Fluorocyclization of Ene-Oximes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kaur, K.; Kumar, V.; Sharma, A.K.; Gupta, G.K. Isoxazoline containing natural products as anticancer agents: A review. Eur. J. Med. Chem. 2014, 77, 121–133. [Google Scholar] [CrossRef]
- Schorderet-Weber, S.; Noack, S.; Selzer, P.M.; Kaminsky, R. Blocking transmission of vector-borne diseases. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Bharathi, E.V.; Azhar, M.A.; Sultana, F.; Pushpavalli, S.; Pal-Bhadra, M.; Juvekar, A.; et al. Design, synthesis and biological evaluation of 3,5-diaryl-isoxazoline/isoxazole-pyrrolobenzodiazepine conjugates as potential anticancer agents. Eur. J. Med. Chem. 2010, 45, 3924–3937. [Google Scholar] [CrossRef]
- Kamal, A.; Bharathi, E.V.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Reddy, M.K.; Viswanath, A.; Reddy, T.L.; Shaik, T.B.; Pushpavalli, S.; et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem. 2011, 46, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.L.; Ellis, C.D.; Liauw, A.Y.; Alexander, R.S.; Knabb, R.M.; Lam, G.; Wright, M.R.; Wong, P.C.; Wexler, R.R. Design and Synthesis of Isoxazoline Derivatives as Factor Xa Inhibitors. 2. J. Med. Chem. 1999, 42, 2760–2773. [Google Scholar] [CrossRef] [PubMed]
- Ozoe, Y.; Asahi, M.; Ozoe, F.; Nakahira, K.; Mita, T. The antiparasitic isoxazoline A1443 is a potent blocker of insect ligand-gated chloride channels. Biochem. Biophys. Res. Commun. 2010, 391, 744–749. [Google Scholar] [CrossRef]
- Gracia-Reynaga, P.; Zhao, C.; Sarpong, R.; Casida, J.E. New GABA/Glutamate Receptor Target for [3H]Isoxazoline Insecticide. Chem. Res. Toxicol. 2013, 26, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Shoop, W.L.; Hartline, E.J.; Gould, B.R.; Waddell, M.E.; McDowell, R.G.; Kinney, J.B.; Lahm, G.P.; Long, J.K.; Xu, M.; Wagerle, T.; et al. Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs. Vet. Parasitol. 2014, 201, 179–189. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Zhou, Y.; Akama, T.; Bu, W.; White, W.H.; Defauw, J.M.; Winkle, J.R.; Balko, T.W.; et al. Discovery of an orally bioavailable isoxazoline benzoxaborole (AN8030) as a long acting animal ectoparasiticide. Bioorg. Med. Chem. Lett. 2015, 25, 5589–5593. [Google Scholar] [CrossRef]
- Madavu, K.; Rai, L. Heterocycles via Oxime Cycloadditions. In Synthesis of Heterocycles via Cycloadditions II; Springer: Berlin/Heidelberg, Germany, 2008; Volume 13, pp. 1–69. [Google Scholar]
- Kawai, H.; Shibata, N. Attractive Trifluoromethylated Dihydroxazoles and Related Compounds under Organocatalysis. Chem. Rec. 2014, 14, 1024–1040. [Google Scholar] [CrossRef]
- Kumer, V.; Kaur, K. Fluorinated isoxazolines and isoxazoles: A synthetic perspective. J. Fluor. Chem. 2015, 180, 55–97. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Oxidative cyclizations of oximes using hypervalent iodine reagents. Arkivoc 2017, i, 99–116. [Google Scholar] [CrossRef]
- Zhu, M.; Fun, W.; Guo, W.; Tian, Y.; Wang, Z.; Xu, C.; Ji, B. Visible-Light-Induced Radical Di- and Trifluoromethylation of β,γ-Unsaturated Oximes: Synthesis of Di- and Trifluoromethylated Isoxazolines. Eur. J. Org. Chem. 2019, 2019, 1614–1619. [Google Scholar] [CrossRef]
- Wei, Q.; Chen, J.-R.; Hu, X.-Q.; Yang, X.-C.; Lu, B.; Xiao, W.-J. Photocatalytic Radical Trifluoromethylation/Cyclization Cascade: Synthesis of CF3-Containing Pyrazolines and Isoxazolines. Org. Lett. 2015, 17, 4464–4467. [Google Scholar] [CrossRef]
