
Fusion of Lysostaphin to an Albumin Binding Domain Prolongs Its Half-Life and Bactericidal Activity in the Systemic Circulation

 , ,
, ,
Abstract
1. Introduction
2. Results
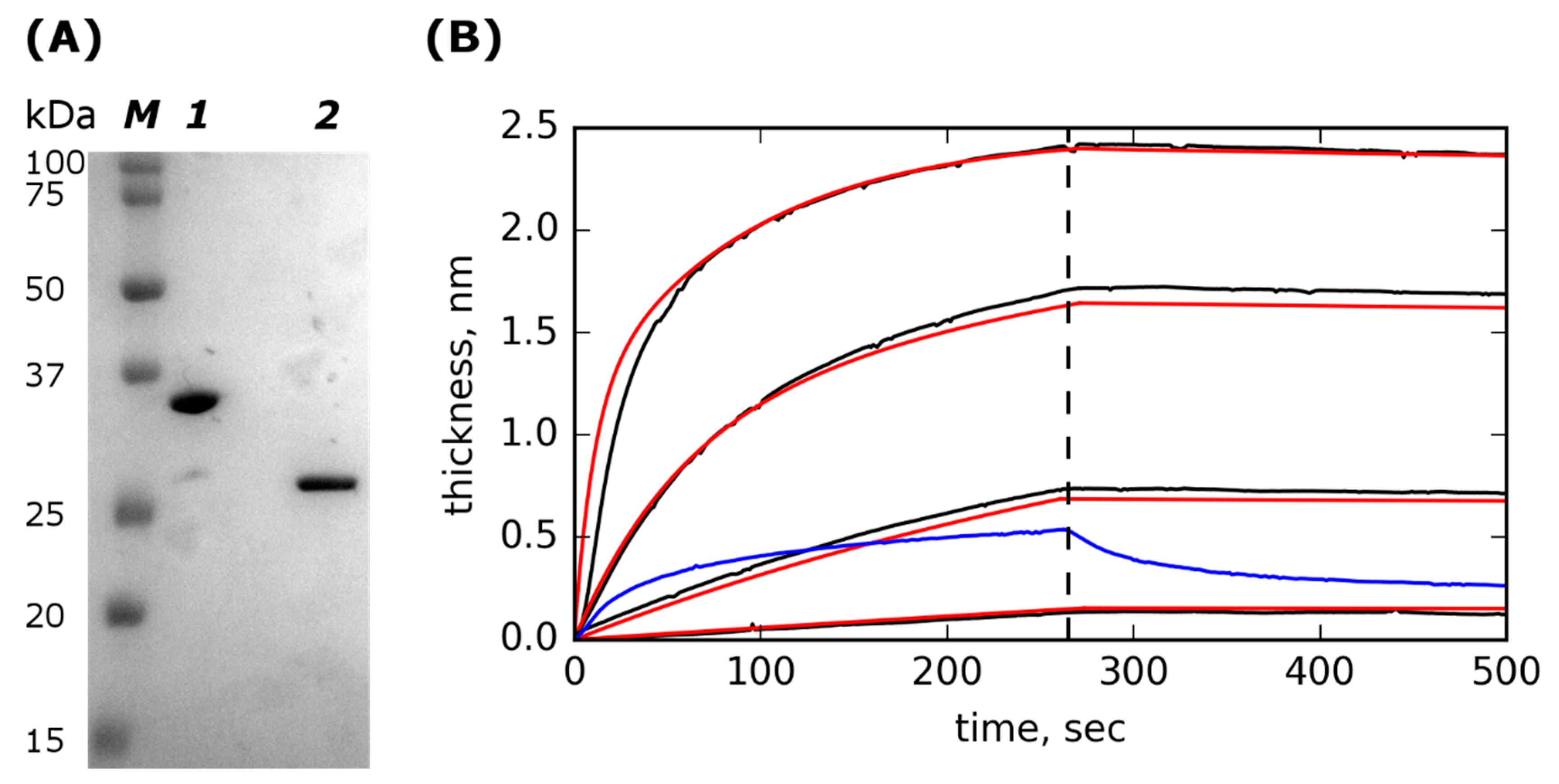
2.1. Lst-ABD Construction and Purification
2.2. Lst-ABD Binds Rat Serum Albumin with Nanomolar Affinity
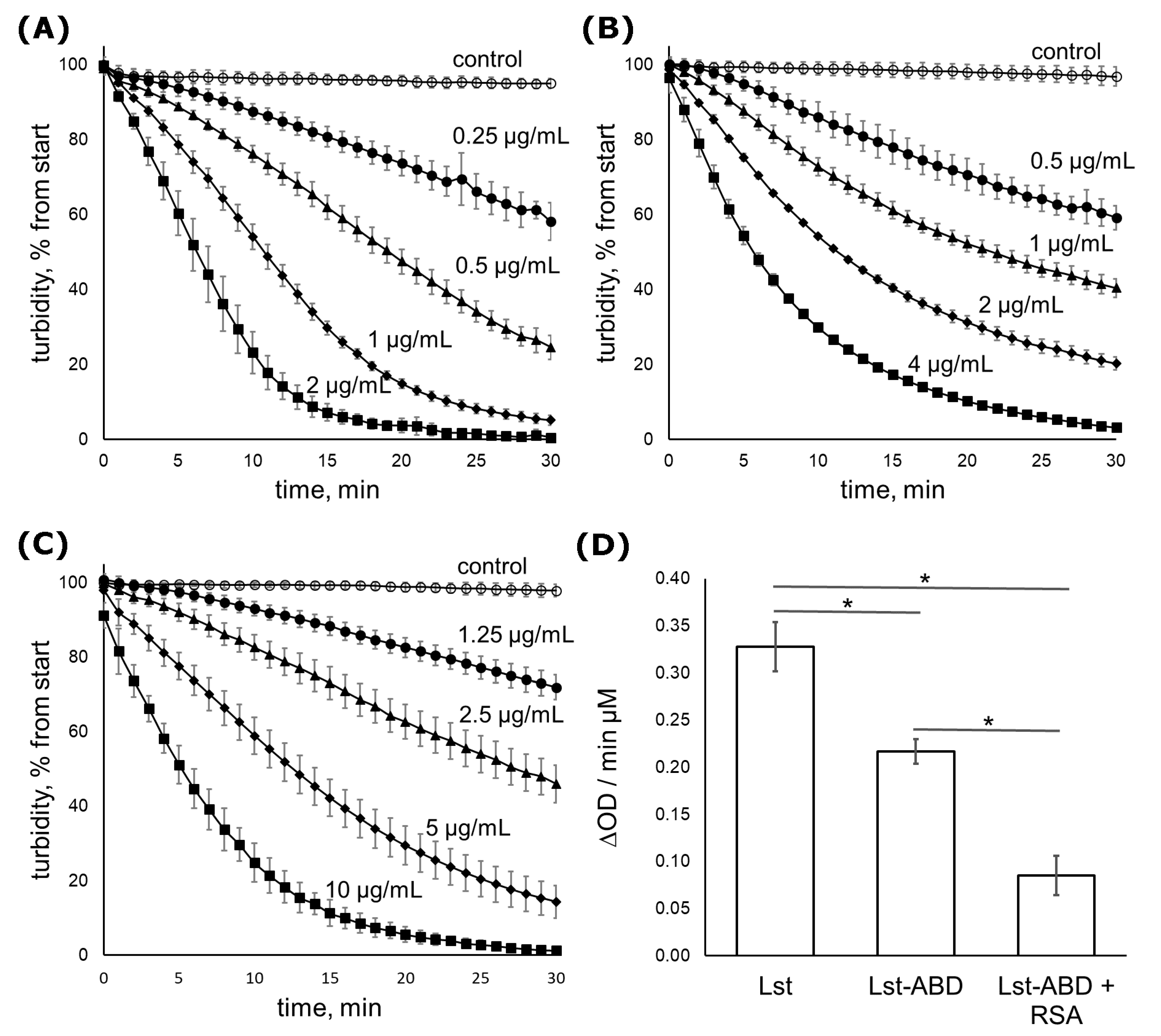
2.3. Lst-ABD Kills Staphylococci Both in the Absence and in the Presence of Albumin
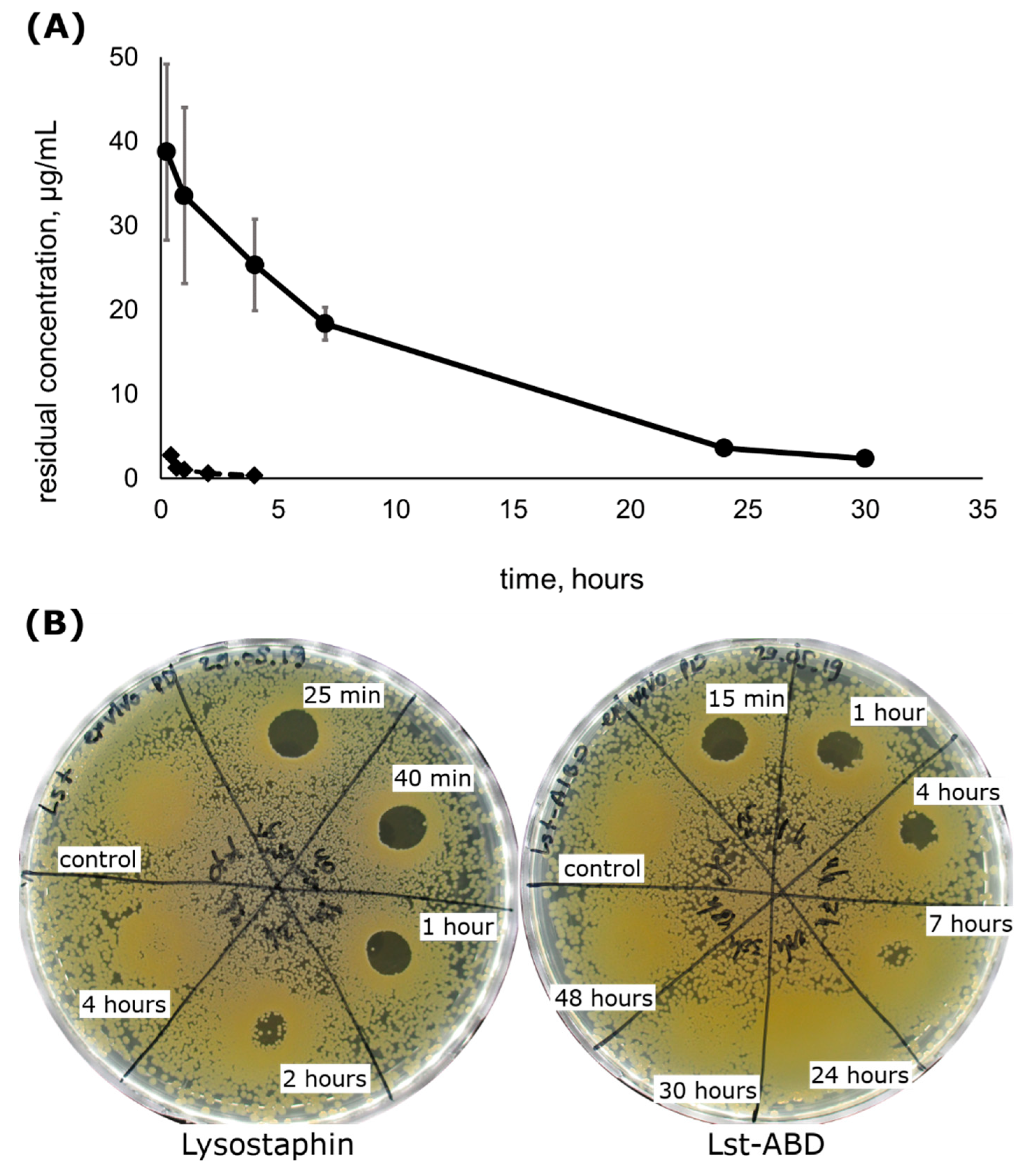
2.4. Lst-ABD Has an Improved Pharmacokinetic Profile
2.5. Lst-ABD Has Improved Pharmacodynamics Ex Vivo
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification
4.2. Binding to Albumin
4.3. Antibacterial Activity
4.4. Pharmacokinetics
4.5. Ex Vivo Pharmacodynamics
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed]
- Pastagia, M.; Schuch, R.; Fischetti, V.A.; Huang, D.B. Lysins: The arrival of pathogen-directed anti-infectives. J. Med. Microbiol. 2013, 62, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Schmelcher, M.; Rodriguez-Rubio, L.; Klumpp, J.; Pritchard, D.G.; Dong, S.; Donovan, D.M. Endolysins as Antimicrobials. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2012; Volume 83, pp. 299–365. ISBN 9780123944382. [Google Scholar]
- São-José, C. Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials. Antibiotics 2018, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Haddad Kashani, H.; Schmelcher, M.; Sabzalipoor, H.; Seyed Hosseini, E.; Moniri, R. Recombinant Endolysins as Potential Therapeutics against Antibiotic-Resistant Staphylococcus aureus: Current Status of Research and Novel Delivery Strategies. Clin. Microbiol. Rev. 2017, 31, e00071-17. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.Y.; Jang, I.J.; Yoon, S.; Jang, K.; Yu, K.S.; Cho, J.Y.; Seong, M.W.; Jung, G.M.; Yoon, S.J.; Kang, S.H. Pharmacokinetics and Tolerance of the Phage Endolysin-Based Candidate Drug SAL200 after a Single Intravenous Administration among Healthy Volunteers. Antimicrob. Agents Chemother. 