
The Role of Charge Transfer in the Formation of Type I Deep Eutectic Solvent-Analogous Ionic Liquid Mixtures

 ,
,  , ,
, ,  , and
, and
Abstract
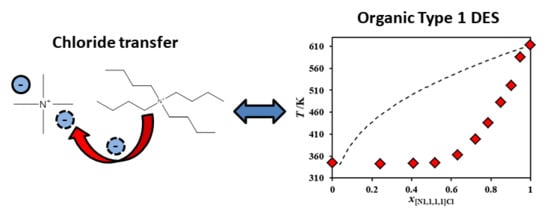
1. Introduction
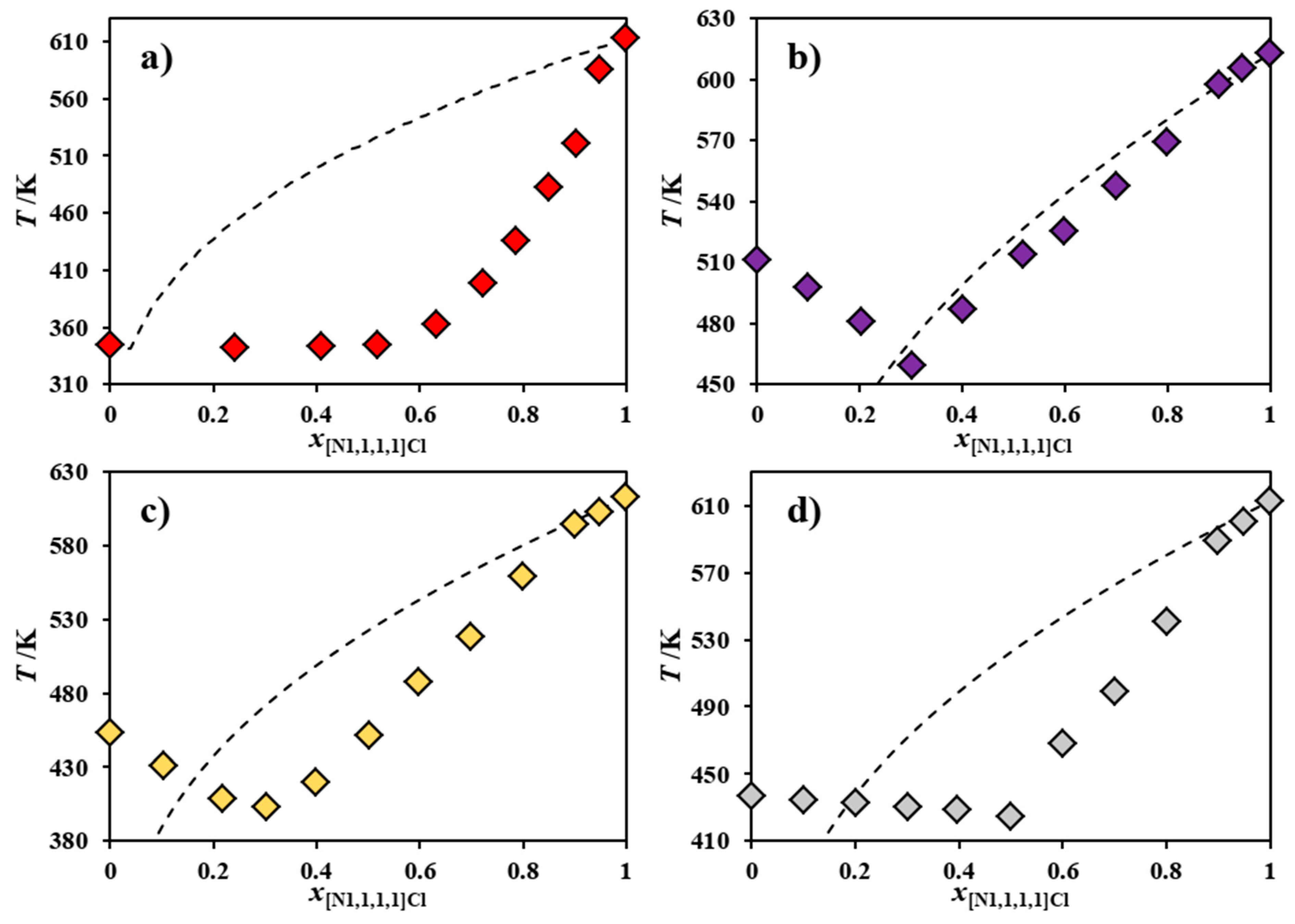
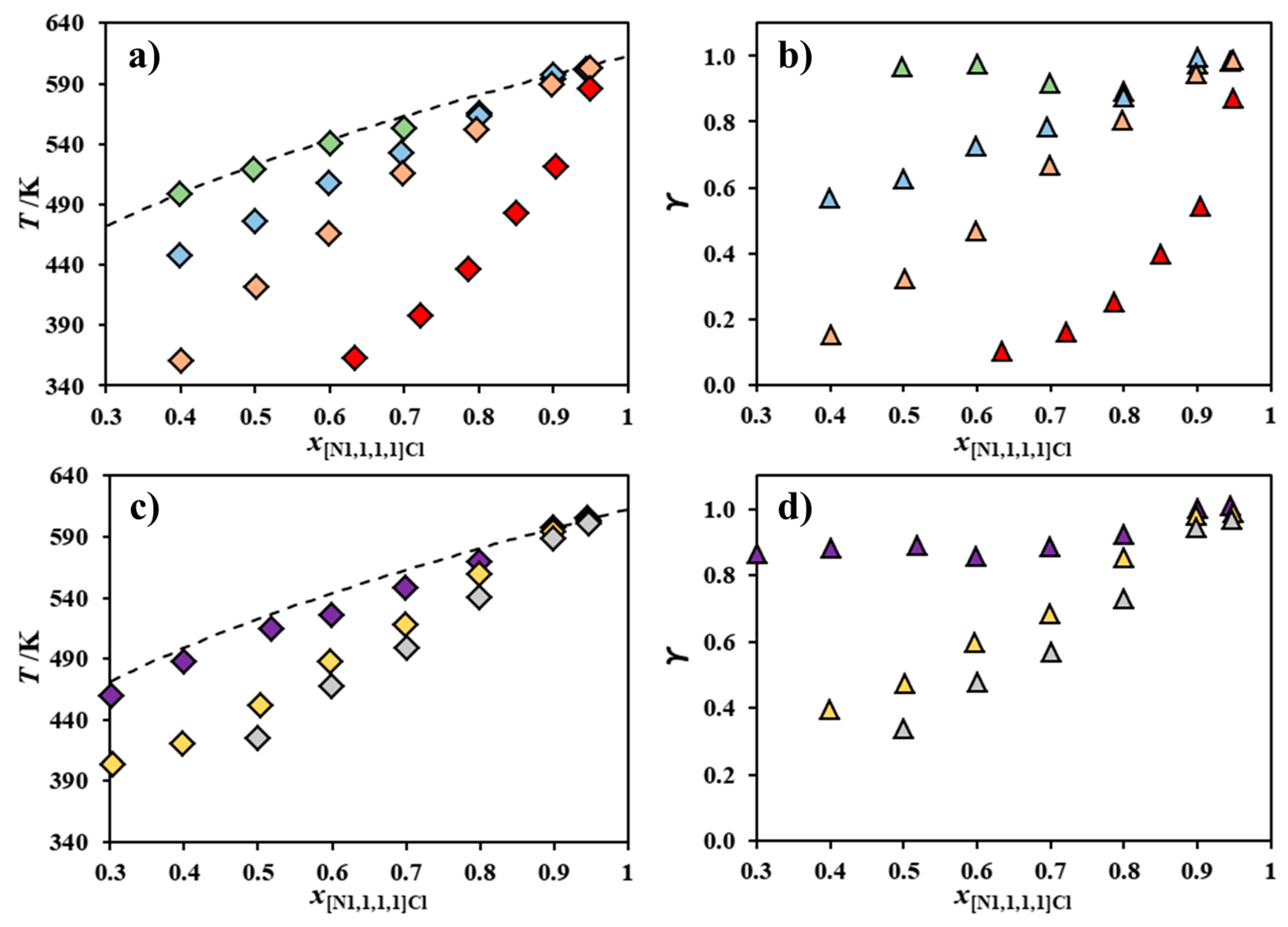
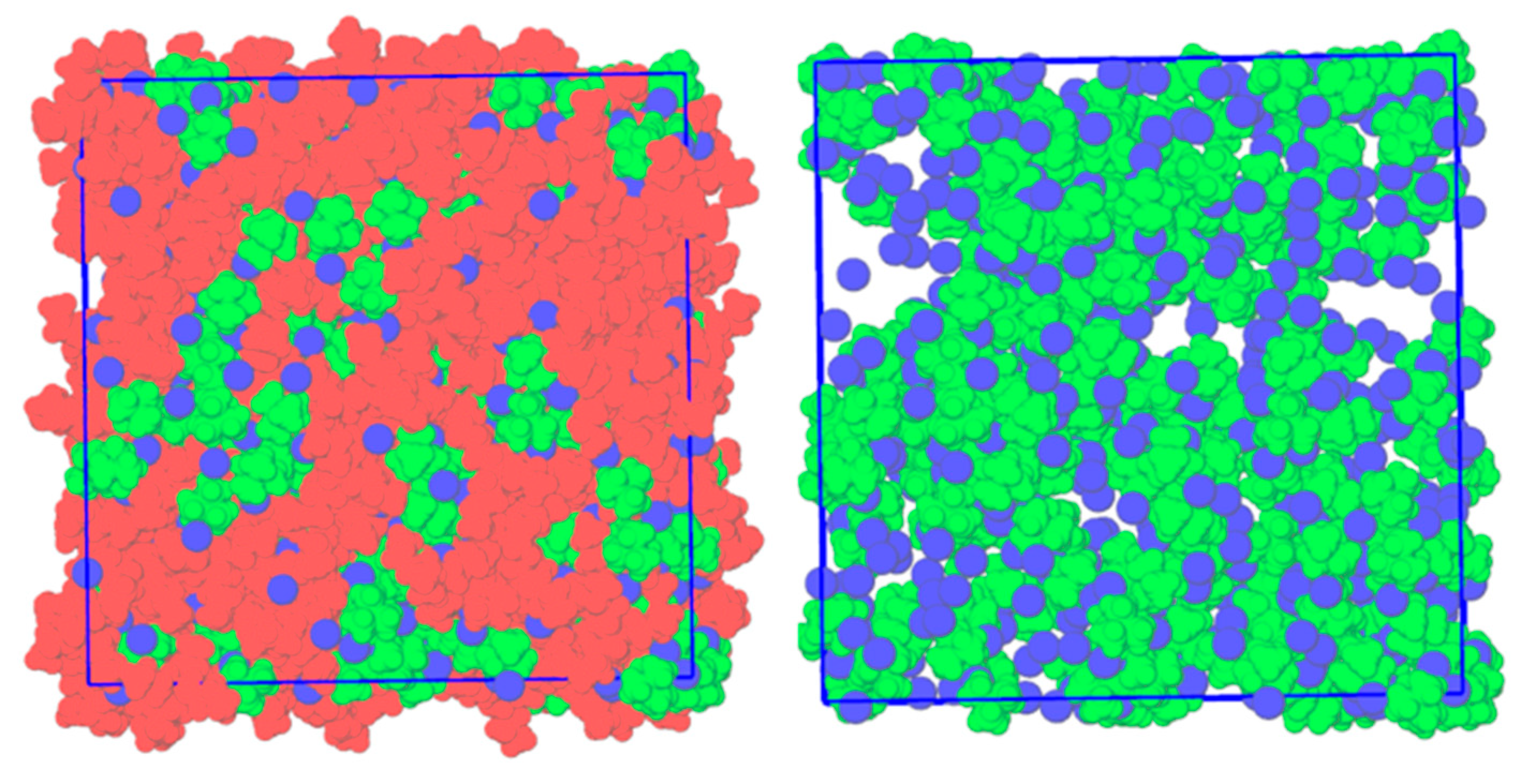
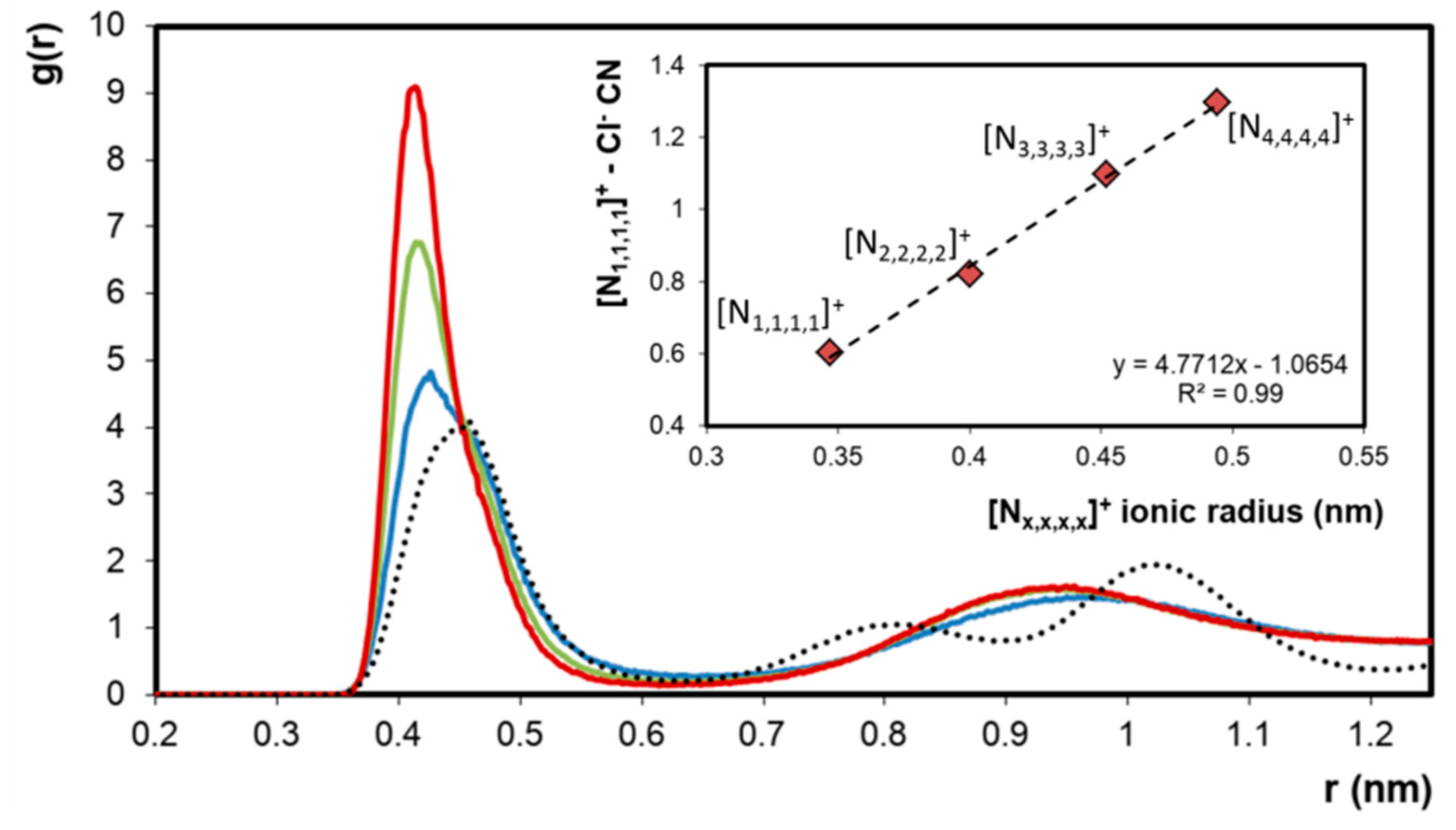
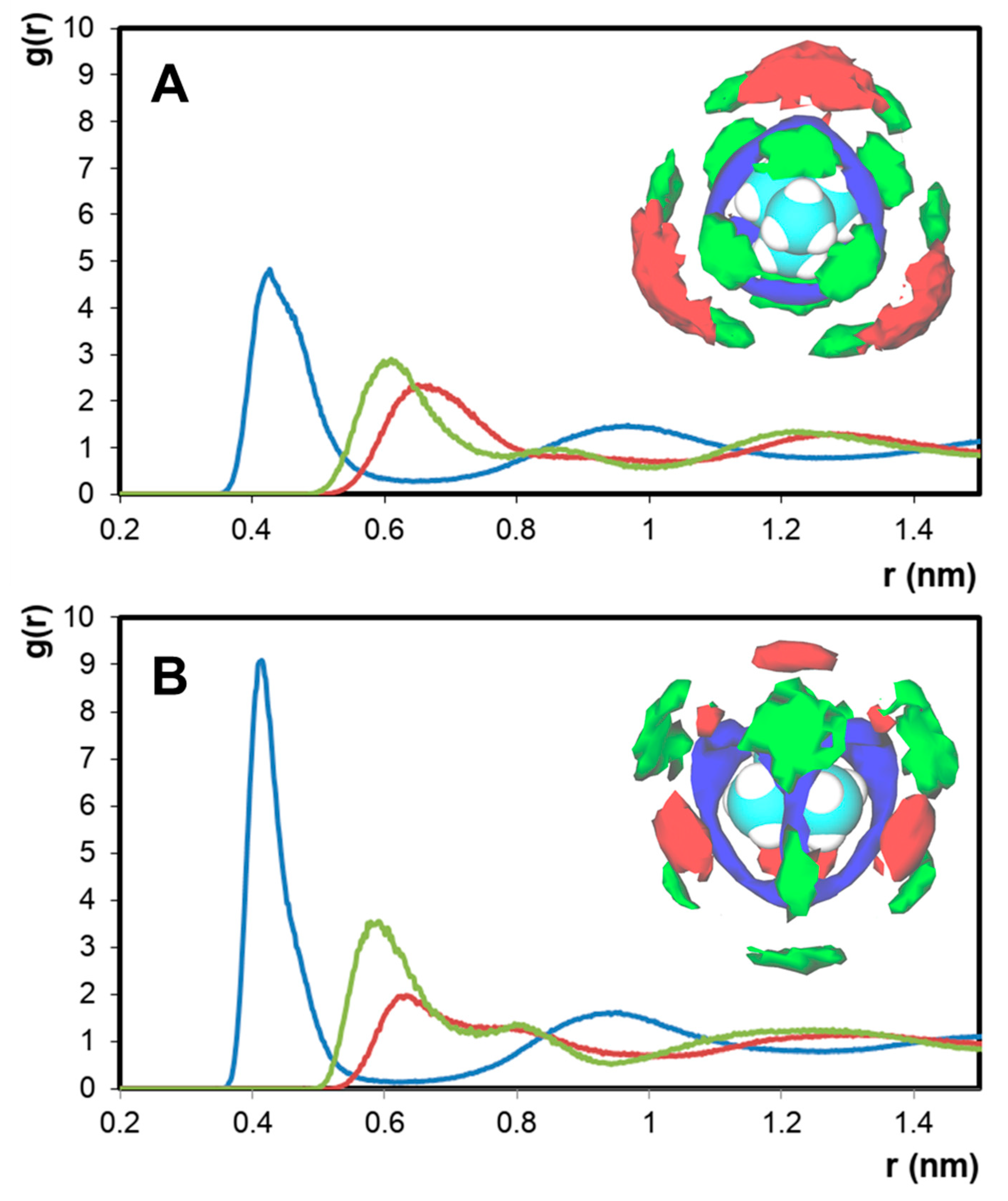
2. Results
3. Materials and Methods
3.1. Experimental Details
3.1.1. Chemicals
3.1.2. Experimental Protocol and Characterization
3.2. Computational Details
3.2.1. Thermodynamic Framework
3.2.2. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hallet, J.P.; Welton, T. Room-Temperature Ionic Liquids: Solvents for Synthesis and Catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, N.; Passos, H.; Billard, I.; Papaiconomou, N.; Coutinho, J.A.P. Recovery of Metals from Waste Electrical and Electronic Equipment (WEEE) Using Unconventional Solvents Based on Ionic Liquid. Crit. Rev. Env. Sci. Technol. 2018, 48, 859–922. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Dong, H.; Zhao, Z.; Zhang, S.; Huang, Y. Carbon capture with ionic liquids: Overview and progress. Energy Environ. Sci. 2012, 5, 6668–6681. [Google Scholar] [CrossRef]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of Cellose with Ionic Liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef] [PubMed]
- Lui, M.Y.; Crowhurst, L.; Hallett, J.P.; Hunt, P.A.; Niedermeyer, H.; Welton, T. Salts dissolved in salts: Ionic liquid mixtures. Chem. Sci. 2011, 2, 1491. [Google Scholar] [CrossRef]
