
Proteasome Activation to Combat Proteotoxicity

Abstract
1. Introduction
2. Intrinsically Disordered Proteins
2.1. Defining “Disorder”
- Any functional protein or protein domain possessed of a unique 3D structure described by minimal fluctuation around their equilibrium Ramachandran angles is termed a “structured protein”.
- Any functional protein or protein domain that exists as a dynamic ensemble lacking specific equilibrium Ramachandran angles with backbone atomic positions that naturally undertake non-cooperative conformational changes is termed an “intrinsically disordered protein or region”.
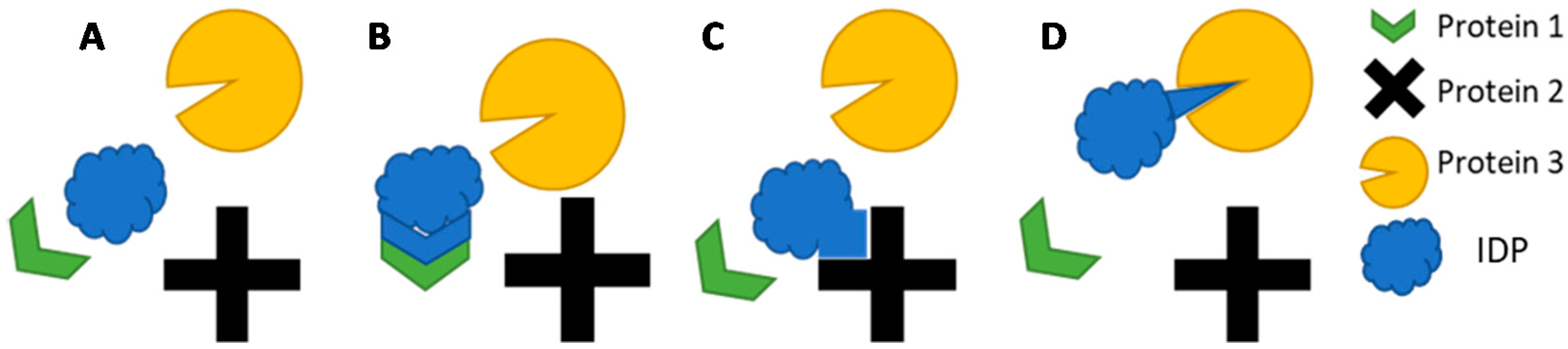
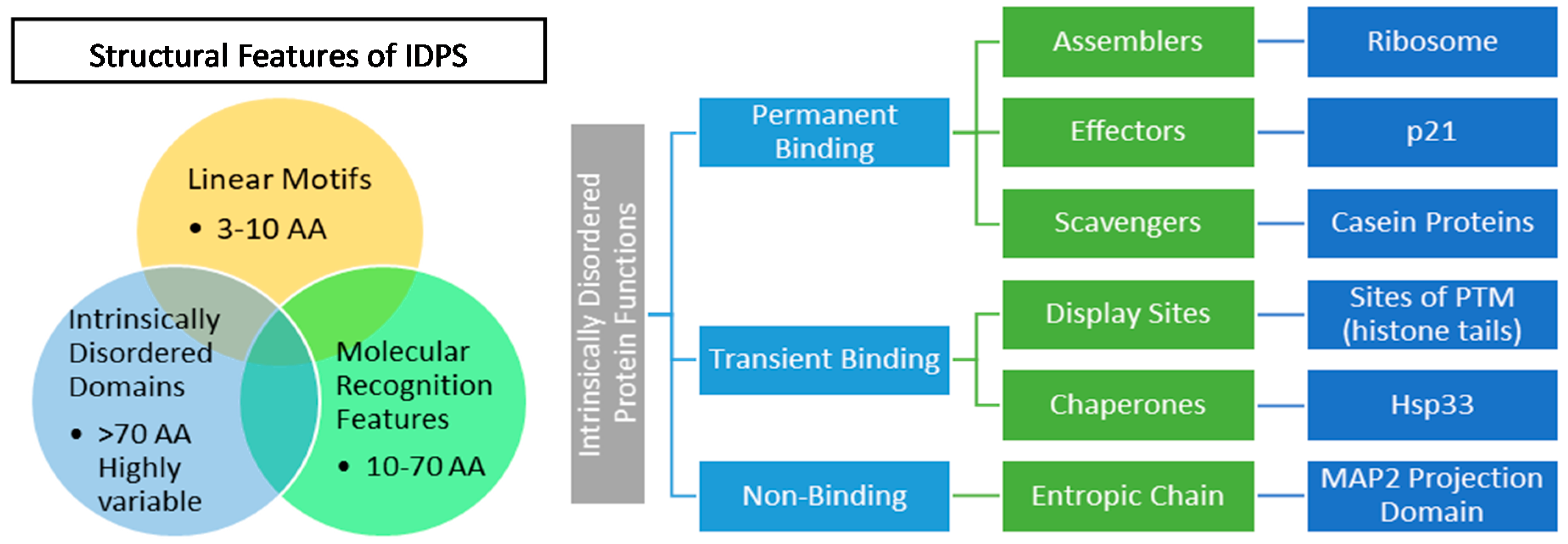
