Quantum Chemistry Insight into the Interactions Between Deep Eutectic Solvents and SO2




Abstract
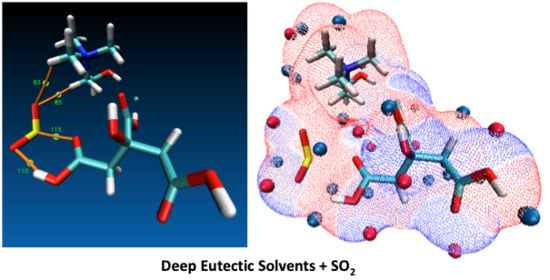
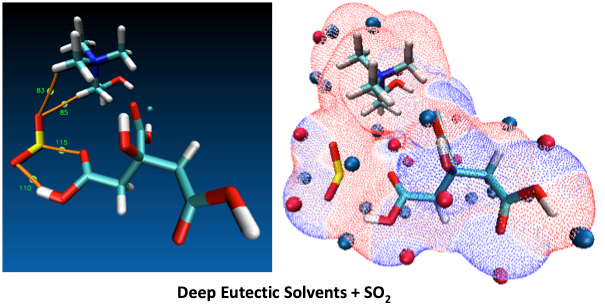
1. Introduction
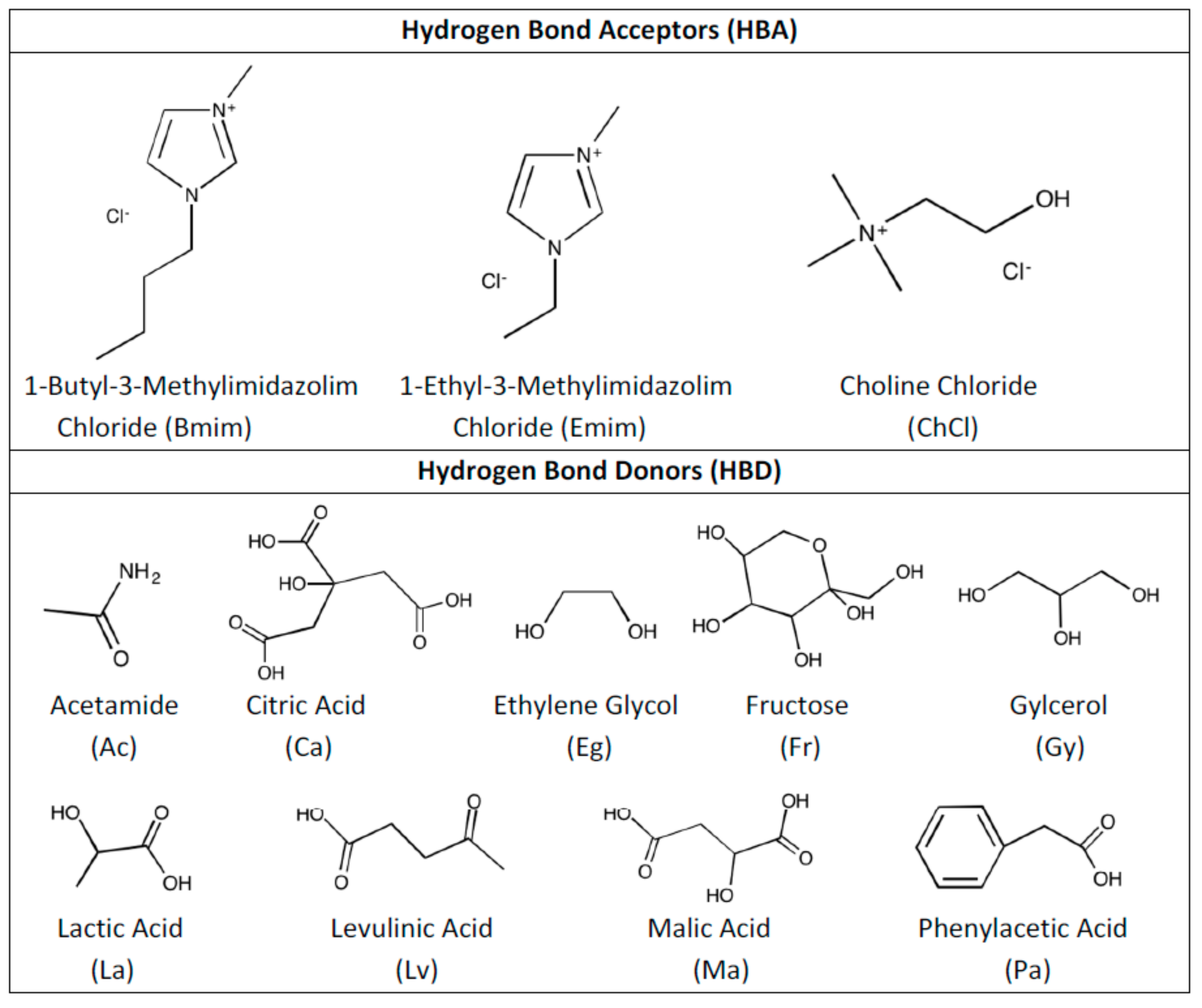
2. Methodology
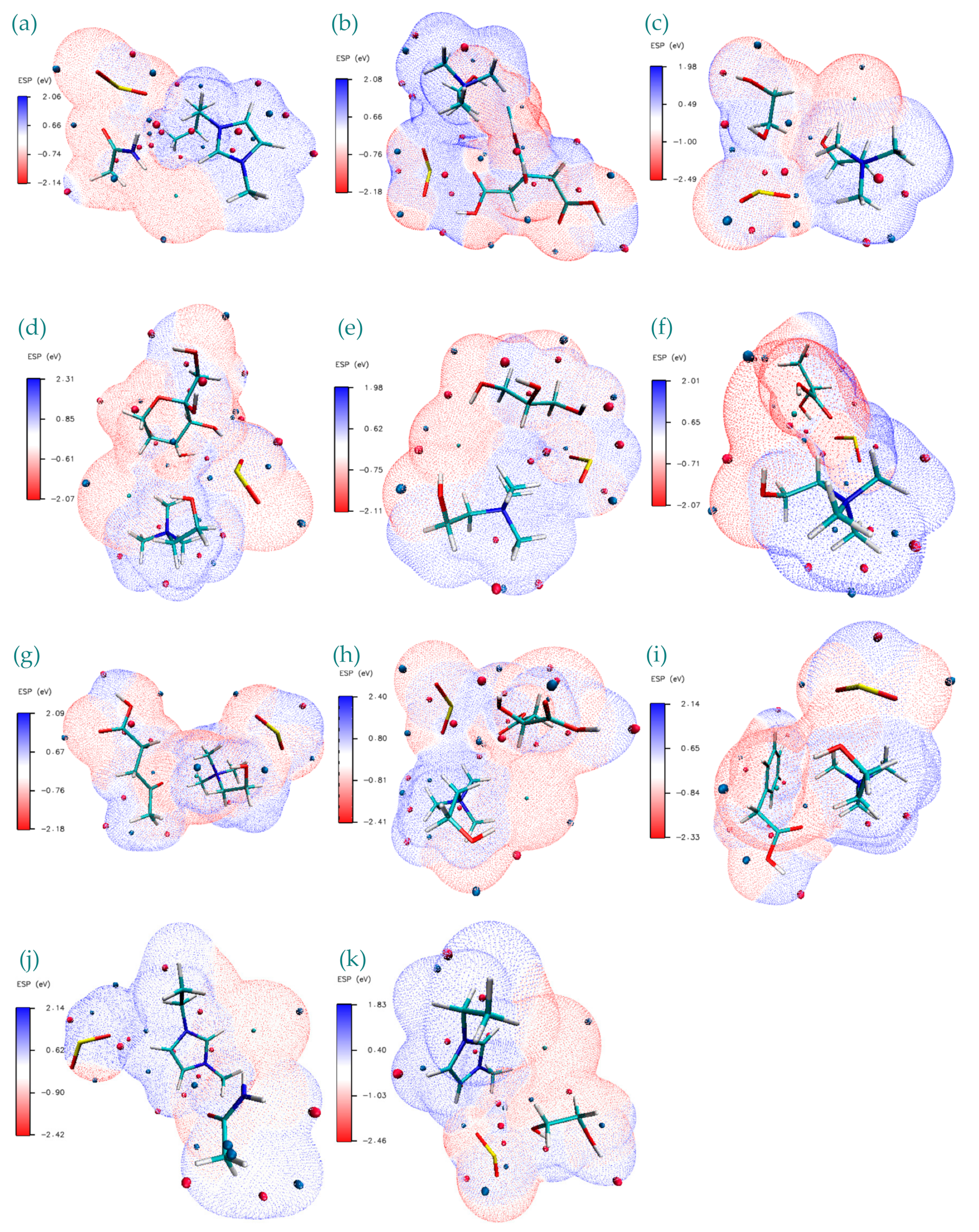
3. Results and Discussion
4. Conclusions
- (i)
- These results are quite useful in order to show the Lewis model interactions and acidity of DESs as absorbents that are considered for SO2 absorption. Furthermore, the chemical and kinetic stability of the DES structures was confirmed when