2017, 61, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cassino, C.; Murphy, M.G.; Boyle, J.; Rotolo, J.; Wittekind, M. Results of the First In Human Study of Lysin CF-301 Evaluating the Safety, Tolerability and Pharmacokinetic Profile in Healthy Volunteers. In Proceedings of the 26th European Congress of Clinical Microbiology and Infectious Diseases, Amsterdam, The Netherlands, 9–12 April 2016. [Google Scholar]
- Wittekind, M.; Schuch, R. Cell wall hydrolases and antibiotics: Exploiting synergy to create efficacious new antimicrobial treatments. Curr. Opin. Microbiol. 2016, 33, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Desbois, A.P.; Coote, P.J. Bactericidal synergy of lysostaphin in combination with antimicrobial peptides. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 1015–1021. [Google Scholar] [CrossRef]
- Sharma, U.; Vipra, A.; Channabasappa, S. Phage-derived lysins as potential agents for eradicating biofilms and persisters. Drug Discov. Today 2018, 23, 848–856. [Google Scholar] [CrossRef]
- Gerstmans, H.; Criel, B.; Briers, Y. Synthetic biology of modular endolysins. Biotechnol. Adv. 2018, 36, 624–640. [Google Scholar] [CrossRef]
- Kokai-Kun, J.F. Lysostaphin: A silver bullet for staph. In Antimicrobial Drug Discovery: Emerging Strategies; CAB International: Wallingford, UK, 2012; pp. 147–165. ISBN 978-1-84593-943-4. [Google Scholar]
- Gajdács, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar] [CrossRef]
- Kusuma, C.M.; Kokai-Kun, J.F. Comparison of Four Methods for Determining Lysostaphin Susceptibility of Various Strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 3256–3263. [Google Scholar] [CrossRef] [PubMed]
- Oluola, O.; Kong, L.; Fein, M.; Weisman, L.E. Lysostaphin in treatment of neonatal Staphylococcus aureus infection. Antimicrob. Agents Chemother. 2007, 51, 2198–2200. [Google Scholar] [CrossRef]
