Design, Synthesis, and Biological Evaluation of Novel Thienopyrimidine Derivatives as PI3Kα Inhibitors


 and
and
Abstract
:
1. Introduction
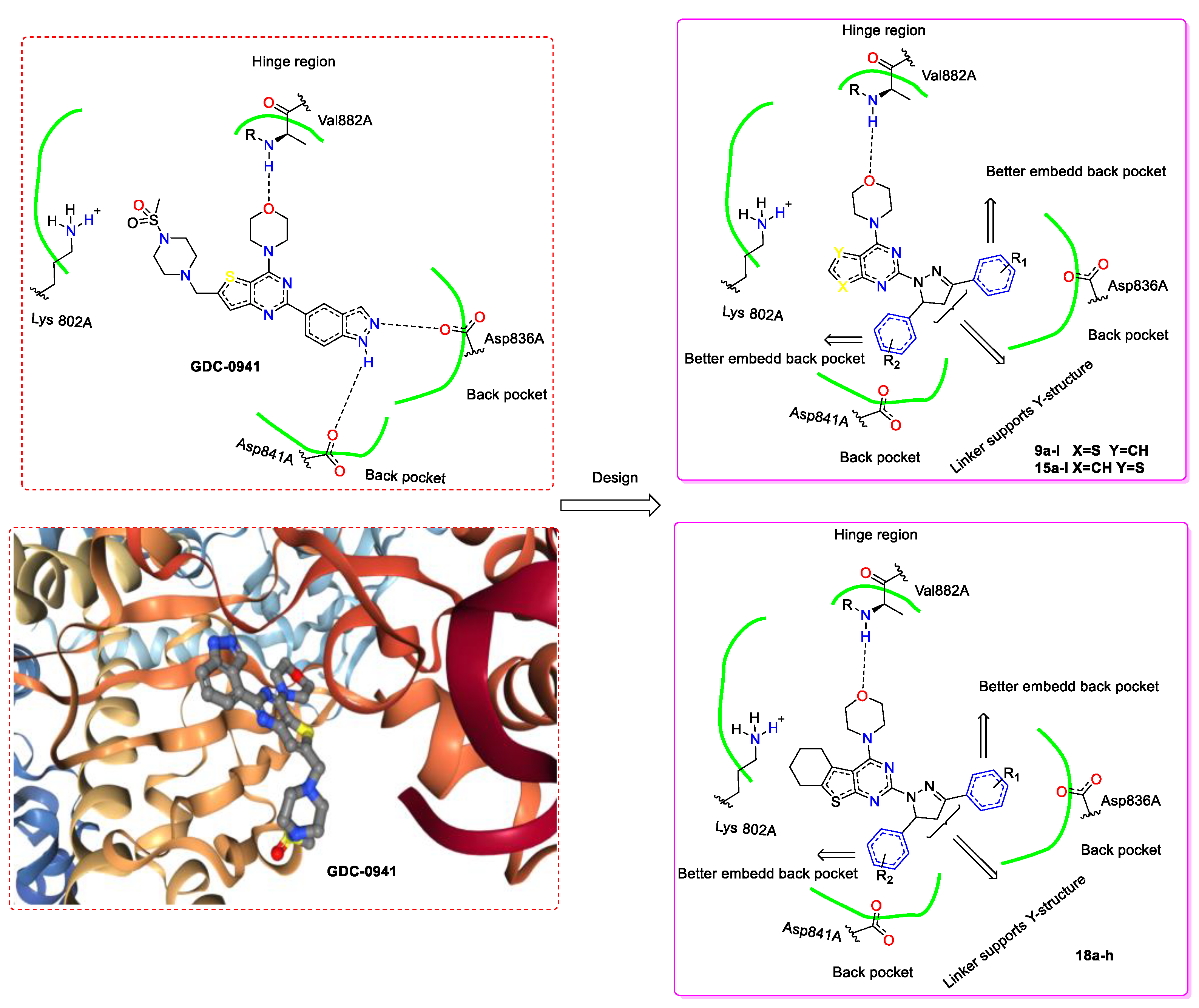
2. Results and Discussion
2.1. Chemistry
2.2. Biological Discussion
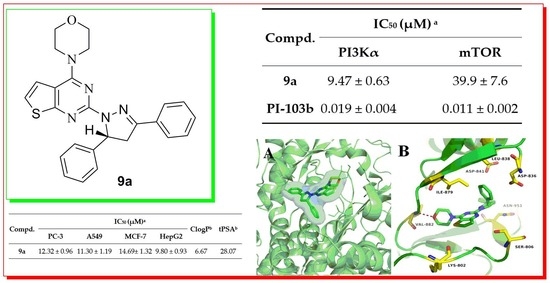
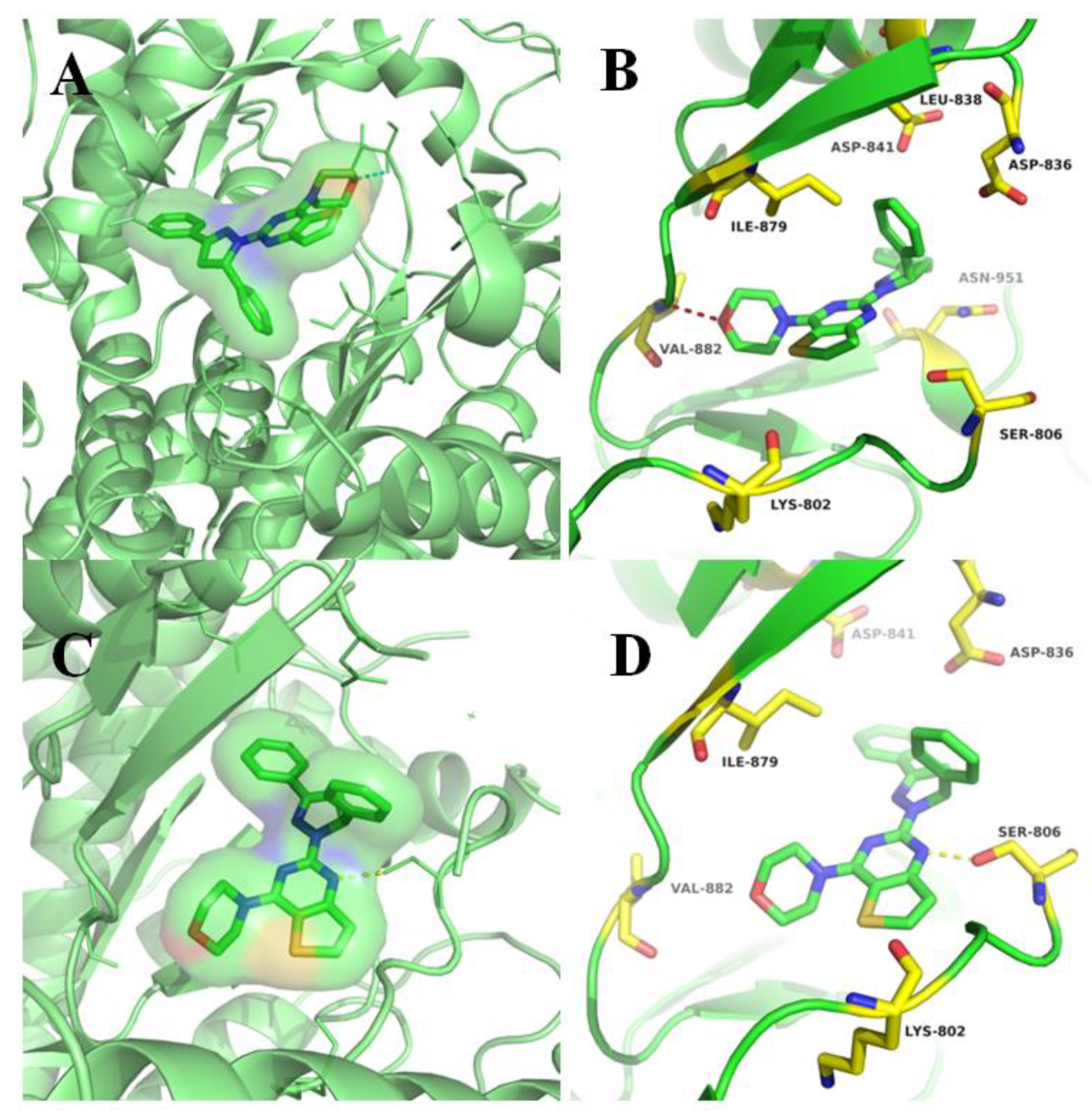
2.3. Molecular Docking Study of Compounds 9a and 15a.
3. Experimental Section
3.1. General Information
3.2. Chemistry
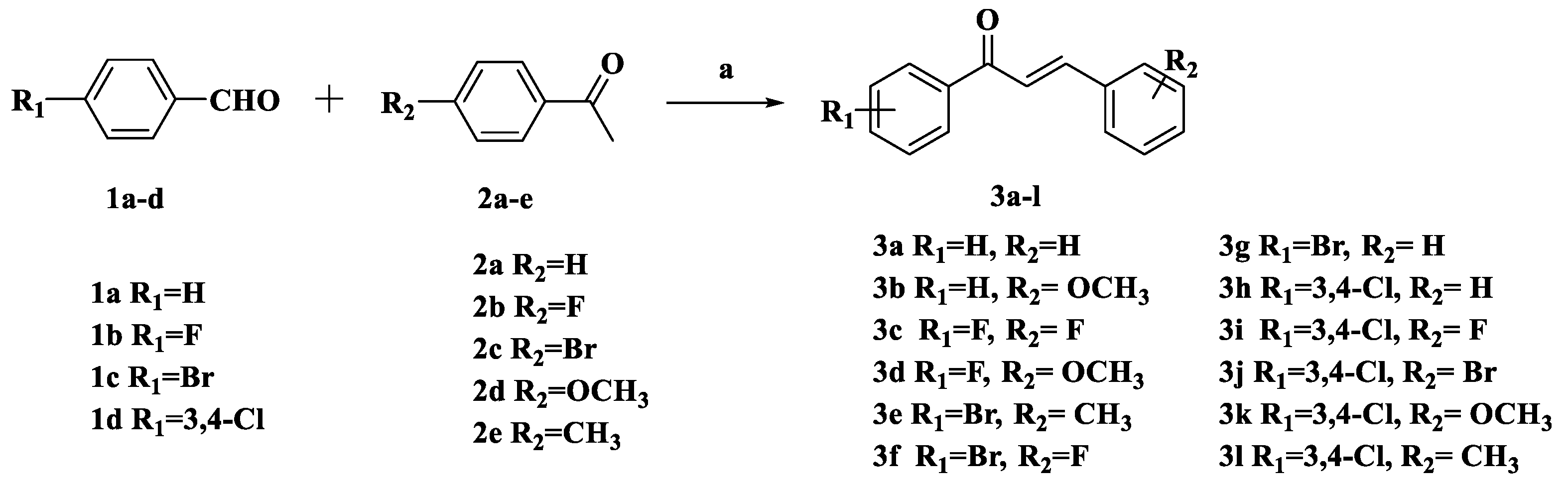
3.2.1. General Procedure for the Preparation of Compounds 3a–l
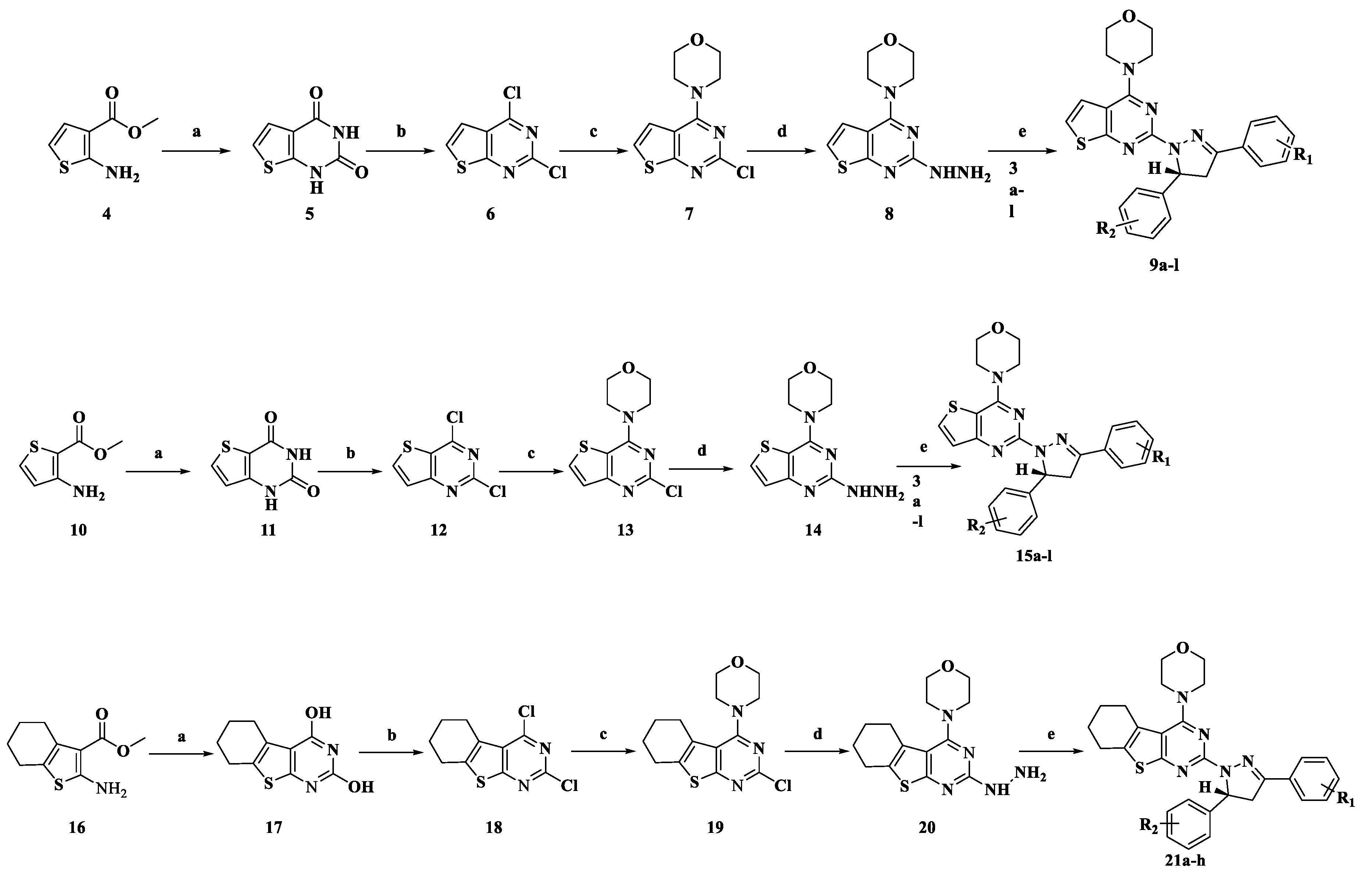
3.2.2. General Procedure for the Preparation of Compounds 8, 14 and 20
3.2.3. General Procedure for the Preparation of Target Compounds 9a–l, 15a–l, and 21a–h.
3.3. Cytotoxicity Assay In Vitro
3.4. mTOR Kinase Assay
3.5. PI3Kα Kinase Assay
3.6. Docking Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dehnhardt, C.M.; Venkatesan, A.M.; Delos Santos, E.; Chen, Z.; Santos, O.; Ayral-Kaloustian, S.; Brooijmans, N.; Mallon, R.; Hollander, I.; Feldberg, L.; et al. Lead optimization of N-3-substituted 7-morpholinotriazolopyrimidines as dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors: Discovery of PKI-402. J. Med. Chem. 2009, 53, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Marone, R.; Cmiljanovic, V.; Giese, B.; Wymann, M.P. Targeting phosphoinositide 3-kinase—Moving towards therapy. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1784, 159–185. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Ma, X.; Hu, Y. Furthering the design and the discovery of small molecule ATP-competitive mTOR inhibitors as an effective cancer treatment. Expert Opin. Drug Dis. 2013, 8, 991–1012. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Tu, Z.C.; Long, Z.J.; Liu, Q.; Lu, G. Discovery of 2-(2-aminopyrimidin-5-yl)-4-morpholino-N-(pyridin-3-yl) quinazolin-7-amines as novel PI3K/mTOR inhibitors and anticancer agents. Eur. J. Med. Chem. 2016, 108, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Mader, M.M.; Cook, J.A.; Iversen, P.; Ajamie, R.; Perkins, E.; Bloem, L.; Yip, Y.Y.; Barda, D.A.; Waid, P.P.; et al. Characterization of LY3023414, a novel PI3K/mTOR dual inhibitor eliciting transient target modulation to impede tumor growth. Mol. Cancer ther. 2016, 15, 2344–2356. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan–class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.; Yoon, S.S.; Harrison, S.J.; Morris, S.; Smith, D.; Freedman, S.J.; Brigandi, R.; Oliff, A.; Opalinska, J.B.; Chen, C. Novel AKT inhibitor GSK2110183 shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Preliminary results from a phase I first-time-in-human study. Blood. 2011, 118, 1856. [Google Scholar]