- Niedermeyer, H.; Hallett, J.P.; Villar-Garcia, I.J.; Hunt, P.A.; Welton, T. Mixtures of ionic liquids. Chem. Soc. Rev. 2012, 41, 7780–7802. [Google Scholar] [CrossRef]
- Chatel, G.; Pereira, J.F.B.; Debbeti, V.; Wang, H.; Rogers, R.D. Mixing ionic liquids—“Simple mixtures” or “double salts”? Green Chem. 2014, 16, 2051–2083. [Google Scholar] [CrossRef]
- Maximo, G.J.; Santos, R.J.B.N.; Brandão, P.; Esperança, J.M.S.S.; Costa, M.C.; Meirelles, A.J.A.; Freire, M.G.; Coutinho, J.A.P. Generating Ionic Liquids from Ionic Solids: An Investigation of the Melting Behavior of Binary Mixtures of Ionic Liquids. Cryst. Growth Des. 2014, 14, 4270–4277. [Google Scholar] [CrossRef]
- Stolarska, O.; Soto, A.; Rodríguez, H.; Smiglak, M. Thermal behaviour of mixtures of 1-alkylpyridinium halides with and without a common ion. J. Mol. Liq. 2018, 268, 781–790. [Google Scholar] [CrossRef]
- Teles, A.R.R.; Correia, H.; Maximo, G.J.; Rebelo, L.P.N.; Freire, M.G.; Pereiro, A.B.; Coutinho, J.A.P. Solid–liquid equilibria of binary mixtures of fluorinated ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 25741–25750. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.A.R.; Pinho, S.P.; Coutinho, J.A.P. Insights into the Nature of Eutectic and Deep Eutectic Mixtures. J. Solut. Chem. 2019, 48, 962–982. [Google Scholar] [CrossRef]
- Villar-Garcia, I.J.; Lovelock, K.R.J.; Men, S.; Licence, P. Tuning the electronic environment of cations and anions using ionic liquid mixtures. Chem. Sci. 2014, 5, 2573–2579. [Google Scholar] [CrossRef]