- Kokai-Kun, J.F.; Chanturiya, T.; Mond, J.J. Lysostaphin as a treatment for systemic Staphylococcus aureus infection in a mouse model. J. Antimicrob. Chemother. 2007, 60, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Placencia, F.X.; Kong, L.; Weisman, L.E. Treatment of methicillin-resistant Staphylococcus aureus in neonatal mice: Lysostaphin versus vancomycin. Pediatr. Res. 2009, 65, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Kokai-Kun, J.F.; Chanturiya, T.; Mond, J.J. Lysostaphin eradicates established Staphylococcus aureus biofilms in jugular vein catheterized mice. J. Antimicrob. Chemother. 2009, 64, 94–100. [Google Scholar] [CrossRef]
- Shah, A.; Mond, J.; Walsh, S. Lysostaphin-coated catheters eradicate Staphylococcus aureus challenge and block surface colonization. Antimicrob. Agents Chemother. 2004, 48, 2704–2707. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fan, H.; Huang, Y.; Peng, F.; Fan, H.; Yuan, S.; Tong, Y. Recombinant Lysostaphin Protects Mice from Methicillin-Resistant Staphylococcus Aureus Pneumonia. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Dajcs, J.J.; Hume, E.B.H.; Moreau, J.M.; Caballero, A.R.; Cannon, B.M.; O’Callaghan, R.J. Lysostaphin treatment of methicillin-resistant Staphylococcus aureus keratitis in the rabbit. Invest. Ophthalmol. Vis. Sci. 2000, 41, 1432–1437. [Google Scholar]
- Dajcs, J.J.; Thibodeaux, B.A.; Hume, E.B.; Zheng, X.; Sloop, G.D.; O’Callaghan, R.J. Lysostaphin is effective in treating methicillin-resistant Staphylococcus aureus endophthalmitis in the rabbit. Curr. Eye Res. 2001, 22, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Kokai-Kun, J.F.; Walsh, S.M.; Chanturiya, T.; Mond, J.J. Lysostaphin Cream Eradicates Staphylococcus aureus Nasal Colonization in a Cotton Rat Model. Antimicrob. Agents Chemother. 