2.2. IDP Structure and Function
2.3. IDPs and Neurodegeneration
2.3.1. Neurodegeneration
2.3.2. α-synuclein
2.3.3. Amyloid β (Aβ) and Tau
2.3.4. Prion Proteins
2.3.5. Polyglutamine Repeats
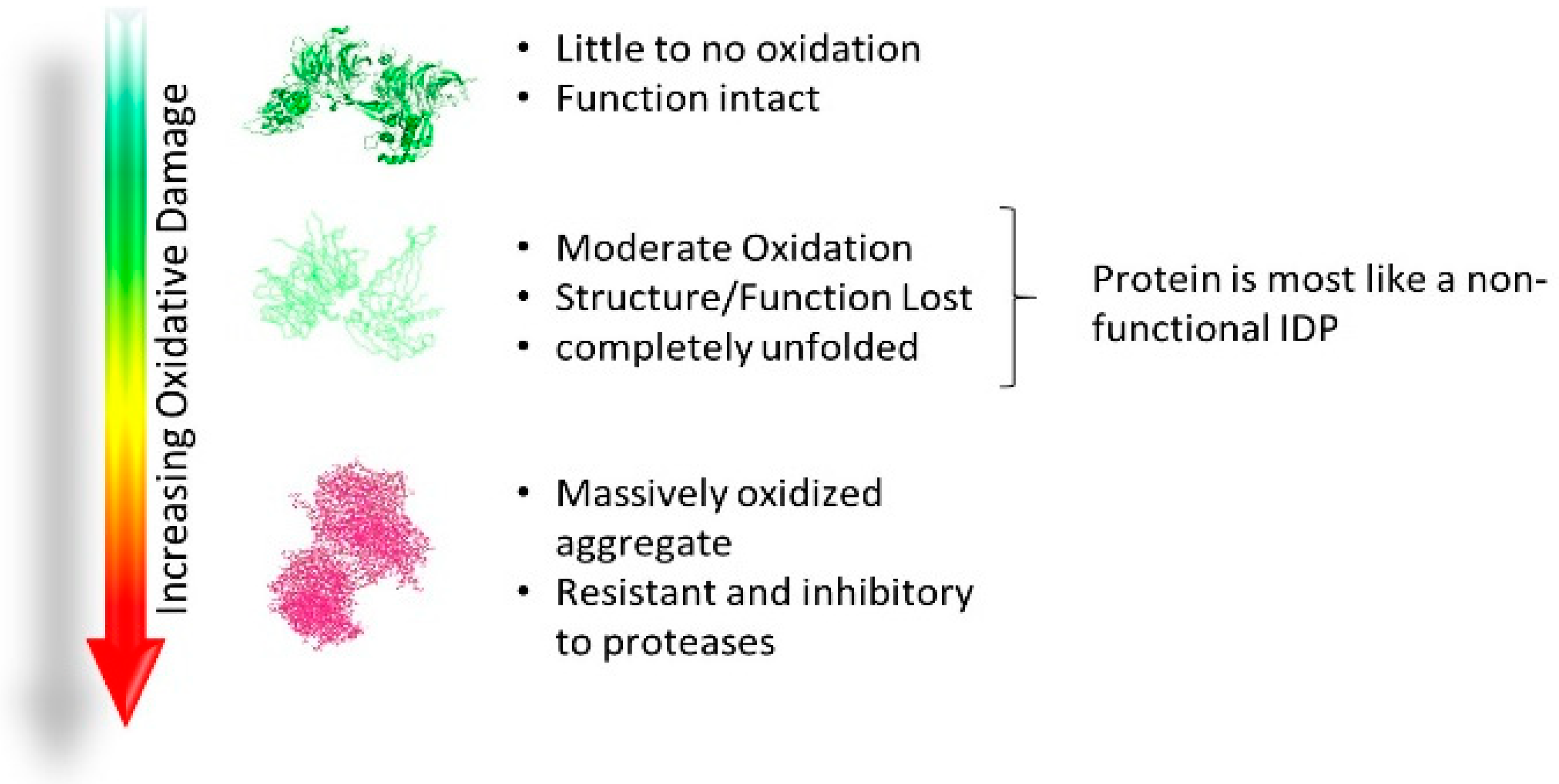
2.4. Acquired Disorder: Oxidative Damage
3. Proteasome Systems
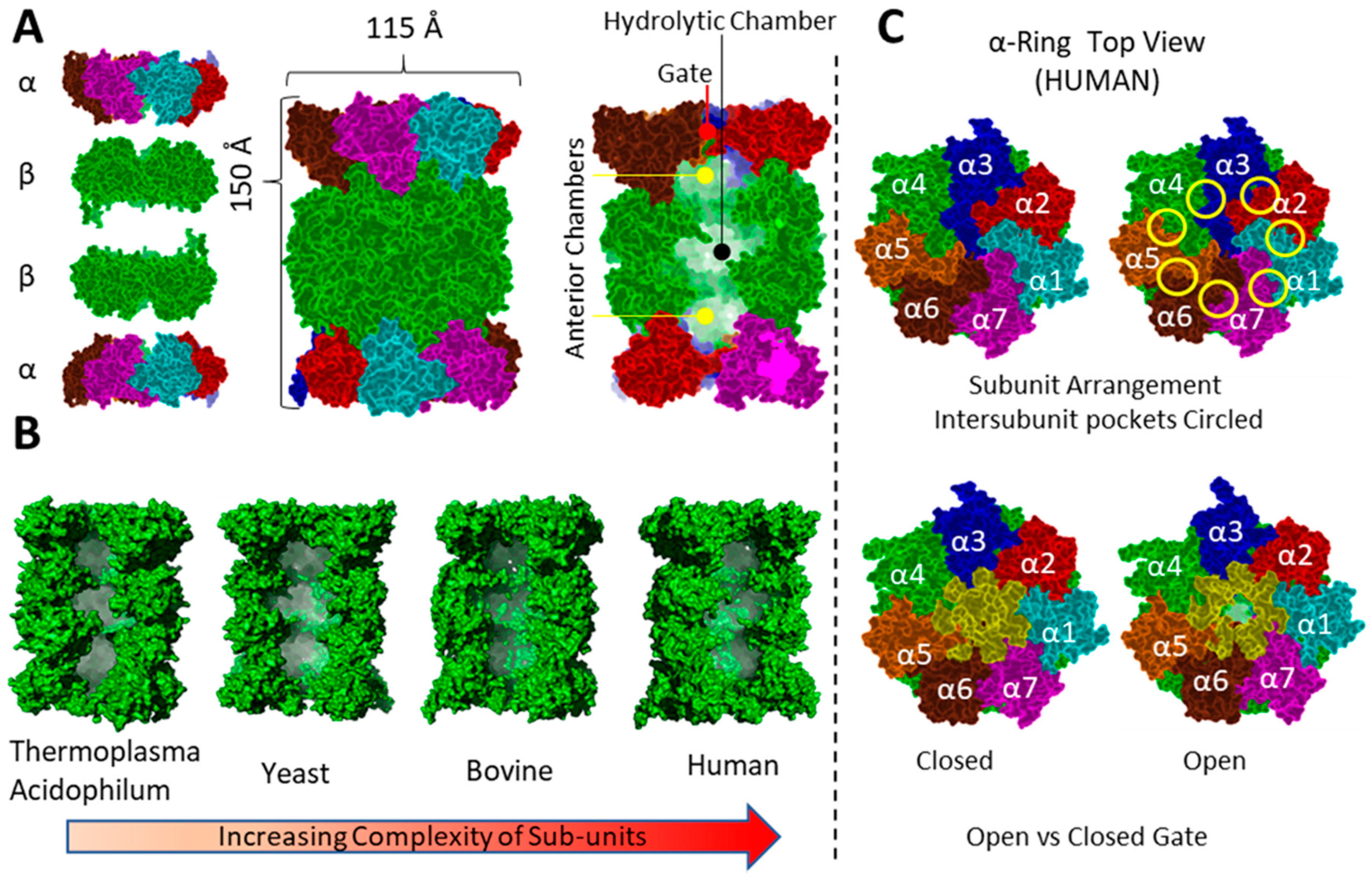
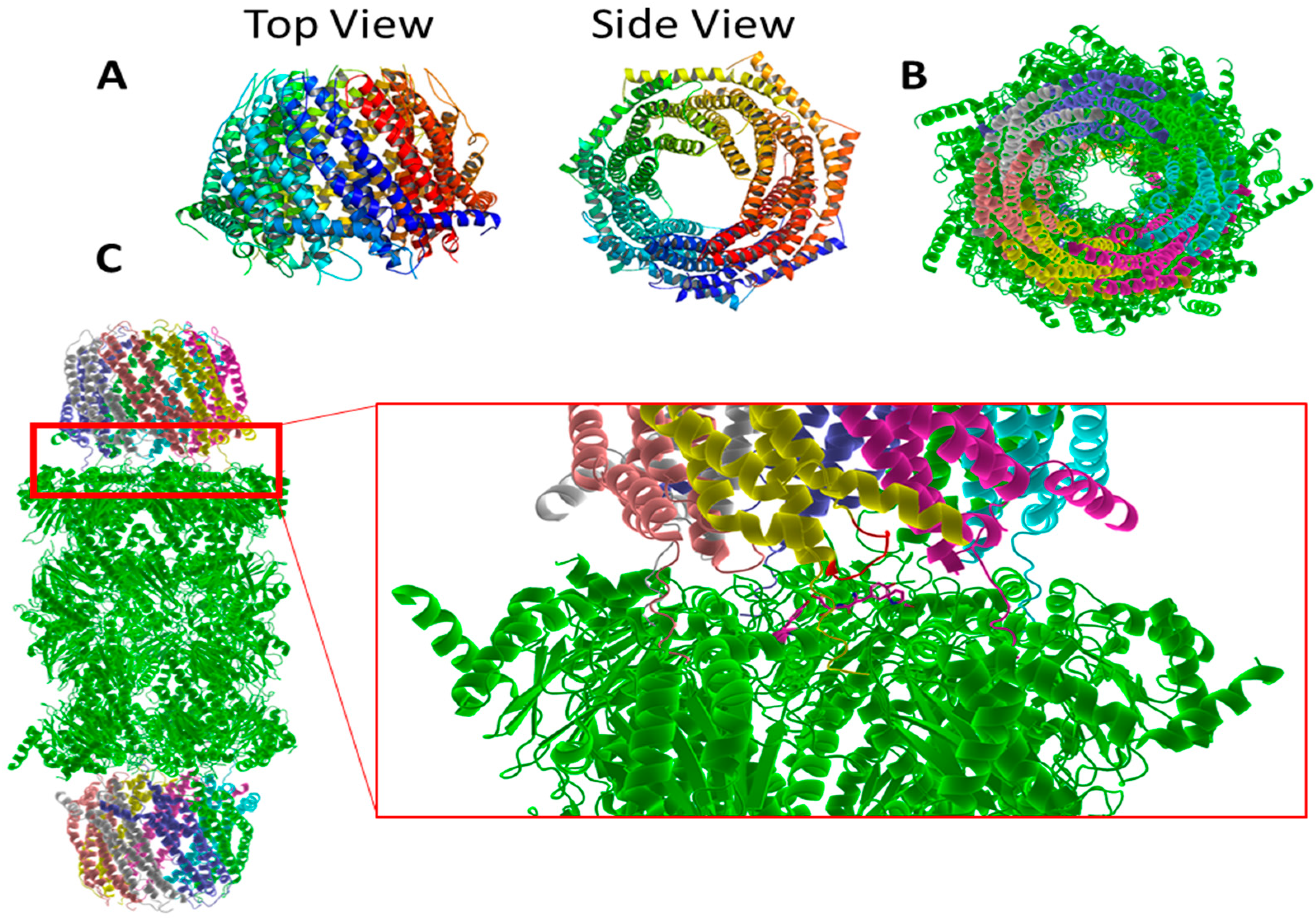
3.1. General Structure
3.2. Function of the Proteasome
- β1: peptidyl-glutamyl-peptide hydrolyzing (more commonly referred to as caspase-like) and cleaves after acidic amino acids
- β2: trypsin-like activity and cleaves after basic amino acids
- β5: chymotrypsin-like activity cleaving after hydrophobic amino acids.