- Li, X.-T.; Lv, L.; Gu, Q.-S.; Liu, X.-Y. Copper-catalyzed radical oxytrifluoromethylation of alkenyl oximes at ambient temperature. Tetrahedron 2018, 74, 6041–6046. [Google Scholar] [CrossRef]
- Li, X.-T.; Gu, Q.-S.; Dong, X.-Y.; Meng, X.; Liu, X.-Y. A Copper Catalyst with a Cinchona-Alkaloid-Based Sulfonamide Ligand for Asymmetric Radical Oxytrifluoromethylation of Alkenyl Oximes. Angew. Chem. Int. Ed. 2018, 57, 7668–7672. [Google Scholar] [CrossRef]
- Zhu, M.-K.; Zhao, J.-F.; Loh, T.-P. Palladium-Catalyzed Oxime Assisted Intramolecular Dioxygenation of Alkenes with 1 atm of Air as the Sole Oxidant. J. Am. Chem. Soc. 2010, 132, 6284–6285. [Google Scholar] [CrossRef]
- Yamamoto, D.; Oguro, T.; Tashiro, Y.; Soga, M.; Miyashita, K.; Aso, Y.; Makino, K. Manganese-Promoted Oxidative Cyclization of Unsaturated Oximes Using Molecular Oxygen in Air under Ambient Conditions. Eur. J. Org. Chem. 2016, 2016, 5216–5219. [Google Scholar] [CrossRef]
- Ji, F.; Fan, Y.; Yang, R.; Yang, Y.; Yu, D.; Wang, M.; Li, Z. Regioselective synthesis of thiocyanate-containing isoxazolines via Fe(III)/K2S2O8-mediated radical thiocyanation/cyclization cascade reaction of β,γ-unsaturated oximes. Asian J. Org. Chem. 2017, 6, 682–685. [Google Scholar] [CrossRef]
- Tripathi, C.B.; Mukherjee, S. Catalytic Enantioselective Iodoetherification of Oximes. Angew. Chem. Int. Ed. 2013, 52, 8450–8453. [Google Scholar] [CrossRef]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VHC: Weinheim, Germany, 2013. [Google Scholar]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Oxford, UK, 2009. [Google Scholar]
- Gouverneur, V.; Müller, K. Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications; Imperial College Press: London, UK, 2012. [Google Scholar]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Haranahalli, K.; Honda, T.; Ojima, I. Resent progress in the strategic incorporation of fluorine into medicinally active compounds. J. Fluor. Chem. 2019, 217, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed]
- Champagne, P.A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev. 2015, 115, 9073–9174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Wolstenhulme, J.R.; Gouverneur, V. Asymmetric Fluorocyclizations of Alkenes. Acc. Chem. Res. 2014, 47, 3560–3570. [Google Scholar] [CrossRef]
- Ishimaru, T.; Shibata, N.; Horikawa, T.; Yasuda, N.; Nakamura, S.; Toru, T.; Shiro, M. Cinchona Alkaloid Catalyzed Enantioselective Fluorination of Allyl Silanes, Silyl Enol Ethers, and Oxindoles. Angew. Chem. Int. Ed. 2008, 47, 4157–4161. [Google Scholar] [CrossRef]
- Rauniyar, V.; Lackner, A.D.; Hamilton, G.L.; Toste, F.D. Asymmetric Electrophilic Fluorination Using an Anionic Chiral Phase-Transfer Catalyst. Science 2011, 334, 1681–1684. [Google Scholar] [CrossRef]
- Kong, W.; Feige, P.; de Haro, T.; Nevado, C. Regio- and Enantioselective Aminofluorination of Alkenes. Angew. Chem. Int. Ed. 2013, 52, 2469–2473. [Google Scholar] [CrossRef]
- Wolstenhulme, J.R.; Rosenqvist, J.; Lozano, O.; Ilupeju, J.; Wurz, N.; Engle, K.M.; Pidgeon, G.W.; Moore, P.R.; Sandford, G.; Gouverneur, V. Asymmetric Electrophilic Fluorocyclization with Carbon Nucleophiles. Angew. Chem. Int. Ed. 2013, 52, 9796–9800. [Google Scholar] [CrossRef] [PubMed]