- Huo, H.; Zhou, Z.; Wang, B.; Qin, J.; Liu, W.Y.; Gu, Y. Dramatic suppression of colorectal cancer cell growth by the dual mTORC1 and mTORC2 inhibitor AZD-2014. Biochem. Bioph. Res. Commun. 2014, 443, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Sutherlin, D.P.; Bao, L.; Berry, M.; Castanedo, G.; Chuckowree, I.; Dotson, J.; Folks, A.; Friedman, L.; Goldsmith, R.; Gunzner, J.; et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J. Med. Chem. 2011, 54, 7579–7587. [Google Scholar] [CrossRef] [PubMed]
- Heffron, T.P.; Berry, M.; Castanedo, G.; Chang, C.; Chuckowree, I.; Dotson, J.; Folkes, A.; Gunzner, J.; Lesnick, J.D.; Lewis, C.; et al. Identification of GNE-477, a potent and efficacious dual PI3K/mTOR inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 2408–2411. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhai, X.; Fu, Q.; Guo, F.; Bai, M.; Wang, J.; Wang, H.; Gong, P. Design, synthesis and anticancer activity of 4-morpholinothieno [3, 2-d] pyrimidine derivatives bearing arylmethylene hydrazine moiety. Chem. Pharm. Bull. 2012, 60, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, C.; Sun, C.; Xu, S.; Wu, C.; Lei, F.; Xia, H.; Tu, Q.; Zheng, P. Design, synthesis and docking studies of novel thienopyrimidine derivatives bearing chromone moiety as mTOR/PI3Kα