they are in contact with SO2, which is a pre-indicator of these solvents for cyclic use and their regenerable nature.
- (ii)
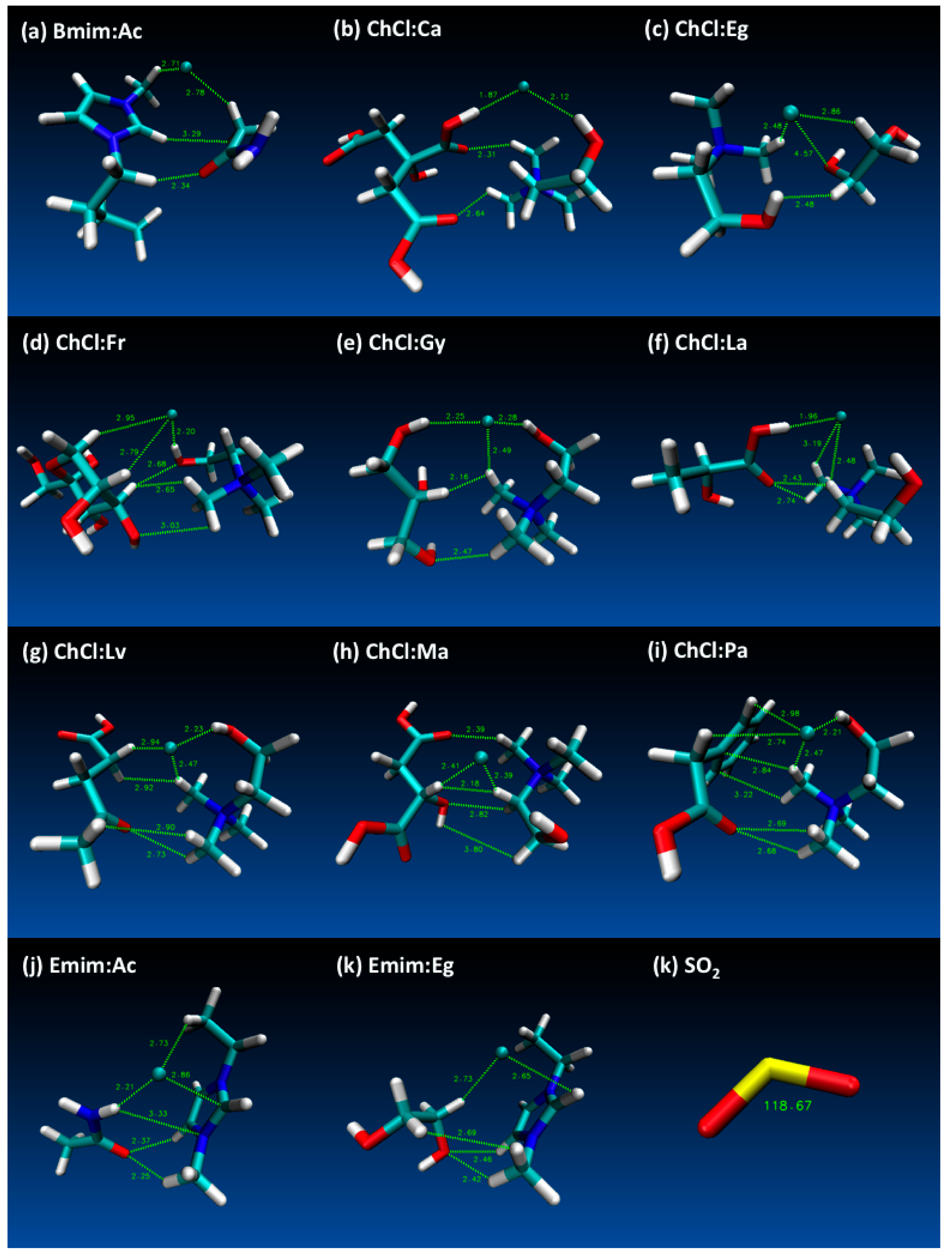
- Both QTAIM and structure analysis results show that strong interactions contribute to SO2 absorption, which are controlled mainly with the cation component of the HBA. These interactions are mostly localized between the O atom of the SO2 and the cation part of the IL. In contrast with this, when HBD dominates (just a couple of cases) the SO2 affinity, O atom in the HBD interacts with the S atom in the SO2 molecule.