- Clough, M.T.; Crick, C.R.; Gräsvik, J.; Hunt, P.A.; Niedermeyer, H.; Welton, T.; Whitaker, O.P. A physicochemical investigation of ionic liquid mixtures. Chem. Sci. 2015, 6, 1101–1114. [Google Scholar] [CrossRef]
- Matthews, R.P.; Villar-Garcia, I.J.; Weber, C.C.; Griffith, J.; Cameron, F.; Hallett, J.P.; Hunt, P.A.; Welton, T. A structural investigation of ionic liquid mixtures. Phys. Chem. Chem. Phys. 2016, 18, 8608–8624. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.J.; Castiglione, F.; Doherty, C.M.; Dolan, A.; Hill, A.J.; Hunt, P.A.; Matthews, R.P.; Mauri, M.; Mele, A.; Simonutti, R.; et al. Linking the structures, free volumes, and properties of ionic liquid mixtures. Chem. Sci. 2017, 8, 6359–6374. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef]
- Abbott, A.P.; Barron, J.C.; Ryder, K.S.; Wilson, D. Eutectic-Based Ionic Liquids with Metal-Containing Anions and Cations. Chemistry 2007, 13, 6495–6501. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R. Ionic Liquids Based upon Metal Halide/Substituted Quaternary Ammonium Salt Mixtures. Inorg. Chem. 2004, 43, 3447–3452. [Google Scholar] [CrossRef]
- Fannin, A.A., Jr.; Floreani, D.A.; King, L.A.; Landers, J.S.; Piersma, B.J.; Stech, D.J.; Vaughn, R.L.; Wilkes, J.S.; Williams, J.L. Properties of 1,3-dialkylimidazolium chloride-aluminum chloride ionic liquids. 2. Phase transitions, densities, electrical conductivities, and viscosities. J. Phys. Chem. 1984, 88, 2614–2621. [Google Scholar] [CrossRef]