inhibitors. Eur. J. Med. Chem. 2015, 93, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liu, Y.; Zhai, X.; Wang, X.; Zhu, Y.; Wu, D.; Zhou, H.; Gong, P.; Zhao, Y. Design, synthesis and 3D-QSAR analysis of novel 2-hydrazinyl-4-morpholinothieno [3, 2-d] pyrimidine derivatives as potential antitumor agents. Eur. J. Med. Chem. 2012, 57, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, X.; Sun, C.; Zhang, B.; Zheng, P.; Zhu, W.; Xu, S. Synthesis and Structure–Activity Relationships of 4-Morpholino-7, 8-Dihydro-5H-Thiopyrano [4, 3-d] pyrimidine Derivatives Bearing Pyrazoline Scaffold. Molecules 2017, 22, 1870. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 9a–l, 15a–l, and 21a–h are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | R1 | R2 | IC50 (μM) a | ClogP b | tPSA b | |||
|---|---|---|---|---|---|---|---|---|
| PC-3 | A549 | MCF-7 | HepG2 | |||||
| 9a | 4-H | 4-H | 12.32 ± 0.96 | 11.30 ± 1.19 | 14.69 ± 1.32 | 9.80 ± 0.93 | 6.67 | 28.07 |
| 9b | 4-H | 4-OCH3 | 15.36 ± 1.26 | 21.22 ± 2.75 | 14.12 ± 2.1 | 13.08 ± 1.20 | 6.59 | 37.3 |
| 9c | 4-F | 4-F | 18.21 ± 1.55 | 24.23 ± 0.15 | 22.37 ± 0.17 | 17.36 ± 0.98 | 6.95 | 28.07 |
| 9d | 4-F | 4-OCH3 | 22.15 ± 1.67 | >50 | 20.62 ± 1.90 | 17.98 ± 2.12 | 6.73 | 37.3 |
| 9e | 4-Br | 4-CH3 | 23.16 ± 1.96 | >50 | 30.06 ± 2.86 | 21.63 ± 2.35 | 8.03 | 28.07 |