- Mennie, K.M.; Banik, S.M.; Reichert, E.C.; Jacobsen, E.N. Catalytic Diastero- and Enantioselective Fluoroamination of Alkenes. J. Am. Chem. Soc. 2018, 140, 4797–4802. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, F.; Schäfer, M.; Sarie, J.C.; Daniliuc, C.G.; Molloy, J.J.; Gilmour, R. Enantioselective, Catalytic Vicinal Difluorination of Alkenes. Angew. Chem. Int. Ed. 2018, 57, 16431–16435. [Google Scholar] [CrossRef]
- Zhao, J.; Jiang, M.; Liu, J.-T. Synthesis of Fluoromethyl-Substituted Isoxazolines via Transition Metal-Free Oxyfluorination of Alkenyl Oximes. Adv. Synth. Catal. 2017, 359, 1626–1630. [Google Scholar] [CrossRef]
- Egami, H.; Asada, J.; Sato, K.; Hashizume, D.; Kawato, Y.; Hamashima, Y. Asymmetric Fluorolactonization with a Bifunctional Hydroxyl Carboxylate Catalyst. J. Am. Chem. Soc. 2015, 137, 10132–10135. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Niwa, T.; Sato, H.; Hotta, R.; Rouno, D.; Kawato, Y.; Hamashima, Y. Dianionic Phase-Transfer Catalyst for Asymmetric Fluorocyclization. J. Am. Chem. Soc. 2018, 140, 2785–2788. [Google Scholar] [CrossRef]
- Niwa, T.; Ujiie, K.; Sato, H.; Egami, H.; Hamashima, Y. Asymmetric Fluorination of Cyclic Tetrasubstituted Alkenes with a Pendant Amide Groups under Dianionic Phase-Transfer Catalysis. Chem. Pharm. Bull. 2018, 66, 920–922. [Google Scholar] [CrossRef]
- Begum, S.A.; Terao, J.; Kambe, N. Conversion of (sp3)C–F Bonds of Alkyl Fluorides to (sp3)C–Heteroaton (Heteroatom = I, SR, SeR, TeR) Bonds by the Use of Magnesium Reagents Having Heteroatom Substituents. Chem. Lett. 2007, 36, 196–197. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-Disproportionation of Enantiomers of Chiral. Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Precatalyst | Solvent | Base | Yield (%) 2 | Ee (%) 3 |
|---|---|---|---|---|---|
| 1 4 | 1 | toluene | Na3PO4 | 39 | 58 |
| 2 4 | 1 | chlorobenzene | Na3PO4 | 26 | 58 |
| 3 4 | 1 | benzene | Na3PO4 | 30 | 57 |
| 4 4 | 1 | CH2Cl2 | Na3PO4 | 11 | - |
| 5 4 | 1 | THF | Na3PO4 | 3 | - |
| 6 | 1 | toluene | Na3PO4 | 42 | 59 |
| 7 | 1 | toluene | Na2HPO4 | 19 | 61 |
| 8 | 1 | toluene | Na2CO3 | 35 | 61 |
| 9 | 1 | toluene | K3PO4 | 24 | 59 |
| 10 | 1 | toluene | K2CO3 | 3 | - |
| 11 | 1 | toluene | proton sponge | 17 | 7 |
| 12 5 | 1 | toluene | Na3PO4 | 63 (58) 6 | 69 |
| 13 5 | 1 | toluene | Na2CO3 | 62 (58) 6 | 65 |
| 14 5 | 7 | toluene | Na3PO4 | 2 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rouno, T.; Niwa, T.; Nishibashi, K.; Yamamoto, N.; Egami, H.; Hamashima, Y. Enantioselective 5-exo-Fluorocyclization of Ene-Oximes. Molecules 2019, 24, 3464. https://doi.org/10.3390/molecules24193464
Rouno T, Niwa T, Nishibashi K, Yamamoto N, Egami H, Hamashima Y. Enantioselective 5-exo-Fluorocyclization of Ene-Oximes. Molecules. 2019; 24(19):3464. https://doi.org/10.3390/molecules24193464
Chicago/Turabian StyleRouno, Taiki, Tomoki Niwa, Kousuke Nishibashi, Nobuharu Yamamoto, Hiromichi Egami, and Yoshitaka Hamashima. 2019. "Enantioselective 5-exo-Fluorocyclization of Ene-Oximes" Molecules 24, no. 19: 3464. https://doi.org/10.3390/molecules24193464
APA StyleRouno, T., Niwa, T., Nishibashi, K., Yamamoto, N., Egami, H., & Hamashima, Y. (2019). Enantioselective 5-exo-Fluorocyclization of Ene-Oximes. Molecules, 24(19), 3464. https://doi.org/10.3390/molecules24193464