- Abranches, D.O.; Silva, L.P.; Martins, M.A.R.; Fernandez, L.; Pinho, S.P.; Coutinho, J.A.P. Can cholinium chloride form eutectic solvents with organic chloride-based salts? Fluid Phase Equilib. 2019, 493, 120–126. [Google Scholar] [CrossRef]
- Vidal, C.; García-Álvarez, J.; Hernán-Gómez, A.; Kennedy, A.R.; Hevia, E. Introducing Deep Eutectic Solvents to Polar Organometallic Chemistry: Chemoselective Addition of Organolithium and Grignard Reagents to Ketones in Air. Angew. Chem. Int. 2014, 53, 5969–5973. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Deep Eutectic Solvents: The Organic Reaction Medium of the Century. Eur. J. Org. Chem. 2016, 2016, 612–632. [Google Scholar] [CrossRef]
- García-Álvarez, J.; Hevia, E.; Capriati, V. The Future of Polar Organometallic Chemistry Written in Bio-Based Solvents and Water. Chem. Eur. J. 2018, 24, 14854–14863. [Google Scholar] [CrossRef] [PubMed]
- Sitze, M.S.; Schreiter, E.R.; Patterson, E.V.; Freeman, R.G. Ionic Liquids Based on FeCl3 and FeCl2. Raman Scattering and ab Initio Calculations. Inorg. Chem. 2001, 40, 2298–2304. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, C.R.; Matthews, R.P.; Welton, T.; Hunt, P.A. Doubly ionic hydrogen bond interactions within the choline chloride–urea deep eutectic solvent. Phys. Chem. Chem. Phys. 2016, 18, 18145–18160. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, E.R. Phenomenological Theory of Ion Solvation. Effective Radii of Hydrated Ions. J. Phys. Chem. 1959, 63, 1381–1387. [Google Scholar] [CrossRef]