- (iii)
- Despite the calculation of overall total energies and more importantly binding energies not showing a wide range of variation, it is obvious that the selection of HBD makes the structure sensitive to the SO2 interaction.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mac Kinnon, M.A.; Brouwer, J.; Samuelsen, S. The role of natural gas and its infrastructure in mitigating greenhouse gas emissions, improving regional air quality and renewable resource integration. Prog. Energy Combust. Sci. 2018, 64, 62–92. [Google Scholar] [CrossRef]
- Keeling, C.D.; Piper, S.C.; Bacastow, R.B.; Wahlen, M.; Whorf, T.P.; Heimann, M.; Meijer, H.A. Exchanges of atmospheric CO2 and 13CO2 with the terrestrial biosphere and oceans from 1978 to 2000. I. Global aspects. Available online: https://escholarship.org/uc/item/09v319r9 (accessed on 28 July 2019).
- Rezaei, F.; Rownaghi, A.A.; Monjezi, S.; Lively, R.P.; Jones, C.W. SOx/NOx Removal from Flue Gas Streams by Solid Adsorbents: A Review of Current Challenges and Future Directions. Energy Fuels 2015, 29, 5467–5486. [Google Scholar] [CrossRef]
- Adil, K.; Bhatt, P.M.; Belmabkhout, Y.; Abtab, S.M.T.; Jiang, H.; Assen, A.H.; Mallick, A.; Cadiau, A.; Aqil, J.; Eddaoudi, M. Valuing Metal–Organic Frameworks for Postcombustion Carbon Capture: A Benchmark Study for Evaluating Physical Adsorbents. Adv. Mater. 2017, 29, 1702953. [Google Scholar] [CrossRef] [PubMed]
- Speight, J.G. Chapter Five—Sources and Types of Inorganic Pollutants. In Environmental Inorganic Chemistry for Engineers; Speight, J.G., Ed.; Butterworth-Heinemann: Oxford, UK, 2017; pp. 231–282. [Google Scholar]
- Chen, T.-M.; Kuschner, W.G.; Gokhale, J.; Shofer, S. Outdoor Air Pollution: Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide Health Effects. Am. J. Med. Sci. 2007, 333, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; van Aardenne, J.; Klimont, Z.; Andres, R.J.; Volke, A.; Arias, S.D. Anthropogenic sulfur dioxide emissions: 1850-2005. Atmos. Chem. Phys. 2011, 11, 1101–1116. [Google Scholar] [CrossRef]
- Clarke, A.G.; Radojevic, M. Oxidation of SO2 in rainwater and its role in acid rain chemistry. Atmos. Environ. 1987, 21, 1115–1123. [Google Scholar] [CrossRef]
- Galloway, J.N.; Dianwu, Z.; Jiling, X.; Likens, G.E. Acid Rain: China, United States and a Remote Area. Science 1987, 236, 1559. [Google Scholar] [CrossRef]
- Emmett, E.J.; Willis, M.C. The Development and Application of Sulfur Dioxide Surrogates in Synthetic Organic Chemistry. Asian J. Org. Chem. 2015, 4, 602–611. [Google Scholar] [CrossRef]
- Pandey, R.A.; Biswas, R.; Chakrabarti, T.; Devotta, S. Flue Gas Desulfurization: Physicochemical and Biotechnological Approaches. Crit. Rev. Environ. Sci. Technol. 2005, 35, 571–622. [Google Scholar] [CrossRef]