| 9f | 4-Br | 4-F | >50 | 15.37 ± 0.85 | 27.72 ± 2.71 | 13.49 ± 1.69 | 7.67 | 28.07 |
| 9g | 4-Br | 4-H | 30.12 ± 3.01 | 22.78 ± 2.15 | 28.46 ± 3.9 | 28.85 ± 2.34 | 7.53 | 28.07 |
| 9h | 3,4-di Cl | 4-H | 32.13 ± 2.96 | 38.53 ± 3.14 | 26.81 ± 2.13 | 23.70 ±2.08 | 7.97 | 28.07 |
| 9i | 3,4-di Cl | 4-F | 27.65± 2.34 | 17.75 ± 1.64 | 29.53 ± 1.87 | 23.70 ± 2.11 | 8.12 | 28.07 |
| 9j | 3,4-di Cl | 4-Br | 28.32 ± 2.16 | >50 | 39.34 ± 2.51 | >50 | 8.83 | 28.07 |
| 9k | 3,4-di Cl | 4-OCH3 | 33.54 ± 3.26 | 40.65 ± 3.85 | 25.30 ± 2.20 | 20.88 ± 1.98 | 7.89 | 37.3 |
| 9l | 3,4-di Cl | 4-CH3 | 28.36 ± 2.58 | >50 | >50 | 30.70 ± 0.15 | 8.47 | 28.07 |
| 15a | 4-H | 4-H | 15.53 ± 1.21 | 16.90 ± 1.61 | 17.03 ± 1.68 | 13.14 ± 1.48 | 6.67 | 28.07 |
| 15b | 4-H | 4-OCH3 | 25.45 ± 2.32 | >50 | >50 | 34.62 ± 2.82 | 6.58 | 37.3 |
| 15c | 4-F | 4-F | 25.31 ± 2.39 | 39.03 ± 3.32 | 37.12 ± 2.99 | 18.90 ± 1.86 | 6.95 | 28.07 |
| 15d | 4-F | 4-OCH3 | 22.13 ± 2.13 | 25.83 ± 1.99 | 35.15 ± 2.04 | 18.02 ± 1.30 | 6.73 | 37.3 |
| 15e | 4-Br | 4-CH3 | 37.12 ± 3.16 | 45.78 ± 0.81 | >50 | 24.41 ± 1.14 | 8.03 | 28.07 |
| 15f | 4-Br | 4-F | 26.45 ± 2.57 | 42.09 ± 0.08 | 36.17 ± 1.13 | 19.90 ± 1.10 | 7.67 | 28.07 |
| 15g | 4-Br | 4-H | 24.22 ± 2.26 | 21.73 ± 1.47 | 32.14 ± 0.89 | 21.36 ± 1.86 | 7.53 | 28.07 |
| 15h | 3,4-di Cl | 4-H | 27.15 ± 2.53 | 33.23 ± 2.14 | >50 | 21.15 ± 1.97 | 7.97 | 28.07 |