- Hammond, O.S.; Bowron, D.T.; Edler, K.J. Structure and Properties of “Type IV” Lanthanide Nitrate Hydrate: Urea Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2019, 7, 4932–4940. [Google Scholar] [CrossRef]
- Estager, J.; Holbrey, J.D.; Swadźba-Kwaśny, M. Halometallate ionic liquids—Revisited. Chem. Soc. Rev. 2014, 43, 847–886. [Google Scholar] [CrossRef]
- Badyal, Y.S.; Howe, R.A. Structural modification in molten metal chloride and alkali chloride mixtures. J. Phys. 1993, 5, 7189–7202. [Google Scholar] [CrossRef]
- Badyal, Y.S.; Allen, D.A.; Howe, R.A. The structure of liquid AlCl3 and structural modification in AlCl3-MCl (M = Li, Na) molten salt mixtures. J. Phys. 1994, 6, 10193–10220. [Google Scholar] [CrossRef]
- Hammond, O.S.; Bowron, D.T.; Edler, K.J. Liquid structure of the choline chloride-urea deep eutectic solvent (reline) from neutron diffraction and atomistic modelling. Green Chem. 2016, 18, 2736–2744. [Google Scholar] [CrossRef]
- Pontes, P.V.A.; Crespo, E.A.; Martins, M.A.R.; Silva, L.P.; Neves, C.M.S.S.; Maximo, G.J.; Hubinger, M.D.; Batista, E.A.C.; Pinho, S.P.; Coutinho, J.A.P.; et al. Measurement and PC-SAFT modeling of solid-liquid equilibrium of deep eutectic solvents of quaternary ammonium chlorides and carboxylic acids. Fluid Phase Equilib. 2017, 448, 69–80. [Google Scholar] [CrossRef]
- Janz, G.J. Nonaqueous Electrolytes Handbook; Academic Press: New York, NY, USA, 1974; Volume 1. [Google Scholar]