- Zheng, Y.; Kiil, S.; Johnsson, J.E. Experimental investigation of a pilot-scale jet bubbling reactor for wet flue gas desulphurisation. Chem. Eng. Sci. 2003, 58, 4695–4703. [Google Scholar] [CrossRef]
- Kikkawa, H.; Nakamoto, T.; Morishita, M.; Yamada, K. New Wet FGD Process Using Granular Limestone. Ind. Eng. Chem. Res. 2002, 41, 3028–3036. [Google Scholar] [CrossRef]
- Zhao, T.; Liang, J.; Zhang, Y.; Wu, Y.; Hu, X. Unexpectedly efficient SO2 capture and conversion to sulfur in novel imidazole-based deep eutectic solvents. Chem. Commun. (Camb) 2018, 54, 8964–8967. [Google Scholar] [CrossRef] [PubMed]
- Kanan, S.M.; El-Kadri, O.M.; Abu-Yousef, I.A.; Kanan, M.C. Semiconducting metal oxide based sensors for selective gas pollutant detection. Sensors (Basel) 2009, 9, 8158–8196. [Google Scholar] [CrossRef] [PubMed]
- Berger, F.; Fromm, M.; Chambaudet, A.; Planade, R. Tin dioxide-based gas sensors for SO2 detection: A chemical interpretation of the increase in sensitivity obtained after a primary detection. Sens. Actuators B Chem. 1997, 45, 175–181. [Google Scholar] [CrossRef]
- Torvela, H.; Huusko, J.; Lantto, V. Reduction of the interference caused by NO and SO2 in the CO response of Pd-catalysed SnO2 combustion gas sensors. Sens. Actuators B Chem. 1991, 4, 479–484. [Google Scholar] [CrossRef]
- Gardon, M.; Guilemany, J.M. A review on fabrication, sensing mechanisms and performance of metal oxide gas sensors. J. Mater. Sci. Mater. Electron. 2013, 24, 1410–1421. [Google Scholar] [CrossRef]
- Barsan, N.; Koziej, D.; Weimar, U. Metal oxide-based gas sensor research: How to? Sens. Actuators B Chem. 2007, 121, 18–35. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, S.; Jiang, D.E.; Dai, S. SO2 absorption in EmimCl-TEG deep eutectic solvents. Phys. Chem. Chem. Phys. 2018, 20, 15168–15173. [Google Scholar] [CrossRef]
- Zhang, K.; Ren, S.; Yang, X.; Hou, Y.; Wu, W.; Bao, Y. Efficient absorption of low-concentration SO 2 in simulated flue gas by functional deep eutectic solvents based on imidazole and its derivatives. Chem. Eng. J. 2017, 327, 128–134. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, Y.; Feng, X.; Zhu, X. A detailed study of physicochemical properties and microstructure of EmimCl-EG deep eutectic solvents: Their influence on SO2 absorption behavior. J. Ind. Eng. Chem. 2018, 67, 148–155. [Google Scholar] [CrossRef]