3.3. Proteasome Distribution and Aging
3.3.1. The UPS
3.3.2. The UIPS
3.3.3. Mechanism of Proteasome Gating
3.4. The 11S and Proteasome Binding
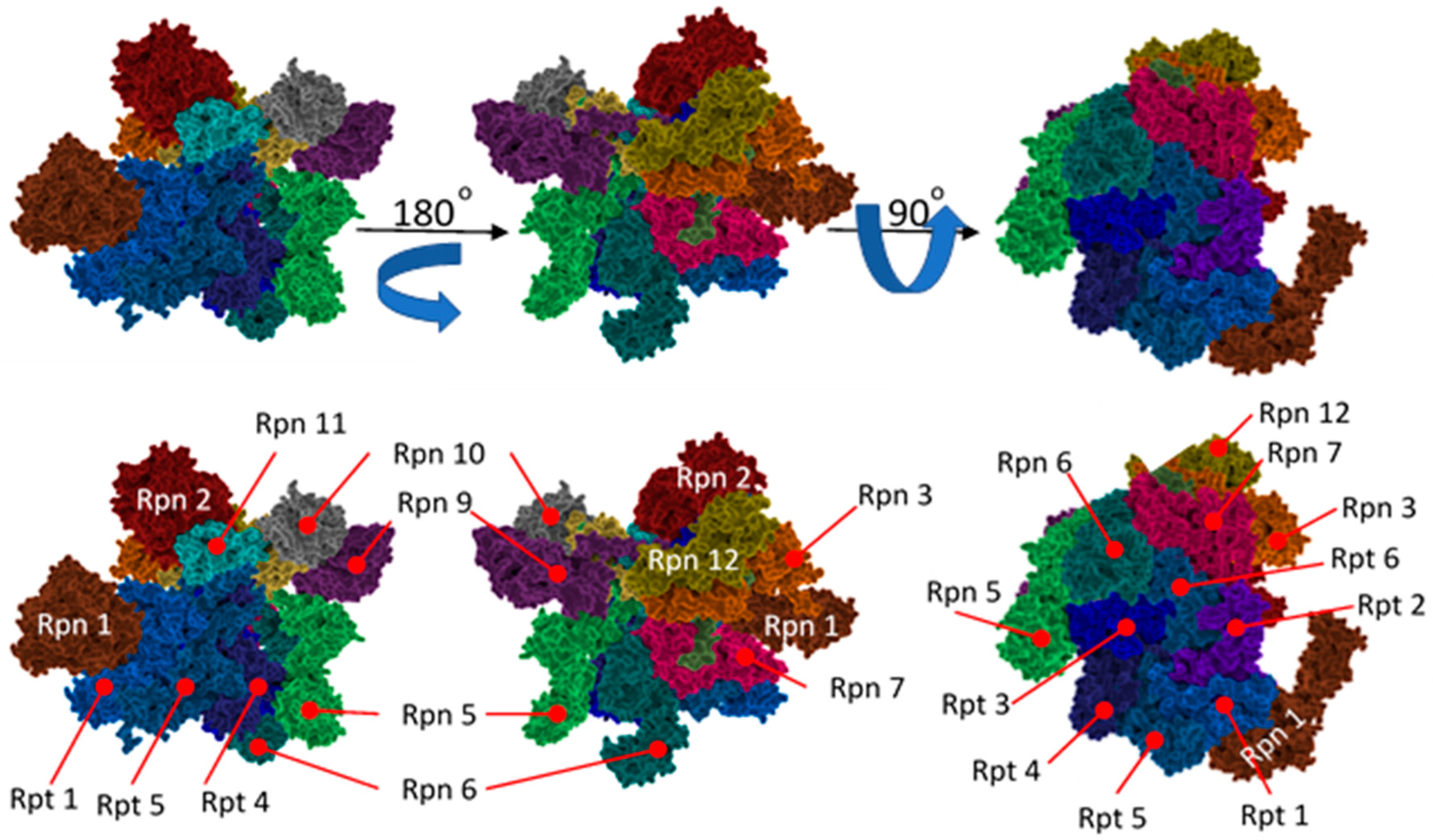
3.5. The 19S
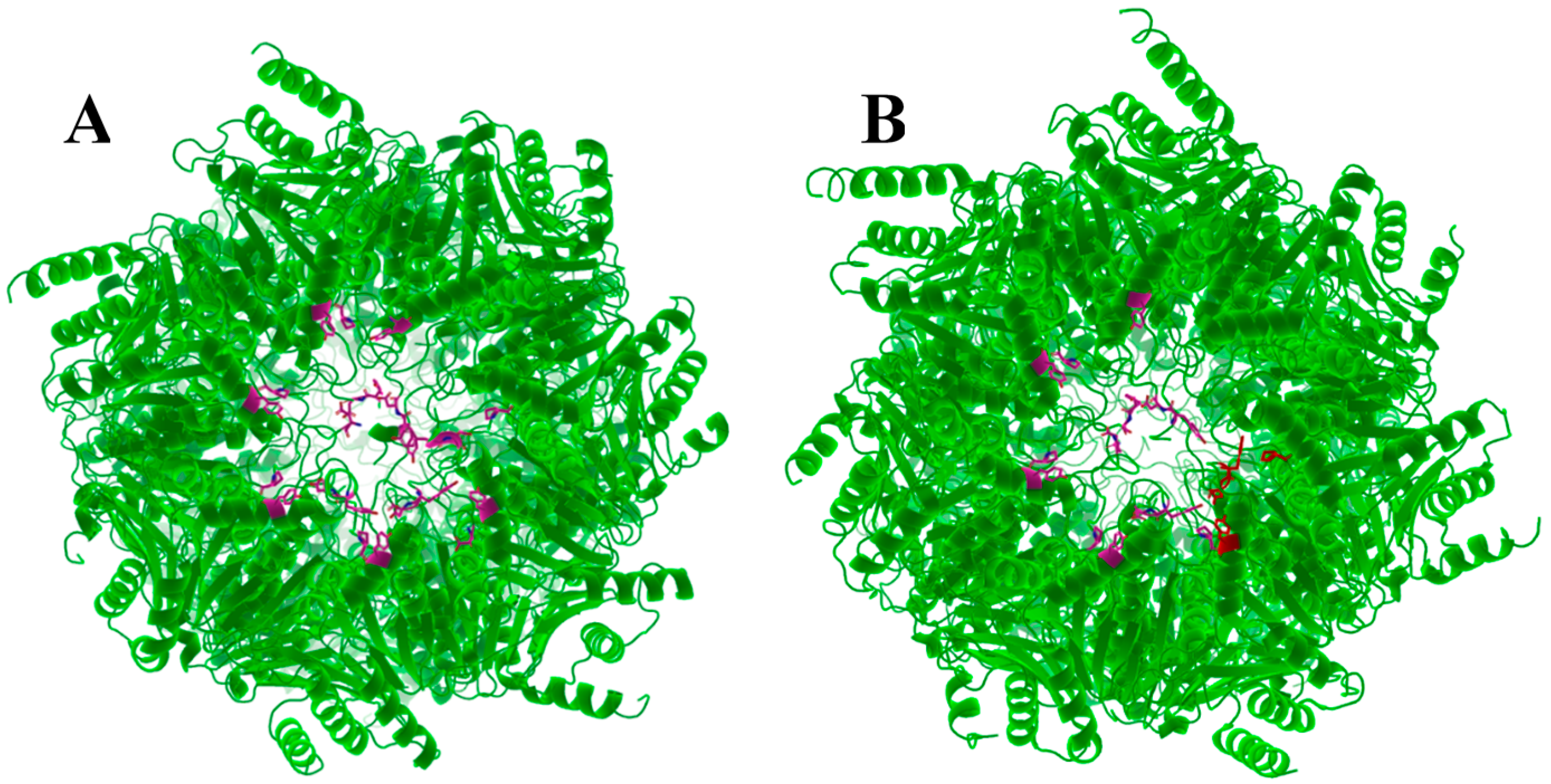
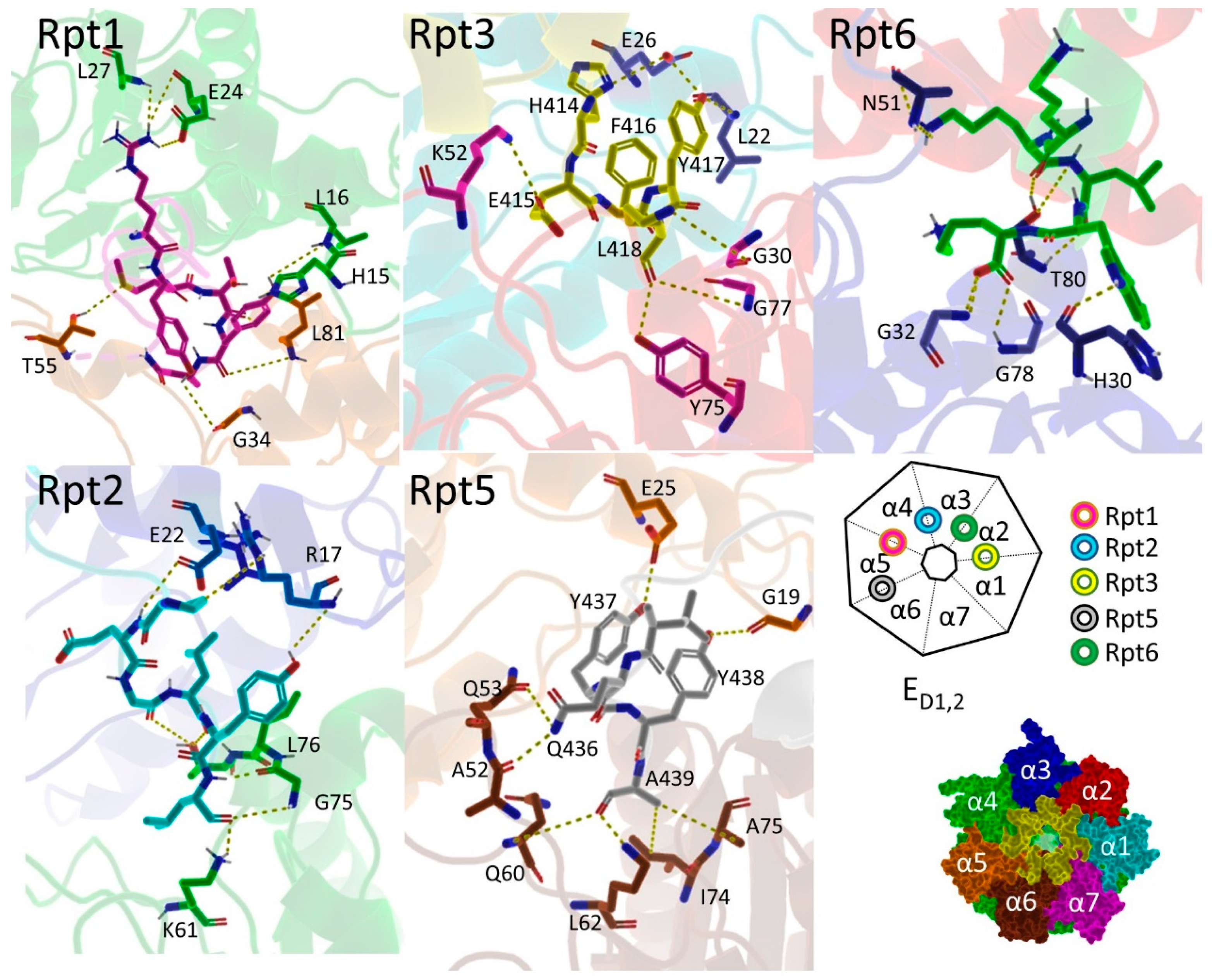
3.6. Hb-Y-X Motif
3.7. Gating of the Proteasome
4. Proteasome Activators
4.1. Denaturants
4.2. Peptides
4.3. Small Molecules
4.3.1. Direct 20S Activators
4.3.2. Indirect Proteasome Activators
4.4. Conclusions
Funding
Conflicts of Interest
References
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 2018, 198, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Crews, C.M. The Proteasome in Modern Drug Discovery: Second Life of a Highly Valuable Drug Target. ACS Cent. Sci. 2017, 3, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.P.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B.; Rutschmann, M.; Kuehn, L.; Reinauer, H. Activation of the multicatalytic proteinase from rat skeletal muscle by fatty acids or sodium dodecyl sulphate. Biochem. J. 1985, 228, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Etlinger, J.D.; Goldberg, A.L. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Hough, R.; Pratt, G.; Rechsteiner, M. Purification of two high molecular weight proteases from rabbit reticulocyte lysate. J. Biol. Chem. 1987, 262, 8303–8313. [Google Scholar] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 2014, 1843, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Medinas, D.B.; Valenzuela, V.; Hetz, C. Proteostasis disturbance in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, R91–R104. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Geyer, P.E.; Mann, M. Loss-less Nano-fractionator for High Sensitivity, High Coverage Proteomics. Mol. Cell. Proteom. 2017, 16, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016. [Google Scholar] [CrossRef]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Bartlett, A.I.; Radford, S.E. An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 582–588. [Google Scholar] [CrossRef]
- Ellis, R.J.; Minton, A.P. Protein aggregation in crowded environments. Biol. Chem. 2006, 387, 485–497. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Kumar, V.; Sami, N.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, M.I. Protein aggregation and neurodegenerative diseases: From theory to therapy. Eur. J. Med. Chem. 2016, 124, 1105–1120. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Lee, S.J.; Lim, H.S.; Masliah, E.; Lee, H.J. Protein aggregate spreading in neurodegenerative diseases: Problems and perspectives. Neurosci. Res. 2011, 70, 339–348. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Hohn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Halliwell, B.; Hu, M.L.; Louie, S.; Duvall, T.R.; Tarkington, B.K.; Motchnik, P.; Cross, C.E. Interaction of nitrogen dioxide with human plasma. Antioxidant depletion and oxidative damage. FEBS Lett. 1992, 313, 62–66. [Google Scholar] [CrossRef]
- Menzel, D.B. The toxicity of air pollution in experimental animals and humans: The role of oxidative stress. Toxicol. Lett. 1994, 72, 269–277. [Google Scholar] [CrossRef]
- Hu, M.L.; Tappel, A.L. Potentiation of oxidative damage to proteins by ultraviolet-A and protection by antioxidants. Photochem. Photobiol. 1992, 56, 357–363. [Google Scholar] [CrossRef]
- Kappus, H. Oxidative stress in chemical toxicity. Arch. Toxicol. 1987, 60, 144–149. [Google Scholar] [CrossRef]
- Davies, K.J. The Oxygen Paradox, oxidative stress, and ageing. Arch. Biochem. Biophys. 2016, 595, 28–32. [Google Scholar] [CrossRef]
- Uversky, V.N. Functions of short lifetime biological structures at large: The case of intrinsically disordered proteins. Brief. Funct. Genom. 2018. [Google Scholar] [CrossRef]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Intrinsically disordered proteins in crowded milieu: When chaos prevails within the cellular gumbo. Cell. Mol. Life Sci. 2018, 75, 3907–3929. [Google Scholar] [CrossRef]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztanyi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Uversky, V.N. Flexible nets of malleable guardians: Intrinsically disordered chaperones in neurodegenerative diseases. Chem. Rev. 2011, 111, 1134–1166. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320. [Google Scholar] [CrossRef]
- Perkins, J.R.; Diboun, I.; Dessailly, B.H.; Lees, J.G.; Orengo, C. Transient protein-protein interactions: Structural, functional, and network properties. Structure 2010, 18, 1233–1243. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic Disorder, Protein-Protein Interactions, and Disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Uversky, V.N.; Na, I.; Landau, K.S.; Schenck, R.O. Highly Disordered Proteins in Prostate Cancer. Curr. Protein Pept. Sci. 2017, 18, 453–481. [Google Scholar] [CrossRef]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef]
- Brucale, M.; Schuler, B.; Samori, B. Single-molecule studies of intrinsically disordered proteins. Chem. Rev. 2014, 114, 3281–3317. [Google Scholar] [CrossRef]
- Jensen, M.R.; Zweckstetter, M.; Huang, J.R.; Blackledge, M. Exploring free-energy landscapes of intrinsically disordered proteins at atomic resolution using NMR spectroscopy. Chem. Rev. 2014, 114, 6632–6660. [Google Scholar] [CrossRef]
- Wei, G.; Xi, W.; Nussinov, R.; Ma, B. Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev. 2016, 116, 6516–6551. [Google Scholar] [CrossRef]