| 15i | 3,4-di Cl | 4-F | 18.48 ± 1.73 | >50 | >50 | 15.56 ± 1.30 | 8.12 | 28.07 |
| 15j | 3,4-di Cl | 4-Br | 32.97 ±3.22 | 41.78 ± 0.81 | >50 | 28.97 ± 2.49 | 8.83 | 28.07 |
| 15k | 3,4-di Cl | 4-OCH3 | 26.57 ± 2.38 | 31.05 ± 0.39 | >50 | 19.86 ± 1.88 | 7.89 | 37.3 |
| 15l | 3,4-di Cl | 4-CH3 | 23.68 ±1.94 | 31.79 ± 0.52 | >50 | 21.59 ± 1.86 | 8.47 | 28.07 |
| GDC-0941c | - | - | 4.35 ± 0.33 | 6.99 ± 0.21 | 0.20 ± 0.08 | 0.07 ± 0.03 | 3.20 | 76.85 |
| Compd. | R1 | R2 | IC50 (μM) a | ClogP b | tPSA b | |||
|---|---|---|---|---|---|---|---|---|
| PC-3 | A549 | MCF-7 | HepG2 | |||||
| 21a | 3,4-di Cl | 4-OCH3 | 31.75 ± 0.95 | 27.68 ± 0.10 | >501 | 32.37 ± 1.84 | 8.23 | 28.07 |
| 21b | 4-Br | 4-CH3 | 20.64 ± 0.63 | 11.59 ± 0.11 | 15.29 ± 0.83 | 12.43 ± 0.96 | 7.44 | 28.07 |
| 21c | 4-H | 4-H | >50 | 41.99 ± 1.49 | >50 | 6>50 | 8.56 | 28.07 |
| 21d | 4-F | 4-OCH3 | 25.28 ± 0.75 | 17.75± 1.1 | 29.53 ± 1.87 | 23.70 ± 0.11 | 8.79 | 28.07 |
| 21e | 4-Br | 4-F | >50 | >50 | >50 | 38.71 ± 1.72 | 7.86 | 28.07 |
| 21f | 4-Br | 4-H | >50 | >50 | >50 | >50 | 8.84 | 28.07 |
| 21g | 3,4-diCl | 4-Br | >50 | >50 | >50 | >50 | 8.63 | 28.07 |
| 21h | 3,4-diCl | 4-H | >50 | 23.53 ± 0.82 | >50 | >50 | 7.95 | 28.07 |
| GDC-0941 c | - | - | 4.35 ± 0.33 | 6.99 ± 0.21 | 0.20 ± 0.08 | 0.07 ± 0.03 | 3.20 | 76.85 |