- Cui, G.; Wang, C.; Zheng, J.; Guo, Y.; Luo, X.; Li, H. Highly efficient SO2 capture by dual functionalized ionic liquids through a combination of chemical and physical absorption. Chem. Commun. (Camb) 2012, 48, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Atilhan, M.; Aparicio, S. A density functional theory insight towards the rational design of ionic liquids for SO2 capture. Phy.s Chem. Chem. Phys. 2015, 17, 13559–13574. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Qin, L.; Mu, T.; Xue, Z.; Gao, G. Are Ionic Liquids Chemically Stable? Chem. Rev. 2017, 117, 7113–7131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Chen, Y.; Zhang, L.; Tantai, X.; Dou, H.; Sun, Y. Design of multiple-site imidazole derivative aqueous solution for SO2 capture in low concentration. J. Taiwan Inst. Chem. Eng. 2018, 91, 441–448. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, J.; Wei, F. Characterization of caprolactam based eutectic ionic liquids and their application in SO2 absorption. J. Mol. Liq. 2013, 180, 19–25. [Google Scholar] [CrossRef]
- Zeng, S.; He, H.; Gao, H.; Zhang, X.; Wang, J.; Huang, Y.; Zhang, S. Improving SO2 capture by tuning functional groups on the cation of pyridinium-based ionic liquids. RSC Adv. 2015, 5, 2470–2478. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 1, 70–71. [Google Scholar] [CrossRef]
- Zhang, Q.; De Oliveira Vigier, K.; Royer, S.; Jérôme, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef]
- Altamash, T.; Atilhan, M.; Aliyan, A.; Ullah, R.; Nasser, M.; Aparicio, S. Rheological, Thermodynamic and Gas Solubility Properties of Phenylacetic Acid-Based Deep Eutectic Solvents. Chem. Eng. Technol. 2017, 40, 778–790. [Google Scholar] [CrossRef]
- Elhamarnah, Y.A.; Nasser, M.; Qiblawey, H.; Benamor, A.; Atilhan, M.; Aparicio, S. A comprehensive review on the rheological behavior of imidazolium based ionic liquids and natural deep eutectic solvents. J. Mol. Liq. 2019, 277, 932–958. [Google Scholar] [CrossRef]
- Garcia, G.; Aparicio, S.; Ullah, R.; Atilhan, M. Deep Eutectic Solvents: Physicochemical Properties and Gas Separation Applications. Energy Fuels 2015, 29, 2616–2644. [Google Scholar] [CrossRef]
- Dai, Y.T.; van Spronsen, J.; Witkamp, G.J.; Verpoorte, R.; Choi, Y.H. Natural deep eutectic solvents as new potential media for green technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Friesen, J.B.; McAlpine, J.B.; Lankin, D.C.; Chen, S.N.; Pauli, G.F. Natural Deep Eutectic Solvents: Properties, Applications, and Perspectives. J. Nat. Prod. 2018, 81, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.; Atilhan, M.; Aparicio, S. Interfacial Properties of Deep Eutectic Solvents Regarding to CO2 Capture. J. Phys. Chem. C 2015, 119, 21413–21425. [Google Scholar] [CrossRef]
- Garcia, G.; Atilhan, M.; Aparicio, S. A theoretical study on mitigation of CO2 through advanced deep eutectic solvents. Int. J. Greenh. Gas Control 2015, 39, 62–73. [Google Scholar] [CrossRef]
- Gutierrez, M.C.; Carriazo, D.; Ania, C.O.; Parra, J.B.; Ferrer, M.L.; del Monte, F. Deep eutectic solvents as both precursors and structure directing agents in the synthesis of nitrogen doped hierarchical carbons highly suitable for CO2 capture. Energy Environ. Sci. 2011, 4, 3535–3544. [Google Scholar] [CrossRef]