2003, 47, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Shah, A.; Mond, J. Improved Pharmacokinetics and Reduced Antibody Reactivity of Lysostaphin Conjugated to Polyethylene Glycol. Antimicrob. Agents Chemother. 2003, 47, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.V.; Lavrova, N.V.; Lyashchuk, A.M.; Strukova, N.V.; Generalova, M.S.; Ryazanova, A.V.; Shestak, N.V.; Boksha, I.S.; Polyakov, N.B.; Galushkina, Z.M.; et al. The Influence of Dimerization on the Pharmacokinetics and Activity of an Antibacterial Enzyme Lysostaphin. Molecules 2019, 24, 1879. [Google Scholar] [CrossRef] [PubMed]
- Swierczewska, M.; Lee, K.C.; Lee, S. What is the future of PEGylated therapies? Expert Opin. Emerg. Drugs 2015, 20, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Resch, G.; Moreillon, P.; Fischetti, V.A. PEGylating a bacteriophage endolysin inhibits its bactericidal activity. AMB Express 2011, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.; Dong, D.; Li, Z.; Li, Z. Recombinant human serum albumin fusion proteins and novel applications in drug delivery and therapy. Curr. Pharm. Des. 2015, 21, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Frejd, F. Half-Life Extension by Binding to Albumin through an Albumin Binding Domain. In Therapeutic Proteins: Strategies to Modulate Their Plasma Half-Lives; Wiley-VCH: Weinheim, Germany, 2012; pp. 269–283. ISBN 9783527644827. [Google Scholar]
- Steiner, D.; Merz, F.W.; Sonderegger, I.; Gulotti-Georgieva, M.; Villemagne, D.; Phillips, D.J.; Forrer, P.; Stumpp, M.T.; Zitt, C.; Binz, H.K. Half-life extension using serum albumin-binding DARPin® domains. Protein Eng. Des. Sel. 2017, 30, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.S.; Zhang, M.; Gloria Meng, Y.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, Y.; Chen, Y.; Sang, Y.; Zhou, T.; Qiu, M.; Huang, X.; Zhou, C.; Su, Z. A protease-based strategy for the controlled release of therapeutic peptides. Angew. Chemie Int. Ed. 2010, 49, 4930–4933. [Google Scholar] [CrossRef]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008, 21, 283–288. [Google Scholar] [CrossRef]