- Uversky, V.N. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: Another illustration of the D2 concept. Expert Rev. Proteom. 2010, 7, 543–564. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Functional advantages of dynamic protein disorder. FEBS Lett. 2015, 589, 2433–2440. [Google Scholar] [CrossRef]
- Galea, C.A.; Wang, Y.; Sivakolundu, S.G.; Kriwacki, R.W. Regulation of cell division by intrinsically unstructured proteins: Intrinsic flexibility, modularity, and signaling conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N. Introduction to intrinsically disordered proteins (IDPs). Chem. Rev. 2014, 114, 6557–6560. [Google Scholar] [CrossRef]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef]
- Dyson, H.J. Making Sense of Intrinsically Disordered Proteins. Biophys. J. 2016, 110, 1013–1016. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Uversky, V.N. The most important thing is the tail: Multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013, 587, 1891–1901. [Google Scholar] [CrossRef]
- Gsponer, J.; Babu, M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009, 99, 94–103. [Google Scholar] [CrossRef]
- Drake, J.A.; Pettitt, B.M. Thermodynamics of Conformational Transitions in a Disordered Protein Backbone Model. Biophys. J. 2018, 114, 2799–2810. [Google Scholar] [CrossRef]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef]
- Davey, N.E.; Trave, G.; Gibson, T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011, 36, 159–169. [Google Scholar] [CrossRef]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. Bioessays 2009, 31, 328–335. [Google Scholar] [CrossRef]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder. J. Proteome Res. 2006, 5, 888–898. [Google Scholar] [CrossRef]
- Chen, J.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 1992, 360, 674–677. [Google Scholar] [CrossRef]
- Vise, P.D.; Baral, B.; Latos, A.J.; Daughdrill, G.W. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005, 33, 2061–2077. [Google Scholar] [CrossRef]
- Collins, M.O.; Yu, L.; Campuzano, I.; Grant, S.G.; Choudhary, J.S. Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol. Cell. Proteom. 2008, 7, 1331–1348. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef]
- Tompa, P.; Kovacs, D. Intrinsically disordered chaperones in plants and animals. Biochem. Cell Biol. 2010, 88, 167–174. [Google Scholar] [CrossRef]
- Hegyi, H.; Tompa, P. Intrinsically disordered proteins display no preference for chaperone binding in vivo. PLoS Comput. Biol. 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Bardwell, J.C.; Jakob, U. Conditional disorder in chaperone action. Trends Biochem. Sci. 2012, 37, 517–525. [Google Scholar] [CrossRef]
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: C-Myc as Model System. Cancer Inform. 2017, 16, 1176935117699408. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef]
- Lu, W.J.; Amatruda, J.F.; Abrams, J.M. p53 ancestry: Gazing through an evolutionary lens. Nat. Rev. Cancer 2009, 9, 758–762. [Google Scholar] [CrossRef]
- Du, Z.; Uversky, V.N. A Comprehensive Survey of the Roles of Highly Disordered Proteins in Type 2 Diabetes. Int. J. Mol. Sci. 2017, 18, 2010. [Google Scholar] [CrossRef]
- Al-Jiffri, O.H.; Al-Sharif, F.M.; Al-Jiffri, E.H.; Uversky, V.N. Intrinsic disorder in biomarkers of insulin resistance, hypoadiponectinemia, and endothelial dysfunction among the type 2 diabetic patients. Intrinsically Disord. Proteins 2016, 4, e1171278. [Google Scholar] [CrossRef]
- Uversky, V.N. Wrecked regulation of intrinsically disordered proteins in diseases: Pathogenicity of deregulated regulators. Front. Mol. Biosci. 2014, 1, 6. [Google Scholar] [CrossRef]
- Lansbury, P.T.; Lashuel, H.A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 2006, 443, 774–779. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 2015, 7, 18. [Google Scholar] [CrossRef]
- Radwan, M.; Wood, R.J.; Sui, X.; Hatters, D.M. When proteostasis goes bad: Protein aggregation in the cell. IUBMB Life 2017, 69, 49–54. [Google Scholar] [CrossRef]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef]
- Wear, M.P.; Kryndushkin, D.; O’Meally, R.; Sonnenberg, J.L.; Cole, R.N.; Shewmaker, F.P. Proteins with Intrinsically Disordered Domains Are Preferentially Recruited to Polyglutamine Aggregates. PLoS ONE 2015, 10, e0136362. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. 2009, 14, 5188–5238. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Kingwell, K. Zeroing in on neurodegenerative alpha-synuclein. Nat. Rev. Drug Discov. 2017, 16, 371–373. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Berrocal, R.; Vasquez, V.; Krs, S.R.; Gadad, B.S.; Rao, K.S. alpha-Synuclein Misfolding Versus Aggregation Relevance to Parkinson’s Disease: Critical Assessment and Modeling. Mol. Neurobiol. 2015, 51, 1417–1431. [Google Scholar] [CrossRef]
- Silva, B.A.; Breydo, L.; Uversky, V.N. Targeting the chameleon: A focused look at alpha-synuclein and its roles in neurodegeneration. Mol. Neurobiol. 2013, 47, 446–459. [Google Scholar] [CrossRef]