- Moldoveanu, S. Pyrolysis of Organic Molecules: Applications to Health and Environmental Issues, 1st ed.; Elsevier Science & Technology: Oxford, UK, 2009; Volume 28. [Google Scholar]
- Schiraldi, D.; Zaikov, G.E. Chemical and Biochemical Physics: A Systematic Approach to Experiments, Evaluation, and Modeling, 1st ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Prausnitz, J.M.; Lichtenthaler, R.N.; Azevedo, E.G. Molecular Thermodynamics of Fluid-Phase Equilibria, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 1998. [Google Scholar]
- Elliot, J.R.; Lira, C.T. Introductory Chemical Engineering Thermodynamics; Prentice Hall PTR: Upper Saddle River, NJ, USA, 1999. [Google Scholar]
- Coutinho, J.A.P.; Andersen, S.I.; Stenby, E.H. Evaluation of activity coefficient models in prediction of alkane solid-liquid equilibria. Fluid Phase Equilib. 1995, 103, 23–39. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Hockney, R.; Goel, S.; Eastwood, J. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Deschamps, J.; Pádua, A.A.H. Modeling Ionic Liquids Using a Systematic All-Atom Force Field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids III: Imidazolium, Pyridinium, and Phosphonium Cations; Chloride, Bromide, and Dicyanamide Anions. J. Phys. Chem. B 2006, 110, 19586–19592. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose—Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 (revision D.01); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Perkins, S.L.; Painter, P.; Colina, C.M. Experimental and Computational Studies of Choline Chloride-Based Deep Eutectic Solvents. J. Chem. Eng. Data 2014, 59, 3652–3662. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Brehm, M.; Kirchner, B. TRAVIS—A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef]
- Brehm, M.; Weber, H.; Thomas, M.; Holloczki, O.; Kirchner, B. Domain Analysis in Nanostructured Liquids: A Post-Molecular Dynamics Study at the Example of Ionic Liquids. ChemPhysChem 2015, 16, 3271–3277. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are no longer available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | T (K) | [Nx,x,x,x]+ Ionic Radii (nm) [27] | Domain Analysis | ||
|---|---|---|---|---|---|
| [N1,1,1,1]+ | [N1,1,1,1]+ + Cl− | [Nx,x,x,x]+ | |||
| [N1,1,1,1]Cl + [N2,2,2,2]Cl | 533.15 | 0.400 | 1.7 | 15.3 | 1.0 |
| [N1,1,1,1]Cl + [N3,3,3,3]Cl | 423.15 | 0.452 | 6.0 | 30.2 | 1.0 |
| [N1,1,1,1]Cl + [N4,4,4,4]Cl | 353.15 | 0.494 | 5.3 | 48.5 | 1.0 |
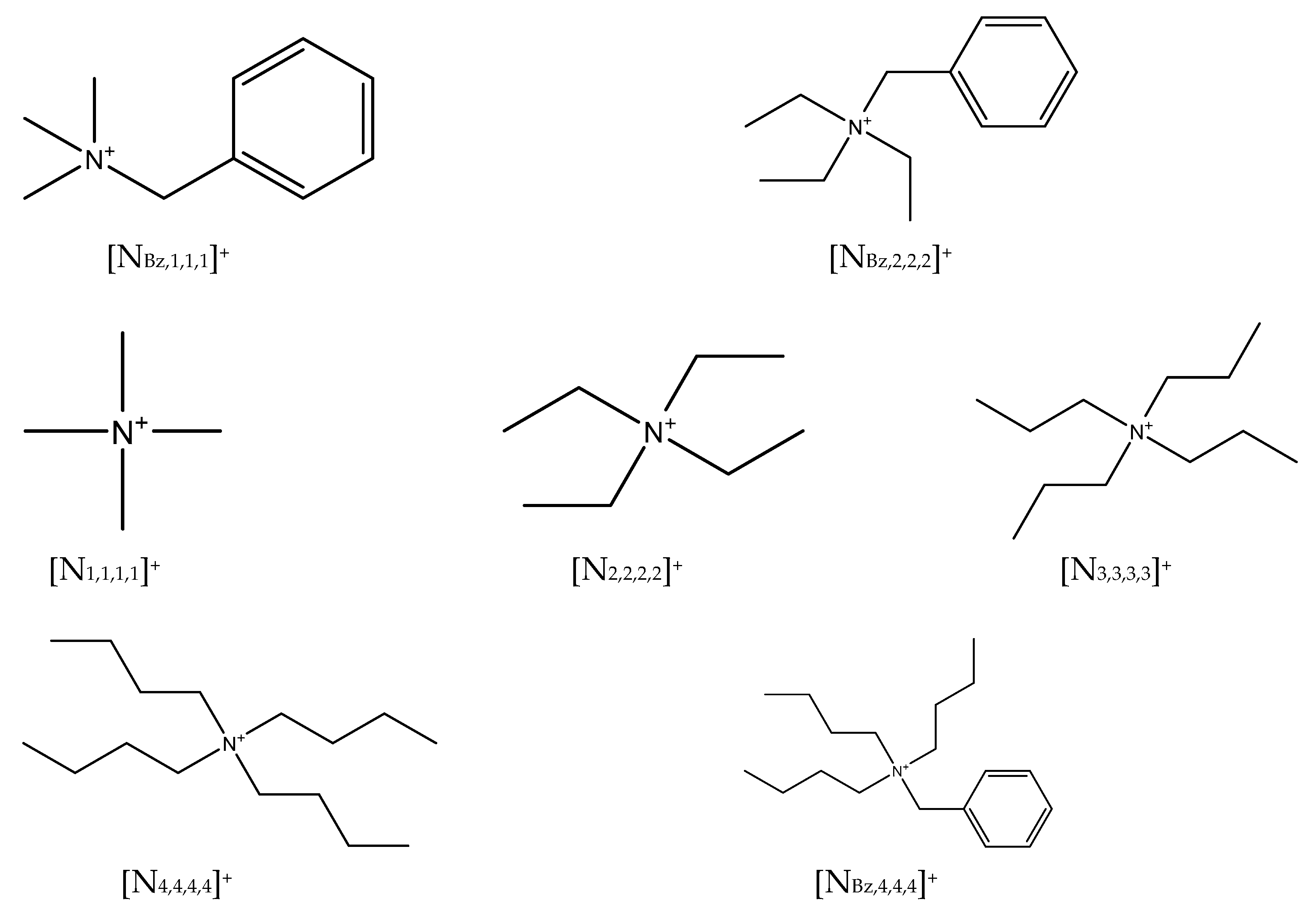
| Substance | CAS Number | Supplier | Purity/wt.% | Tm/K | Tdeg (onset/peak)/K | [H2O]/wt.% | ΔmH/kJ·mol−1 |
|---|---|---|---|---|---|---|---|
| [N1,1,1,1]Cl | 75-57-0 | Sigma-Aldrich | 97 | 612.9 [22] | 602.3/627.5 | 0.30 | 20.49 c |
| [N4,4,4,4]Cl | 1112-67-0 | Sigma-Aldrich | 97 | 344.0 a | 453.7/484.1 | 0.58 | 14.69 a |
| [NBz,1,1,1]Cl | 56-93-9 | Acros Organics | 98 | 511.0 b | 512.1/519.1 | 1.26 | _____ |