- Hao, G.P.; Jin, Z.Y.; Sun, Q.; Zhang, X.Q.; Zhang, J.T.; Lu, A.H. Porous carbon nanosheets with precisely tunable thickness and selective CO2 adsorption properties. Energy Environ. Sci. 2013, 6, 3740–3747. [Google Scholar] [CrossRef]
- Sarmad, S.; Mikkola, J.P.; Ji, X.Y. Carbon Dioxide Capture with Ionic Liquids and Deep Eutectic Solvents: A New Generation of Sorbents. Chemsuschem 2017, 10, 324–352. [Google Scholar] [CrossRef]
- Li, H.; Chang, Y.; Zhu, W.; Wang, C.; Wang, C.; Yin, S.; Zhang, M.; Li, H. Theoretical evidence of charge transfer interaction between SO(2) and deep eutectic solvents formed by choline chloride and glycerol. Phys. Chem. Chem. Phys. 2015, 17, 28729–28742. [Google Scholar] [CrossRef]
- Yang, D.; Han, Y.; Qi, H.; Wang, Y.; Dai, S. Efficient Absorption of SO2 by EmimCl-EG Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2017, 5, 6382–6386. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, B.; Dou, H.; Zhang, L.; Tantai, X.; Sun, Y.; Zhang, H. Highly Efficient and Reversible Capture of Low Partial Pressure SO2 by Functional Deep Eutectic Solvents. Energy Fuels 2018, 32, 10737–10744. [Google Scholar] [CrossRef]
- Korotkevich, A.; Firaha, D.S.; Padua, A.A.H.; Kirchner, B. Ab initio molecular dynamics simulations of SO 2 solvation in choline chloride/glycerol deep eutectic solvent. Fluid Phase Equilibria 2017, 448, 59–68. [Google Scholar] [CrossRef]
- Yang, D.; Hou, M.; Ning, H.; Zhang, J.; Ma, J.; Yang, G.; Han, B. Efficient SO2 absorption by renewable choline chloride–glycerol deep eutectic solvents. Green Chem. 2013, 15, 2261–2265. [Google Scholar] [CrossRef]
- Deng, D.; Liu, X.; Gao, B. Physicochemical Properties and Investigation of Azole-Based Deep Eutectic Solvents as Efficient and Reversible SO2 Absorbents. Ind. Eng. Chem. Res. 2017, 56, 13850–13856. [Google Scholar] [CrossRef]
- Warrag, S.E.E. Capturing impurities from oil and gas using deep eutectic solvents. Phd Thesis, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2018. [Google Scholar]
- Zhang, K.; Ren, S.; Hou, Y.; Wu, W. Efficient absorption of SO2 with low-partial pressures by environmentally benign functional deep eutectic solvents. J. Hazard. Mater. 2017, 324, 457–463. [Google Scholar] [CrossRef]
- Waite, S.L.; Li, H.; Page, A.J. NO2 Solvation Structure in Choline Chloride Deep Eutectic Solvents-The Role of the Hydrogen Bond Donor. J. Phys. Chem. B 2018, 122, 4336–4344. [Google Scholar] [CrossRef]
- Sun, S.; Niu, Y.; Xu, Q.; Sun, Z.; Wei, X. Efficient SO2 Absorptions by Four Kinds of Deep Eutectic Solvents Based on Choline Chloride. Ind. Eng. Chem. Res. 2015, 54, 8019–8024. [Google Scholar] [CrossRef]