- Uversky, V.N. Alpha-synuclein misfolding and neurodegenerative diseases. Curr. Protein Pept. Sci. 2008, 9, 507–540. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Lee, H.J.; Choi, C.; Lee, S.J. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Alim, M.A.; Hossain, M.S.; Arima, K.; Takeda, K.; Izumiyama, Y.; Nakamura, M.; Kaji, H.; Shinoda, T.; Hisanaga, S.; Ueda, K. Tubulin seeds alpha-synuclein fibril formation. J. Biol. Chem. 2002, 277, 2112–2117. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Kokhan, V.S.; Afanasyeva, M.A.; Van’kin, G.I. alpha-Synuclein knockout mice have cognitive impairments. Behav. Brain Res. 2012, 231, 226–230. [Google Scholar] [CrossRef]
- Uversky, V.N. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Korsak, M.; Kozyreva, T. Beta Amyloid Hallmarks: From Intrinsically Disordered Proteins to Alzheimer’s Disease. Adv. Exp. Med. Biol. 2015, 870, 401–421. [Google Scholar]
- Bamburg, J.R.; Bloom, G.S. Cytoskeletal pathologies of Alzheimer disease. Cell Motil. Cytoskelet. 2009, 66, 635–649. [Google Scholar] [CrossRef]
- Ciasca, G.; Campi, G.; Battisti, A.; Rea, G.; Rodio, M.; Papi, M.; Pernot, P.; Tenenbaum, A.; Bianconi, A. Continuous thermal collapse of the intrinsically disordered protein tau is driven by its entropic flexible domain. Langmuir 2012, 28, 13405–13410. [Google Scholar] [CrossRef]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar] [CrossRef]
- Von Bergen, M.; Barghorn, S.; Jeganathan, S.; Mandelkow, E.M.; Mandelkow, E. Spectroscopic approaches to the conformation of tau protein in solution and in paired helical filaments. Neurodegener. Dis. 2006, 3, 197–206. [Google Scholar] [CrossRef]
- Chirita, C.N.; Congdon, E.E.; Yin, H.; Kuret, J. Triggers of full-length tau aggregation: A role for partially folded intermediates. Biochemistry 2005, 44, 5862–5872. [Google Scholar] [CrossRef]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef]
- Orr, M.E.; Sullivan, A.C.; Frost, B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 637–648. [Google Scholar] [CrossRef]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2015, 9, 43–50. [Google Scholar] [CrossRef]
- Purro, S.A.; Nicoll, A.J.; Collinge, J. Prion Protein as a Toxic Acceptor of Amyloid-beta Oligomers. Biol. Psychiatry 2018, 83, 358–368. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Das, A.S.; Zou, W.Q. Prions: Beyond a Single Protein. Clin. Microbiol. Rev. 2016, 29, 633–658. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Kupfer, L.; Hinrichs, W.; Groschup, M.H. Prion protein misfolding. Curr. Mol. Med. 2009, 9, 826–835. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, S.; Li, X.J.; Li, S. The Expanding Clinical Universe of Polyglutamine Disease. Neuroscientist 2019, 1073858418822993. [Google Scholar] [CrossRef]
- Silva, A.; de Almeida, A.V.; Macedo-Ribeiro, S. Polyglutamine expansion diseases: More than simple repeats. J. Struct. Biol. 2018, 201, 139–154. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar]
- Perevozchikova, T.; Stanley, C.B.; McWilliams-Koeppen, H.P.; Rowe, E.L.; Berthelier, V. Investigating the structural impact of the glutamine repeat in huntingtin assembly. Biophys. J. 2014, 107, 411–421. [Google Scholar] [CrossRef]
- De Los Rios, P.; Hafner, M.; Pastore, A. Explaining the length threshold of polyglutamine aggregation. J. Phys. Condens. Matter 2012, 24, 244105. [Google Scholar] [CrossRef]
- Perutz, M.F.; Pope, B.J.; Owen, D.; Wanker, E.E.; Scherzinger, E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc. Natl. Acad. Sci. USA 2002, 99, 5596–5600. [Google Scholar] [CrossRef]
- Polling, S.; Hill, A.F.; Hatters, D.M. Polyglutamine aggregation in Huntington and related diseases. Adv. Exp. Med. Biol. 2012, 769, 125–140. [Google Scholar]
- Zoghbi, H.Y.; Orr, H.T. Polyglutamine diseases: Protein cleavage and aggregation. Curr. Opin. Neurobiol. 1999, 9, 566–570. [Google Scholar] [CrossRef]
- Davies, K.J. Oxidative stress: The paradox of aerobic life. Biochem. Soc. Symp. 1995, 61, 1–31. [Google Scholar] [CrossRef]
- Bader, N.; Grune, T. Protein oxidation and proteolysis. Biol. Chem. 2006, 387, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Englander, S.W.; Mayne, L. The case for defined protein folding pathways. Proc. Natl. Acad. Sci. USA 2017, 114, 8253–8258. [Google Scholar] [CrossRef] [PubMed]
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873–15880. [Google Scholar] [CrossRef] [PubMed]
- De Graff, A.M.; Hazoglou, M.J.; Dill, K.A. Highly Charged Proteins: The Achilles’ Heel of Aging Proteomes. Structure 2016, 24, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid. Redox Signal. 2015, 23, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Grune, T. Protein oxidation and proteolytic signalling in aging. Curr. Pharm. Des. 2014, 20, 3040–3051. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef]
- Hohn, T.J.; Grune, T. The proteasome and the degradation of oxidized proteins: Part III-Redox regulation of the proteasomal system. Redox Biol. 2014, 2, 388–394. [Google Scholar]
- Jung, T.; Hohn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part II-Protein oxidation and proteasomal degradation. Redox Biol. 2013, 2, 99–104. [Google Scholar] [CrossRef]
- Wedel, S.; Manola, M.; Cavinato, M.; Trougakos, I.P.; Jansen-Durr, P. Targeting Protein Quality Control Mechanisms by Natural Products to Promote Healthy Ageing. Molecules 2018, 23, 1219. [Google Scholar] [CrossRef]
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. Eat or be eaten: The autophagic plight of inactive 26S proteasomes. Autophagy 2015, 11, 1927–1928. [Google Scholar] [CrossRef][Green Version]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar]