| [NBz,2,2,2]Cl | 56-37-1 | Acros Organics | 98 | 453.6 b | 464.4/475.1 | 0.68 | _____ |
| [NBz,4,4,4]Cl | 23616-79-7 | Acros Organics | 98 | 437.0 b | 441.6/452.2 | 0.32 | _____ |
| [N2,2,2,2]Br | 71-91-0 | Alfa Aesar | 98 | 568.3 b | 532.7/571.3 | 0.21 | _____ |
| [N3,3,3,3]Br | 1941-30-6 | Sigma-Aldrich | 98 | 535.0 b | 547.3/557.2 | 0.34 | _____ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abranches, D.O.; Schaeffer, N.; Silva, L.P.; Martins, M.A.R.; Pinho, S.P.; Coutinho, J.A.P. The Role of Charge Transfer in the Formation of Type I Deep Eutectic Solvent-Analogous Ionic Liquid Mixtures. Molecules 2019, 24, 3687. https://doi.org/10.3390/molecules24203687
Abranches DO, Schaeffer N, Silva LP, Martins MAR, Pinho SP, Coutinho JAP. The Role of Charge Transfer in the Formation of Type I Deep Eutectic Solvent-Analogous Ionic Liquid Mixtures. Molecules. 2019; 24(20):3687. https://doi.org/10.3390/molecules24203687
Chicago/Turabian StyleAbranches, Dinis O., Nicolas Schaeffer, Liliana P. Silva, Mónia A. R. Martins, Simão P. Pinho, and João A. P. Coutinho. 2019. "The Role of Charge Transfer in the Formation of Type I Deep Eutectic Solvent-Analogous Ionic Liquid Mixtures" Molecules 24, no. 20: 3687. https://doi.org/10.3390/molecules24203687
APA StyleAbranches, D. O., Schaeffer, N., Silva, L. P., Martins, M. A. R., Pinho, S. P., & Coutinho, J. A. P. (2019). The Role of Charge Transfer in the Formation of Type I Deep Eutectic Solvent-Analogous Ionic Liquid Mixtures. Molecules, 24(20), 3687. https://doi.org/10.3390/molecules24203687