- Müller, M.R.; Saunders, K.; Grace, C.; Jin, M.; Piche-Nicholas, N.; Steven, J.; O’Dwyer, R.; Wu, L.; Khetemenee, L.; Vugmeyster, Y.; et al. Improving the pharmacokinetic properties of biologics by fusion to an anti-HSA shark VNAR domain. MAbs 2012, 4, 673–685. [Google Scholar] [CrossRef]
- Van Roy, M.; Ververken, C.; Beirnaert, E.; Hoefman, S.; Kolkman, J.; Vierboom, M.; Breedveld, E.; ’t Hart, B.; Poelmans, S.; Bontinck, L.; et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody® ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Herne, N. Method for Reducing the Immune Response to a Biologically Active Protein. U.S. Patent Application No. 8642743 B2, 4 February 2014. [Google Scholar]
- Johansson, M.U.; Frick, I.M.; Nilsson, H.; Kraulis, P.J.; Hober, S.; Jonasson, P.; Linhult, M.; Nygren, P.Å.; Uhlén, M.; Björck, L.; et al. Structure, specificity, and mode of interaction for bacterial albumin-binding modules. J. Biol. Chem. 2002, 277, 8114–8120. [Google Scholar] [CrossRef] [PubMed]
- Nygren, P.A.; Ljungquist, C.; Tromborg, H.; Nustad, K.; Uhlen, M. Species-dependent binding of serum albumins to the streptococcal receptor protein G. Eur. J. Biochem. 1990, 193, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.; Dogan, J.; Herne, N.; Abrahmsén, L.; Nygren, P.A. Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng. Des. Sel. 2008, 21, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Konopsky, V.N.; Karakouz, T.; Alieva, E.V.; Vicario, C.; Sekatskii, S.K.; Dietler, G. Photonic crystal biosensor based on optical surface waves. Sensors 2013, 13, 2566–2578. [Google Scholar] [CrossRef] [PubMed]