- Anderson, J.L.; Dixon, J.K.; Maginn, E.J.; Brennecke, J.F. Measurement of SO2 Solubility in Ionic Liquids. J. Phys. Chem. B 2006, 110, 15059–15062. [Google Scholar] [CrossRef]
- Deng, D.; Han, G.; Jiang, Y. Investigation of a deep eutectic solvent formed by levulinic acid with quaternary ammonium salt as an efficient SO2 absorbent. New J. Chem. 2015, 39, 8158–8164. [Google Scholar] [CrossRef]
- Adams, S.; de Castro, P.; Echenique, P.; Estrada, J.; Hanwell, M.D.; Murray-Rust, P.; Sherwood, P.; Thomas, J.; Townsend, J.A. The Quixote project: Collaborative and Open Quantum Chemistry data management in the Internet age. J. Cheminformatics 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Xiao, J.; Zhao, Y.-P.; Fan, X.; Cao, J.-P.; Kang, G.-J.; Zhao, W.; Wei, X.-Y. Hydrogen bonding interactions between the organic oxygen/nitrogen monomers of lignite and water molecules: A DFT and AIM study. Fuel Process. Technol. 2017, 168, 58–64. [Google Scholar] [CrossRef]
- Anbu, V.; Vijayalakshmi, K.A.; Karunathan, R.; Stephen, A.D.; Nidhin, P.V. Explosives properties of high energetic trinitrophenyl nitramide molecules: A DFT and AIM analysis. Arab. J. Chem. 2016, 5, 621–632. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683. [Google Scholar] [CrossRef]
- Santiago, A.; Yavuz, C.T.; Mert, A. Molecular Insights into Benzimidazole-Linked Polymer Interactions with Carbon Dioxide and Nitrogen. ChemistrySelect 2018, 3, 3691–3701. [Google Scholar]
- Saleh, G.; Gatti, C.; Lo Presti, L. Non-covalent interaction via the reduced density gradient: Independent atom model vs experimental multipolar electron densities. Comput. Theor. Chem. 2012, 998, 148–163. [Google Scholar] [CrossRef]
- Zupan, A.; Perdew, J.P.; Burke, K.; Causà, M. Density-gradient analysis for density functional theory: Application to atoms. Int. J. Quantum Chem. 1997, 61, 835–845. [Google Scholar] [CrossRef]
- Zupan, A.; Burke, K.; Ernzerhof, M.; Perdew, J.P. Distributions and averages of electron density parameters: Explaining the effects of gradient corrections. J. Chem. Phys. 1997, 106, 10184–10193. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2018, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Aparicio, S.; Atilhan, M. Design of arginine-based therapeutic deep eutectic solvents as drug solubilization vehicles for active pharmaceutical ingredients. Phys. Chem. Chem. Phys. 2019, 21, 10621–10634. [Google Scholar] [CrossRef]
- Altamash, T.; Amhamed, A.I.; Aparicio, S.; Atilhan, M. Combined Experimental and Theoretical Study on High Pressure Methane Solubility in Natural Deep Eutectic Solvents. Ind. Eng. Chem. Res. 2019, 58, 8097–8111. [Google Scholar] [CrossRef]