| Compd. | R1 | R2 | IC50 (µM) a | |
|---|---|---|---|---|
| PI3Kα | mTOR | |||
| 9a | 4-H | 4-H | 9.47 ± 0.63 | 39.9 ± 7.6 |
| 15a | 4-H | 4-H | 25.68 ± 2.33 | >50 |
| PI-103 b | - | - | 0.019 ± 0.004 | 0.011 ± 0.002 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Wang, Q.; Wang, C.; Zhang, B.; Yang, Z.; Fang, Y.; Zhu, W.; Zheng, P. Design, Synthesis, and Biological Evaluation of Novel Thienopyrimidine Derivatives as PI3Kα Inhibitors. Molecules 2019, 24, 3422. https://doi.org/10.3390/molecules24193422
Yu L, Wang Q, Wang C, Zhang B, Yang Z, Fang Y, Zhu W, Zheng P. Design, Synthesis, and Biological Evaluation of Novel Thienopyrimidine Derivatives as PI3Kα Inhibitors. Molecules. 2019; 24(19):3422. https://doi.org/10.3390/molecules24193422
Chicago/Turabian StyleYu, Lide, Qinqin Wang, Caolin Wang, Binliang Zhang, Zunhua Yang, Yuanying Fang, Wufu Zhu, and Pengwu Zheng. 2019. "Design, Synthesis, and Biological Evaluation of Novel Thienopyrimidine Derivatives as PI3Kα Inhibitors" Molecules 24, no. 19: 3422. https://doi.org/10.3390/molecules24193422
APA StyleYu, L., Wang, Q., Wang, C., Zhang, B., Yang, Z., Fang, Y., Zhu, W., & Zheng, P. (2019). Design, Synthesis, and Biological Evaluation of Novel Thienopyrimidine Derivatives as PI3Kα Inhibitors. Molecules, 24(19), 3422. https://doi.org/10.3390/molecules24193422