- Seijsing, J.; Sobieraj, A.M.; Keller, N.; Shen, Y.; Zinkernagel, A.S.; Loessner, M.J.; Schmelcher, M. Improved Biodistribution and Extended Serum Half-Life of a Bacteriophage Endolysin by Albumin Binding Domain Fusion. Front. Microbiol. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Boksha, I.S.; Lavrova, N.V.; Grishin, A.V.; Demidenko, A.V.; Lyashchuk, A.M.; Galushkina, Z.M.; Ovchinnikov, R.S.; Umyarov, A.M.; Avetisian, L.R.; Chernukha, M.I.; et al. Staphylococcus simulans recombinant lysostaphin: Production, purification, and determination of antistaphylococcal activity. Biochemistry 2016, 81, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Meth. 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Lavigne, R.; Volckaert, G.; Hertveldt, K. A standardized approach for accurate quantification of murein hydrolase activity in high-throughput assays. J. Biochem. Biophys. Methods 2007, 70, 531–533. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Weiner, D. Non-compartmental Analysis. In Computational Toxicology: Volume I, Methods in Molecular Biology; Humana Press: New York, NY, USA, 2012; Volume 929, pp. 377–389. ISBN 9781627030502. [Google Scholar]
Sample Availability: Samples of the pL475 plasmid are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Parameter | Mean ± Stdev |
|---|---|
| koff | 5.9 × 10−5 ± 2.0 × 10−5 s−1 |
| kon1 | 2.6 × 104 ± 8.4 × 102 M−1 s−1 |
| kon2 | 3.0 × 103 ± 4.3 × 101 M−1 s−1 |
| KD1 | 2.3 ± 0.8 nM |
| KD2 | 20.0 ± 6.6 nM |
| Strain | MSSA/MRSA | MIC, µg/mL | MBC, µg/mL | ||||
|---|---|---|---|---|---|---|---|
| Lysostaphin | Lst-ABD | Lst-ABD + RSA | Lysostaphin | Lst-ABD | Lst-ABD + RSA | ||
| ATCC 29213 | MSSA | 0.11 | 0.8 | 1.6 | 0.11 | 0.8 | 1.6 |
| Z 715-18 | MSSA | 0.4 | >12.8 | >12.8 | 0.4 | >12.8 | >12.8 |
| Z 76-19 | MSSA | 0.05 | 3.2 | 3.2 | 0.05 | 3.2 | 3.2 |
| Z 88-19 | MSSA | 0.05 | 0.8 | 1.6 | 0.05 | 0.8 | 1.6 |
| F 832-14 | MRSA | 0.1 | 3.2 | 3.2 | 0.1 | 3.2 | 3.2 |
| R 81-19 | MSSA | 0.05 | 3.2 | 3.2 | 0.05 | 3.2 | 3.2 |
| In 0102-19 | MSSA | 0.025 | 1.6 | 1.6 | 0.05 | 1.6 | 3.2 |