- Liebl, M.P.; Hoppe, T. It’s all about talking: Two-way communication between proteasomal and lysosomal degradation pathways via ubiquitin. Am. J. Physiol. Cell Physiol. 2016, 311, C166–C178. [Google Scholar] [CrossRef]
- Wang, X.J.; Yu, J.; Wong, S.H.; Cheng, A.S.; Chan, F.K.; Ng, S.S.; Cho, C.H.; Sung, J.J.; Wu, W.K. A novel crosstalk between two major protein degradation systems: Regulation of proteasomal activity by autophagy. Autophagy 2013, 9, 1500–1508. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Cohen-Rosenzweig, C.; Ciechanover, A. The ubiquitin-proteasome system and autophagy: Coordinated and independent activities. Int. J. Biochem. Cell Biol. 2016, 79, 403–418. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef]
- Kudriaeva, A.A.; Belogurov, A.A. Proteasome: A Nanomachinery of Creative Destruction. Biochemistry 2019, 84, S159–S192. [Google Scholar] [CrossRef]
- Opoku-Nsiah, K.A.; Gestwicki, J.E. Aim for the core: Suitability of the ubiquitin-independent 20S proteasome as a drug target in neurodegeneration. Transl Res. 2018, 198, 48–57. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Yu, H.; Mim, C.; Matouschek, A. Regulated protein turnover: Snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 2014, 15, 122–133. [Google Scholar] [CrossRef]
- Ciechanover, A.; Stanhill, A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Biophys. Acta 2014, 1843, 86–96. [Google Scholar] [CrossRef]
- Jankowska, E.; Stoj, J.; Karpowicz, P.; Osmulski, P.A.; Gaczynska, M. The proteasome in health and disease. Curr. Pharm. Des. 2013, 19, 1010–1028. [Google Scholar]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Smith, D.M.; Benaroudj, N.; Goldberg, A. Proteasomes and their associated ATPases: A destructive combination. J. Struct. Biol. 2006, 156, 72–83. [Google Scholar] [CrossRef]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Lowe, J.; Stock, D.; Jap, B.; Zwickl, P.; Baumeister, W.; Huber, R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. [Google Scholar] [CrossRef]
- Groll, M.; Ditzel, L.; Lowe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. Structure determination of the constitutive 20S proteasome from bovine liver at 2.75 A resolution. J. Biochem. 2002, 131, 171–173. [Google Scholar] [CrossRef]
- Harshbarger, W.; Miller, C.; Diedrich, C.; Sacchettini, J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 2015, 23, 418–424. [Google Scholar] [CrossRef]
- Morimoto, Y.; Mizushima, T.; Yagi, A.; Tanahashi, N.; Tanaka, K.; Ichihara, A.; Tsukihara, T. Ordered structure of the crystallized bovine 20S proteasome. J. Biochem. 1995, 117, 471–474. [Google Scholar] [CrossRef]
- Huber, E.M.; Heinemeyer, W.; Li, X.; Arendt, C.S.; Hochstrasser, M.; Groll, M. A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat. Commun. 2016, 7, 10900. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.; Lu, Y.; Ma, Y.B.; Lee, B.H.; Yu, Z.; Ouyang, Q.; Finley, D.J.; Kirschner, M.W.; Mao, Y. Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, 12991–12996. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Gaczynska, M. Nanoenzymology of the 20S proteasome: Proteasomal actions are controlled by the allosteric transition. Biochemistry 2002, 41, 7047–7053. [Google Scholar] [CrossRef]
- Giletto, M.B.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.E.; Tepe, J.J. Pipecolic esters as minimized templates for proteasome inhibition. Org. Biomol. Chem. 2019, 17, 2734–2746. [Google Scholar] [CrossRef]
- Dahlmann, B. Mammalian proteasome subtypes: Their diversity in structure and function. Arch. Biochem. Biophys. 2016, 591, 132–140. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Tomita, T.; Matouschek, A. Substrate selection by the proteasome through initiation regions. Protein Sci. 2019, 28, 1222–1232. [Google Scholar] [CrossRef]
- Yu, H.; Matouschek, A. Recognition of Client Proteins by the Proteasome. Annu. Rev. Biophys. 2017, 46, 149–173. [Google Scholar] [CrossRef]
- Yu, H.; Singh Gautam, A.K.; Wilmington, S.R.; Wylie, D.; Martinez-Fonts, K.; Kago, G.; Warburton, M.; Chavali, S.; Inobe, T.; Finkelstein, I.J.; et al. Conserved Sequence Preferences Contribute to Substrate Recognition by the Proteasome. J. Biol. Chem. 2016, 291, 14526–14539. [Google Scholar] [CrossRef]
- Yu, H.; Kago, G.; Yellman, C.M.; Matouschek, A. Ubiquitin-like domains can target to the proteasome but proteolysis requires a disordered region. EMBO J. 2016, 35, 1522–1536. [Google Scholar] [CrossRef]
- De Poot, S.A.H.; Tian, G.; Finley, D. Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J. Mol. Biol. 2017, 429, 3525–3545. [Google Scholar] [CrossRef]
- Strickland, E.; Hakala, K.; Thomas, P.J.; DeMartino, G.N. Recognition of misfolding proteins by PA700, the regulatory subcomplex of the 26 S proteasome. J. Biol. Chem. 2000, 275, 5565–5572. [Google Scholar] [CrossRef]
- Sadre-Bazzaz, K.; Whitby, F.G.; Robinson, H.; Formosa, T.; Hill, C.P. Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell 2010, 37, 728–735. [Google Scholar] [CrossRef]
- Li, J.; Rechsteiner, M. Molecular dissection of the 11S REG (PA28) proteasome activators. Biochimie 2001, 83, 373–383. [Google Scholar] [CrossRef]
- Mao, I.; Liu, J.; Li, X.; Luo, H. REGgamma, a proteasome activator and beyond? Cell. Mol. Life Sci. 2008, 65, 3971–3980. [Google Scholar] [CrossRef]
- Whitby, F.G.; Masters, E.I.; Kramer, L.; Knowlton, J.R.; Yao, Y.; Wang, C.C.; Hill, C.P. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 2000, 408, 115–120. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef]