- Yoosefian, M.; Zahedi, M.; Mola, A.; Naserian, S. A DFT comparative study of single and double SO2 adsorption on Pt-doped and Au-doped single-walled carbon nanotube. Appl. Surf. Sci. 2015, 349, 864–869. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Zhao, L.; Schwarz, W.H.E.; Frenking, G. The Lewis electron-pair bonding model: The physical background, one century later. Nat. Rev. Chem. 2019, 3, 35–47. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Johnson, S.; Tang, T.H.; Popelier, P.L.A. The Electron Pair. J. Phys. Chem. 1996, 100, 15398–15415. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and Beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
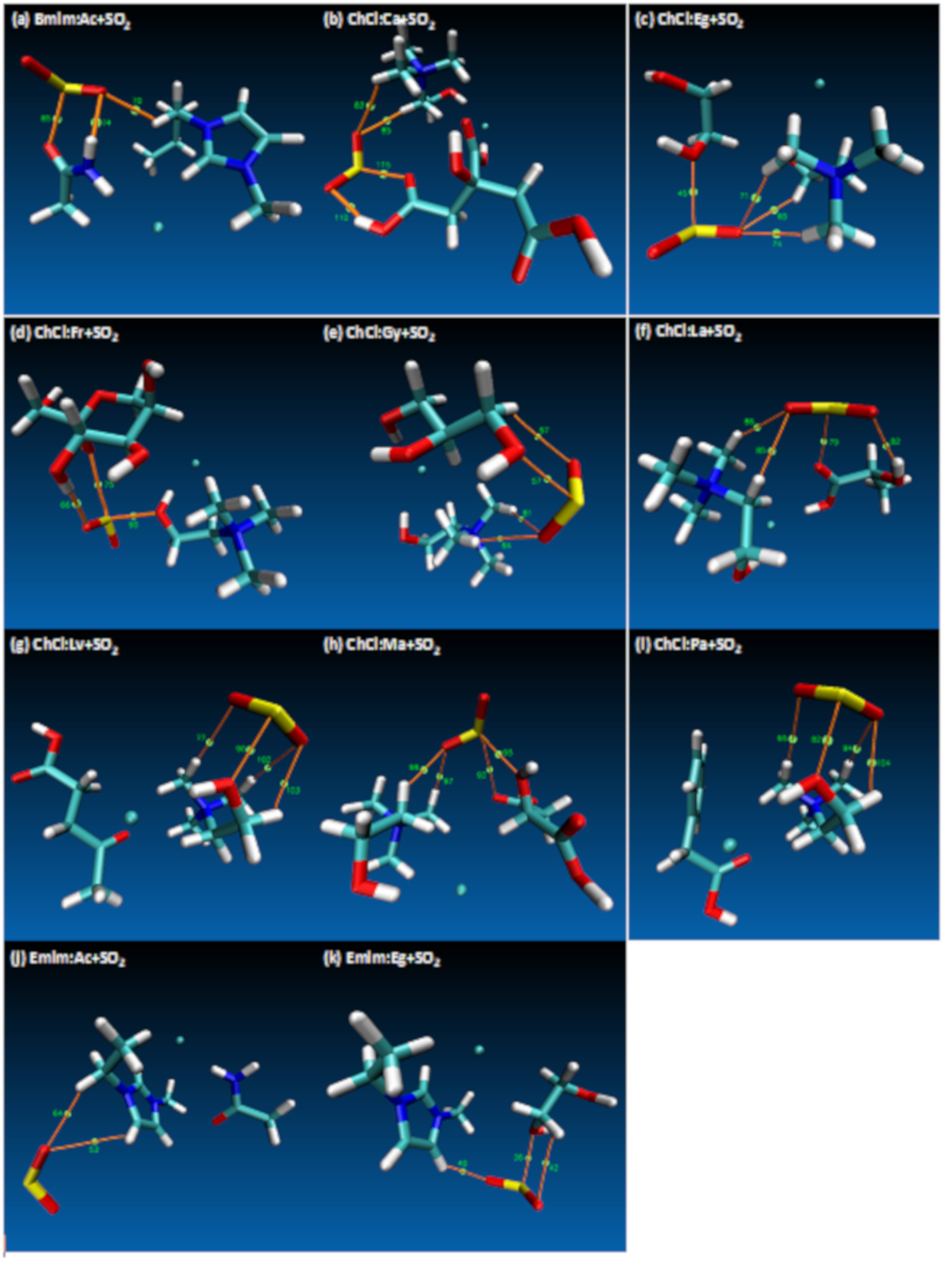{kind=link}
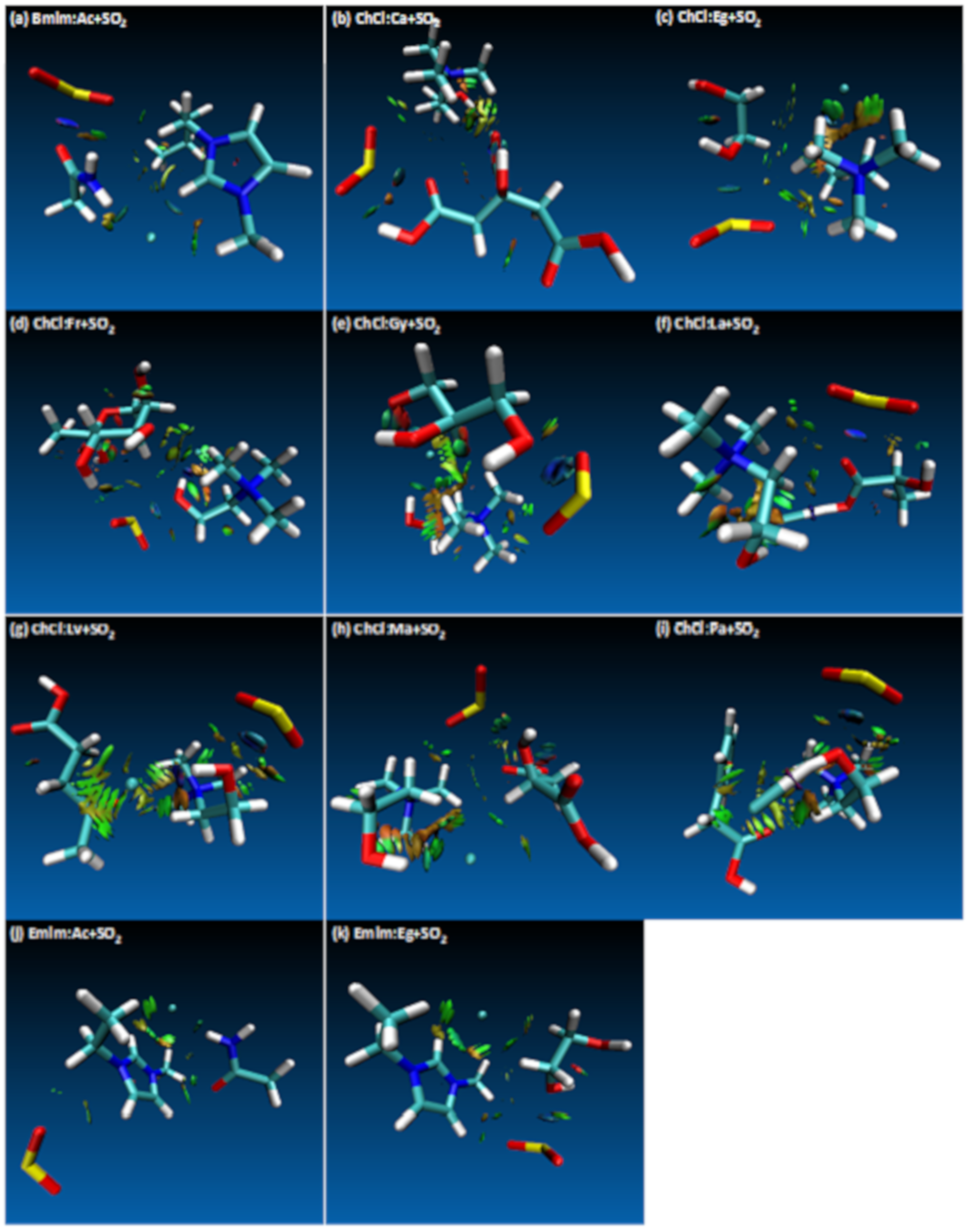{kind=link}
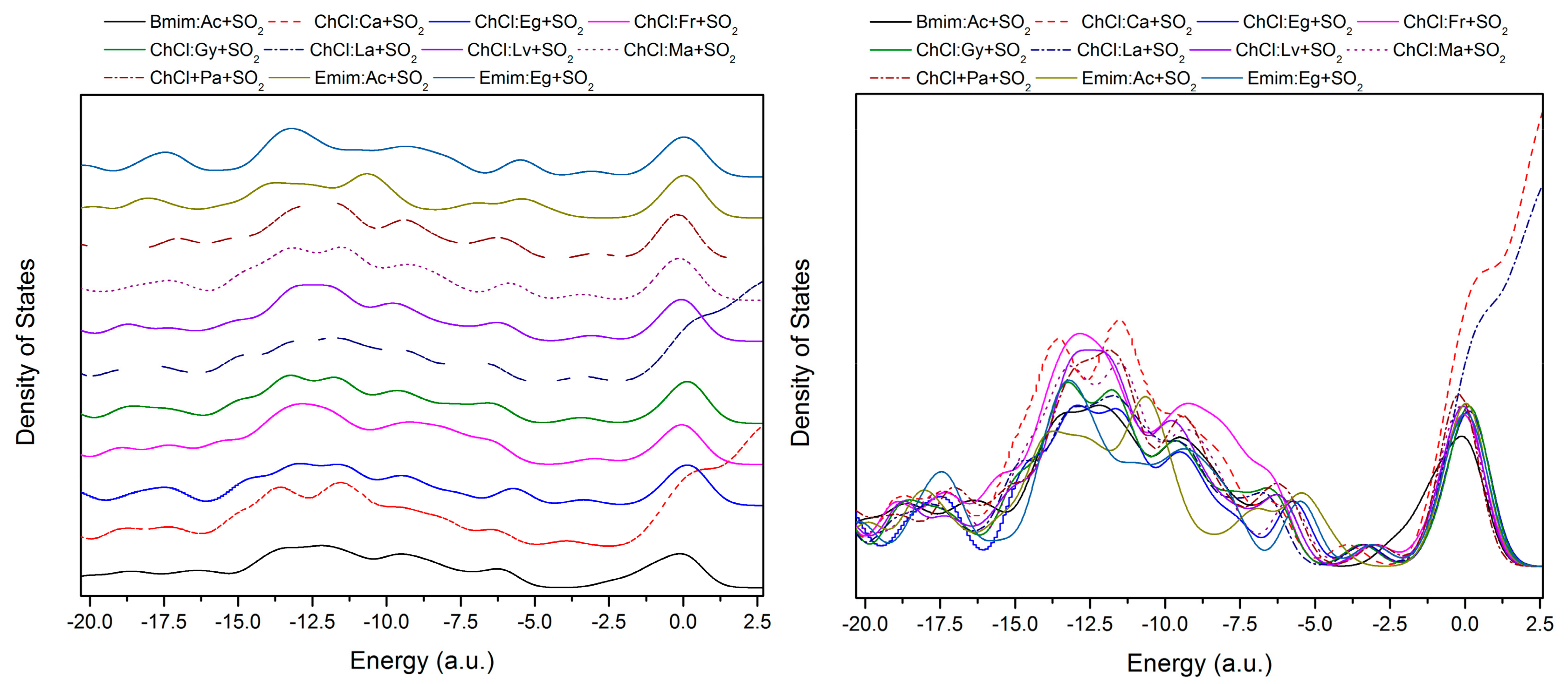{kind=link}
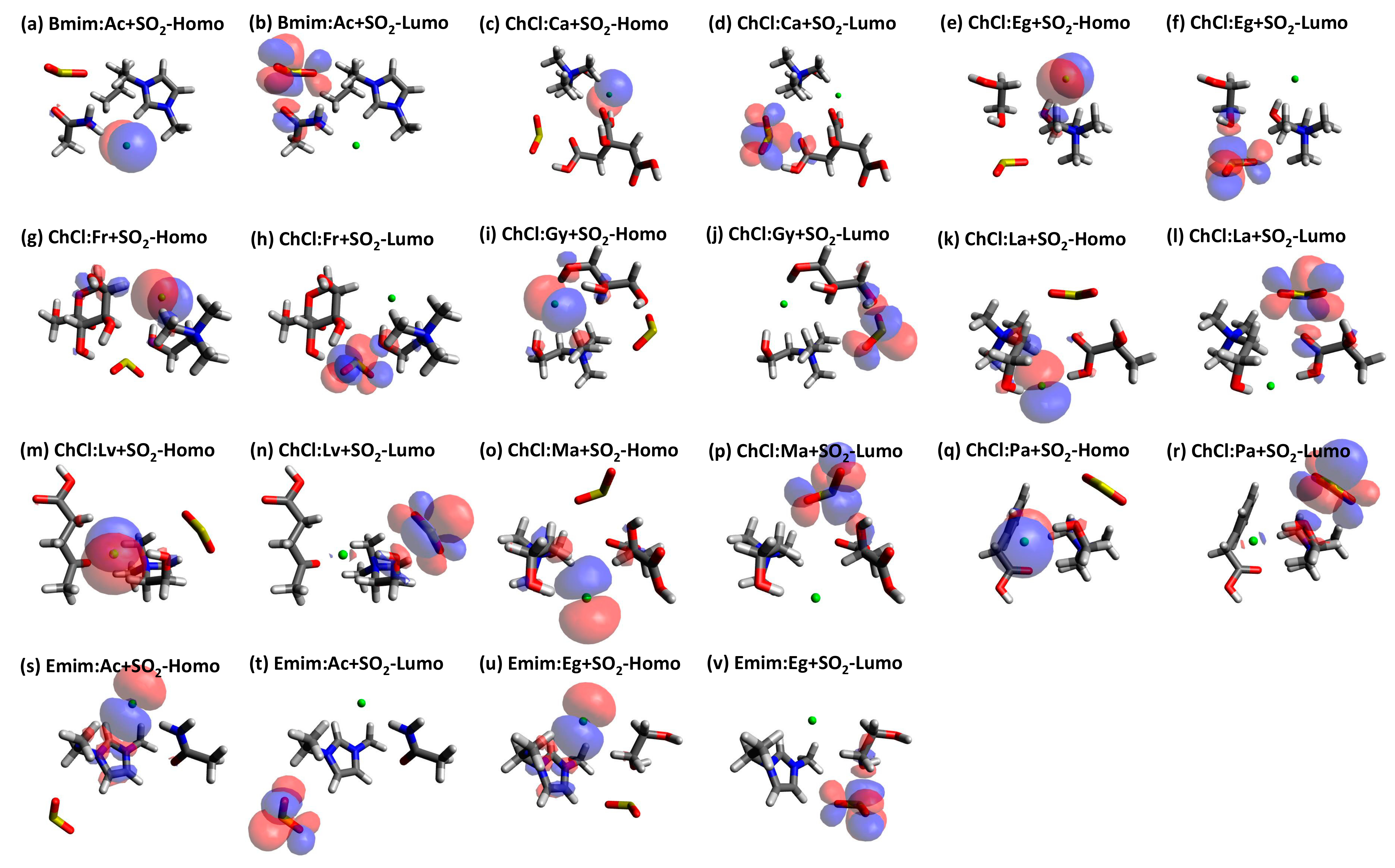{kind=link}
{kind=link}
| Structure | BCP No. | ρ × 103/a.u. | ∇2ρ × 102/a.u. | Structure | BCP No. | ρ × 103/a.u. | ∇2ρ × 102/a.u. |
|---|---|---|---|---|---|---|---|
| Bmim:Ac+SO2 | 70 | 1.24 | 3.11 | ChCl:La+SO2 | 79 | 0.800 | 1.36 |
| 74 | 1.14 | 3.81 | 82 | −283 | −99.0 | ||
| 85 | 0.498 | 12.1 | 85 | 0.965 | 2.10 | ||
| ChCl:Ca+SO2 | 83 | 2.06 | 6.81 | 86 | 0.842 | 2.67 | |
| 85 | 1.46 | 8.87 | ChCl:Lv+SO2 | 77 | 1.07 | 3.02 | |
| 110 | 0.872 | 2.75 | 90 | −322 | 99.9 | ||
| 115 | 0.784 | 2.10 | 102 | −279 | −97.2 | ||
| ChCl:Eg+SO2 | 45 | 1.31 | 8.91 | 103 | 0.866 | 2.74 | |
| 60 | 0.835 | 2.50 | ChCl:Ma+SO2 | 92 | 0.829 | 1.90 | |
| 71 | 0.620 | 1.80 | 93 | 1.50 | 7.00 | ||
| 74 | 1.10 | 2.99 | 97 | 1.31 | 3.65 | ||
| ChCl:Fr+SO2 | 66 | 2.64 | 7.59 | 98 | 1.01 | 3.00 | |
| 75 | 0.889 | 2.26 | ChCl:Pa+SO2 | 66 | 0.679 | 1.84 | |
| 90 | 1.07 | 8.95 | 82 | 1.09 | 10.9 | ||
| ChCl:Gy+SO2 | 57 | −276 | −95.3 | 94 | 1.25 | 3.64 | |
| 64 | 1.11 | 2.67 | 104 | 0.908 | 3.12 | ||
| 67 | 0.796 | 2.22 | Emim:Eg+SO2 | 36 | 0.854 | 9.83 | |
| 81 | −248 | −50.1 | 40 | 1.07 | 2.57 | ||
| Emim:Ac+SO2 | 53 | 0.637 | 1.20 | 42 | 0.660 | 1.13 | |
| 64 | 0.719 | 2.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atilhan, M.; Altamash, T.; Aparicio, S. Quantum Chemistry Insight into the Interactions Between Deep Eutectic Solvents and SO2. Molecules 2019, 24, 2963. https://doi.org/10.3390/molecules24162963
Atilhan M, Altamash T, Aparicio S. Quantum Chemistry Insight into the Interactions Between Deep Eutectic Solvents and SO2. Molecules. 2019; 24(16):2963. https://doi.org/10.3390/molecules24162963
Chicago/Turabian StyleAtilhan, Mert, Tausif Altamash, and Santiago Aparicio. 2019. "Quantum Chemistry Insight into the Interactions Between Deep Eutectic Solvents and SO2" Molecules 24, no. 16: 2963. https://doi.org/10.3390/molecules24162963
APA StyleAtilhan, M., Altamash, T., & Aparicio, S. (2019). Quantum Chemistry Insight into the Interactions Between Deep Eutectic Solvents and SO2. Molecules, 24(16), 2963. https://doi.org/10.3390/molecules24162963