| R 116-14 | MRSA | 0.05 | 1.6 | 3.2 | 0.05 | 3.2 | 3.2 |
| Z 73-14 | MSSA | 0.05 | 3.2 | 6.4 | 0.1 | 3.2 | 6.4 |
| 301 | MRSA | 0.05 | 1.6 | 1.6 | 0.05 | 1.6 | 1.6 |
| 629 | MRSA | 0.05 | 1.6 | 3.2 | 0.05 | 1.6 | 3.2 |
| 247G | MRSA | 0.4 | 12.8 | 12.8 | 0.4 | 12.8 | 12.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grishin, A.V.; Shestak, N.V.; Lavrova, N.V.; Lyashchuk, A.M.; Popova, L.I.; Strukova, N.V.; Generalova, M.S.; Ryazanova, A.V.; Polyakov, N.B.; Galushkina, Z.M.; et al. Fusion of Lysostaphin to an Albumin Binding Domain Prolongs Its Half-Life and Bactericidal Activity in the Systemic Circulation. Molecules 2019, 24, 2892. https://doi.org/10.3390/molecules24162892
Grishin AV, Shestak NV, Lavrova NV, Lyashchuk AM, Popova LI, Strukova NV, Generalova MS, Ryazanova AV, Polyakov NB, Galushkina ZM, et al. Fusion of Lysostaphin to an Albumin Binding Domain Prolongs Its Half-Life and Bactericidal Activity in the Systemic Circulation. Molecules. 2019; 24(16):2892. https://doi.org/10.3390/molecules24162892
Chicago/Turabian StyleGrishin, Alexander V., Nikita V. Shestak, Natalia V. Lavrova, Alexander M. Lyashchuk, Liubov I. Popova, Natalia V. Strukova, Maria S. Generalova, Anna V. Ryazanova, Nikita B. Polyakov, Zoya M. Galushkina, and et al. 2019. "Fusion of Lysostaphin to an Albumin Binding Domain Prolongs Its Half-Life and Bactericidal Activity in the Systemic Circulation" Molecules 24, no. 16: 2892. https://doi.org/10.3390/molecules24162892
APA StyleGrishin, A. V., Shestak, N. V., Lavrova, N. V., Lyashchuk, A. M., Popova, L. I., Strukova, N. V., Generalova, M. S., Ryazanova, A. V., Polyakov, N. B., Galushkina, Z. M., Soboleva, L. A., Boksha, I. S., Karyagina, A. S., & Lunin, V. G. (2019). Fusion of Lysostaphin to an Albumin Binding Domain Prolongs Its Half-Life and Bactericidal Activity in the Systemic Circulation. Molecules, 24(16), 2892. https://doi.org/10.3390/molecules24162892