- Asher, G.; Bercovich, Z.; Tsvetkov, P.; Shaul, Y.; Kahana, C. 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol. Cell 2005, 17, 645–655. [Google Scholar] [CrossRef]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef]
- Eleuteri, A.M.; Cuccioloni, M.; Bellesi, J.; Lupidi, G.; Fioretti, E.; Angeletti, M. Interaction of Hsp90 with 20S proteasome: Thermodynamic and kinetic characterization. Proteins 2002, 48, 169–177. [Google Scholar] [CrossRef]
- Whittier, J.E.; Xiong, Y.; Rechsteiner, M.C.; Squier, T.C. Hsp90 enhances degradation of oxidized calmodulin by the 20 S proteasome. J. Biol. Chem. 2004, 279, 46135–46142. [Google Scholar] [CrossRef]
- Mayer-Kuckuk, P.; Ullrich, O.; Ziegler, M.; Grune, T.; Schweiger, M. Functional interaction of poly(ADP-ribose) with the 20S proteasome in vitro. Biochem. Biophys. Res. Commun. 1999, 259, 576–581. [Google Scholar] [CrossRef]
- Wiggins, C.M.; Tsvetkov, P.; Johnson, M.; Joyce, C.L.; Lamb, C.A.; Bryant, N.J.; Komander, D.; Shaul, Y.; Cook, S.J. BIM(EL), an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation. J. Cell Sci. 2011, 124, 969–977. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010, 17, 103–108. [Google Scholar] [CrossRef]
- Asher, G.; Reuven, N.; Shaul, Y. 20S proteasomes and protein degradation “by default”. Bioessays 2006, 28, 844–849. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Shaul, Y. Determination of IUP based on susceptibility for degradation by default. Methods Mol. Biol. 2012, 895, 3–18. [Google Scholar]
- Pena, M.M.; Melo, S.P.; Xing, Y.Y.; White, K.; Barbour, K.W.; Berger, F.G. The intrinsically disordered N-terminal domain of thymidylate synthase targets the enzyme to the ubiquitin-independent proteasomal degradation pathway. J. Biol. Chem. 2009, 284, 31597–31607. [Google Scholar] [CrossRef]
- Heck, J.W.; Cheung, S.K.; Hampton, R.Y. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA 2010, 107, 1106–1111. [Google Scholar] [CrossRef]
- Fang, N.N.; Mayor, T. Hul5 ubiquitin ligase: Good riddance to bad proteins. Prion 2012, 6, 240–244. [Google Scholar] [CrossRef]
- Fang, N.N.; Ng, A.H.; Measday, V.; Mayor, T. Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol. 2011, 13, 1344–1352. [Google Scholar] [CrossRef]
- Njomen, E.; Tepe, J.J. Proteasome Activation as a New Therapeutic Approach To Target Proteotoxic Disorders. J. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Krahn, J.H.; Kaschani, F.; Kaiser, M. Turning-ON Proteasomes. Cell Chem. Biol. 2017, 24, 653–655. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Voutetakis, K.; Kapetanou, M.; Delitsikou, V.; Papaevgeniou, N.; Sakellari, M.; Lefaki, M.; Filippopoulou, K.; Gonos, E.S. Proteasome activation: An innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res. Rev. 2015, 23, 37–55. [Google Scholar] [CrossRef]
- Gonos, E. Proteasome activation delays aging and protects against proteotoxicity in neurodegenerative disease. Adv. Exp. Med. Biol. 2015, 821, 7. [Google Scholar]
- Gonos, E. Proteasome activation as a novel anti-aging strategy. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S7. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Sakellari, M.; Lefaki, M.; Papaevgeniou, N.; Gonos, E.S. Proteasome activation delays aging in vitro and in vivo. Free Radic. Biol. Med. 2014, 71, 303–320. [Google Scholar] [CrossRef]
- Han, D.H.; Na, H.K.; Choi, W.H.; Lee, J.H.; Kim, Y.K.; Won, C.; Lee, S.H.; Kim, K.P.; Kuret, J.; Min, D.H.; et al. Direct cellular delivery of human proteasomes to delay tau aggregation. Nat. Commun. 2014, 5, 5633. [Google Scholar] [CrossRef]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.P. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef]
- Hwang, J.S.; Hwang, J.S.; Chang, I.; Kim, S. Age-associated decrease in proteasome content and activities in human dermal fibroblasts: Restoration of normal level of proteasome subunits reduces aging markers in fibroblasts from elderly persons. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 490–499. [Google Scholar] [CrossRef]
- Masson, P.; Lundin, D.; Soderbom, F.; Young, P. Characterization of a REG/PA28 proteasome activator homolog in Dictyostelium discoideum indicates that the ubiquitin- and ATP-independent REGgamma proteasome is an ancient nuclear protease. Eukaryot. Cell 2009, 8, 844–851. [Google Scholar] [CrossRef]
- Kish-Trier, E.; Hill, C.P. Structural biology of the proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [CrossRef]
- Stadtmueller, B.M.; Hill, C.P. Proteasome Activators. Mol. Cell 2011, 41, 8–19. [Google Scholar] [CrossRef]
- Knowlton, J.R.; Johnston, S.C.; Whitby, F.G.; Realini, C.; Zhang, Z.; Rechsteiner, M.; Hill, C.P. Structure of the proteasome activator REGalpha (PA28alpha). Nature 1997, 390, 639–643. [Google Scholar] [CrossRef]
- Forster, A.; Masters, E.I.; Whitby, F.G.; Robinson, H.; Hill, C.P. The 1.9 A structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol. Cell 2005, 18, 589–599. [Google Scholar] [CrossRef]
- Forster, A.; Whitby, F.G.; Hill, C.P. The pore of activated 20S proteasomes has an ordered 7-fold symmetric conformation. EMBO J. 2003, 22, 4356–4364. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, W.L.; Yu, D.; Ouyang, Q.; Lu, Y.; Mao, Y. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat. Commun. 2018, 9, 1360. [Google Scholar] [CrossRef]
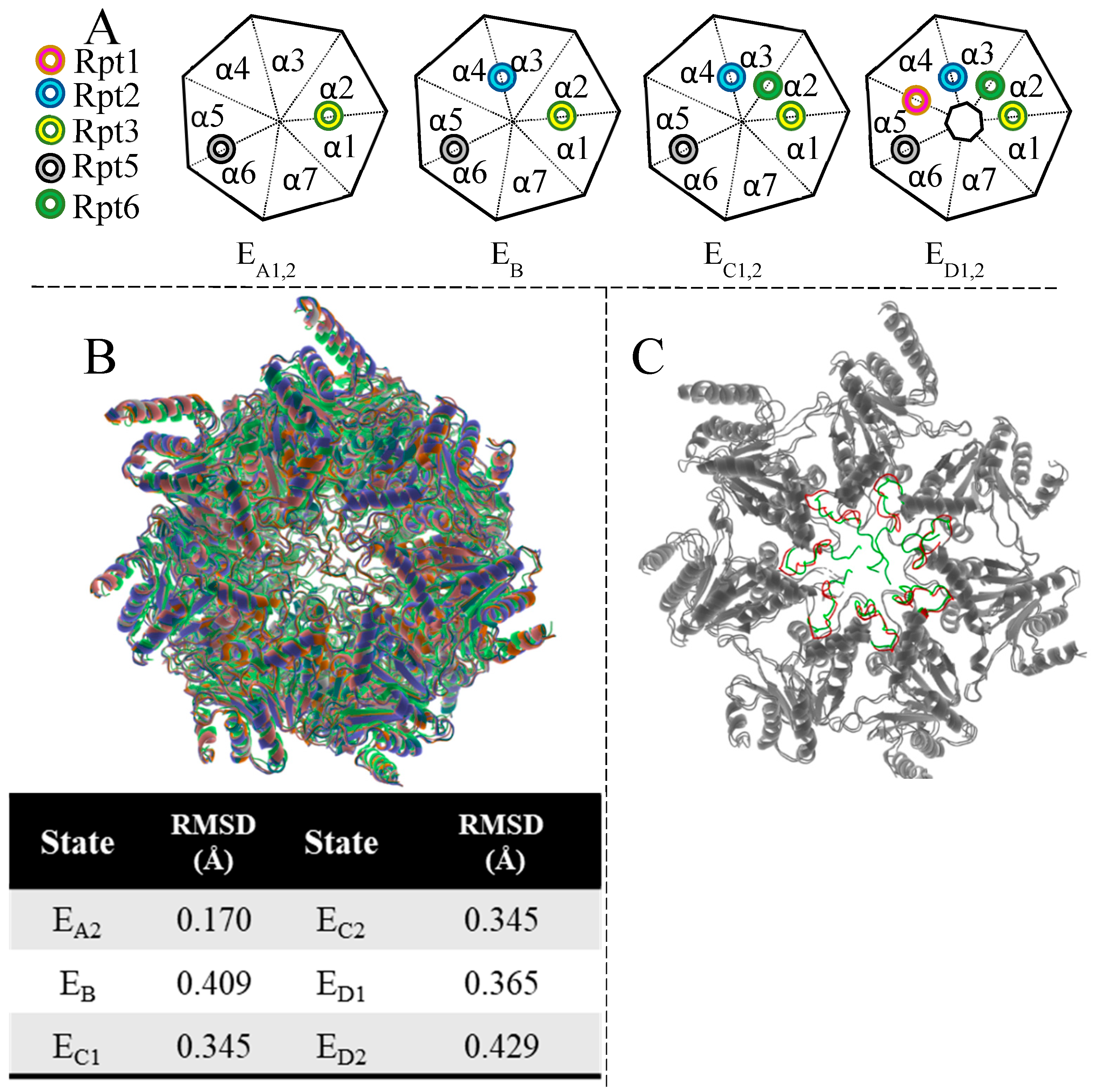
- Eisele, M.R.; Reed, R.G.; Rudack, T.; Schweitzer, A.; Beck, F.; Nagy, I.; Pfeifer, G.; Plitzko, J.M.; Baumeister, W.; Tomko, R.J.; et al. Expanded Coverage of the 26S Proteasome Conformational Landscape Reveals Mechanisms of Peptidase Gating. Cell Rep. 2018, 24, 1301–1315.e5. [Google Scholar] [CrossRef]
- Wehmer, M.; Rudack, T.; Beck, F.; Aufderheide, A.; Pfeifer, G.; Plitzko, J.M.; Forster, F.; Schulten, K.; Baumeister, W.; Sakata, E. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. USA 2017, 114, 1305–1310. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Wang, X.; Cimermancic, P.; Yu, C.; Schweitzer, A.; Chopra, N.; Engel, J.L.; Greenberg, C.; Huszagh, A.S.; Beck, F.; Sakata, E.; et al. Molecular Details Underlying Dynamic Structures and Regulation of the Human 26S Proteasome. Mol. Cell. Proteom. 2017, 16, 840–854. [Google Scholar] [CrossRef]
- Kusmierczyk, A.R.; Kunjappu, M.J.; Kim, R.Y.; Hochstrasser, M. A conserved 20S proteasome assembly factor requires a C-terminal HbYX motif for proteasomal precursor binding. Nat. Struct. Mol. Biol. 2011, 18, 622–629. [Google Scholar] [CrossRef]
- Smith, D.M.; Chang, S.C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell 2007, 27, 731–744. [Google Scholar] [CrossRef]
- Rabl, J.; Smith, D.M.; Yu, Y.; Chang, S.C.; Goldberg, A.L.; Cheng, Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell 2008, 30, 360–368. [Google Scholar] [CrossRef]
- Huang, X.; Luan, B.; Wu, J.; Shi, Y. An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 2016, 23, 778–785. [Google Scholar] [CrossRef]
- Lasker, K.; Forster, F.; Bohn, S.; Walzthoeni, T.; Villa, E.; Unverdorben, P.; Beck, F.; Aebersold, R.; Sali, A.; Baumeister, W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. USA 2012, 109, 1380–1387. [Google Scholar] [CrossRef]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef]
- Enenkel, C. Proteasome dynamics. Biochim. Biophys. Acta 2014, 1843, 39–46. [Google Scholar] [CrossRef]
- Shibatani, T.; Ward, W.F. Sodium dodecyl sulfate (SDS) activation of the 20S proteasome in rat liver. Arch. Biochem. Biophys. 1995, 321, 160–166. [Google Scholar] [CrossRef]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta 2004, 1666, 105–117. [Google Scholar] [CrossRef]
- Trader, D.J.; Simanski, S.; Dickson, P.; Kodadek, T. Establishment of a suite of assays that support the discovery of proteasome stimulators. Biochim. Biophys. Acta 2017, 1861, 892–899. [Google Scholar] [CrossRef]
- Ruiz de Mena, I.; Mahillo, E.; Arribas, J.; Castano, J.G. Kinetic mechanism of activation by cardiolipin (diphosphatidylglycerol) of the rat liver multicatalytic proteinase. Biochem. J. 1993, 296 Pt 1, 93–97. [Google Scholar] [CrossRef]
- Watanabe, N.; Yamada, S. Activation of 20S proteasomes from spinach leaves by fatty acids. Plant Cell Physiol. 1996, 37, 147–151. [Google Scholar] [CrossRef]
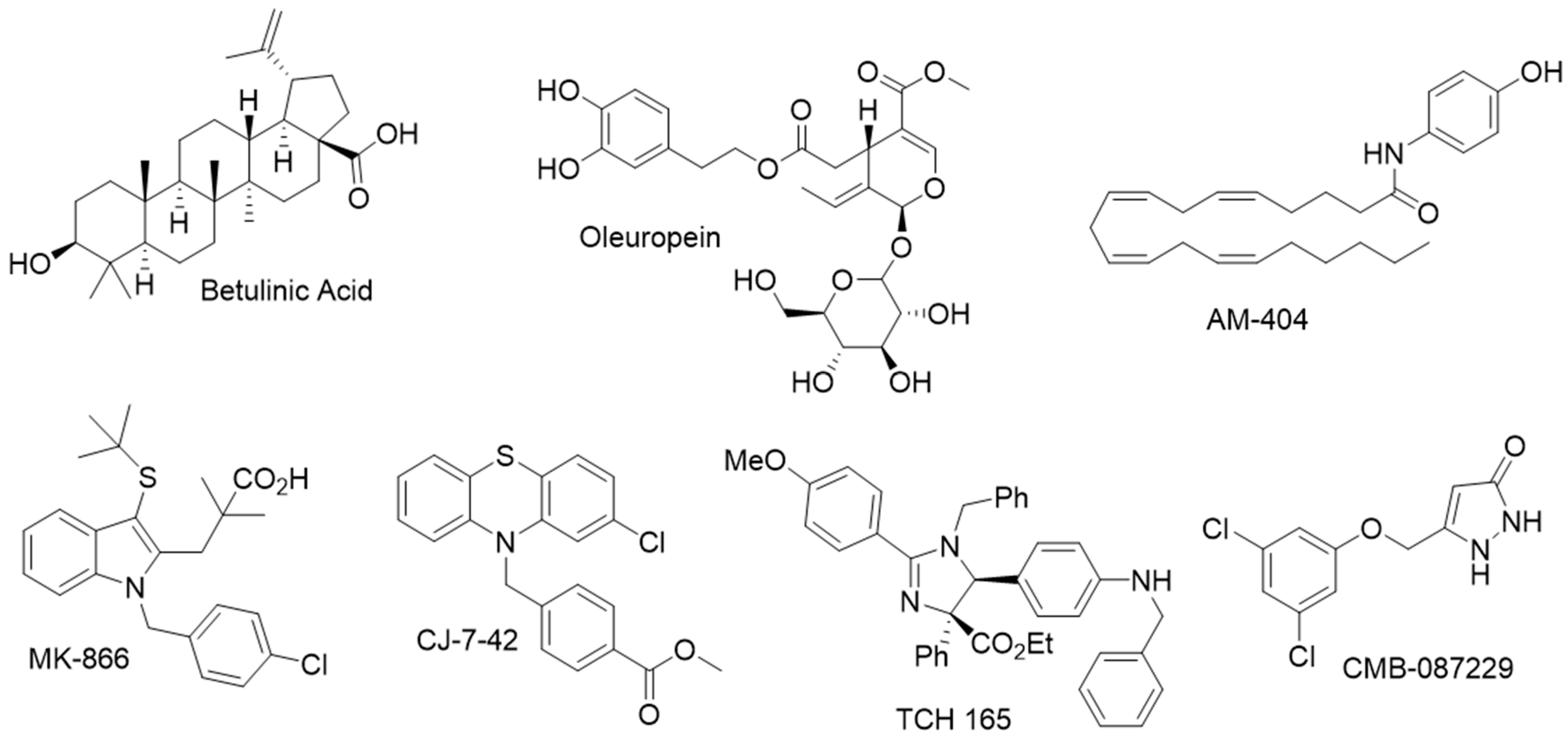
- Katsiki, M.; Chondrogianni, N.; Chinou, I.; Rivett, A.J.; Gonos, E.S. The olive constituent oleuropein exhibits proteasome stimulatory properties in vitro and confers life span extension of human embryonic fibroblasts. Rejuvenation Res. 2007, 10, 157–172. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorgan. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Kim, Y.C.; DeMartino, G.N. C termini of proteasomal ATPases play nonequivalent roles in cellular assembly of mammalian 26 S proteasome. J. Biol. Chem. 2011, 286, 26652–26666. [Google Scholar] [CrossRef]
- Gillette, T.G.; Kumar, B.; Thompson, D.; Slaughter, C.A.; DeMartino, G.N. Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J. Biol. Chem. 2008, 283, 31813–31822. [Google Scholar] [CrossRef]
- Dal Vechio, F.H.; Cerqueira, F.; Augusto, O.; Lopes, R.; Demasi, M. Peptides that activate the 20S proteasome by gate opening increased oxidized protein removal and reduced protein aggregation. Free Radic. Biol. Med. 2014, 67, 304–313. [Google Scholar] [CrossRef]
- Gizynska, M.; Witkowska, J.P.; Karpowicz, P.; Rostankowski, R.; Chocron, E.S.; Pickering, A.M.; Osmulski, P.A.; Gaczynska, M.E.; Jankowska, E. Proline- and arginine-rich peptides as flexible allosteric modulators of human proteasome activity. J. Med. Chem. 2019, 62, 359–370. [Google Scholar] [CrossRef]
- Huang, L.; Ho, P.; Chen, C.H. Activation and inhibition of the proteasome by betulinic acid and its derivatives. FEBS Lett. 2007, 581, 4955–4959. [Google Scholar] [CrossRef]
- Azevedo, L.M.; Lansdell, T.A.; Ludwig, J.R.; Mosey, R.A.; Woloch, D.K.; Cogan, D.P.; Patten, G.P.; Kuszpit, M.R.; Fisk, J.S.; Tepe, J.J. Inhibition of the human proteasome by imidazoline scaffolds. J. Med. Chem. 2013, 56, 5974–5978. [Google Scholar] [CrossRef]
- Lansdell, T.A.; Hewlett, N.M.; Skoumbourdis, A.P.; Fodor, M.D.; Seiple, I.B.; Su, S.; Baran, P.S.; Feldman, K.S.; Tepe, J.J. Palau’amine and Related Oroidin Alkaloids Dibromophakellin and Dibromophakellstatin Inhibit the Human 20S Proteasome. J. Nat. Prod. 2012, 75, 980–985. [Google Scholar] [CrossRef]
- Gallastegui, N.; Beck, P.; Arciniega, M.; Huber, R.; Hillebrand, S.; Groll, M. Hydroxyureas as noncovalent proteasome inhibitors. Angew. Chem. Int. Ed. 2012, 51, 247–249. [Google Scholar] [CrossRef]
- Coleman, R.A.; Muli, C.S.; Zhao, Y.; Bhardwaj, A.; Newhouse, T.R.; Trader, D.J. Analysis of chain length, substitution patterns, and unsaturation of AM-404 derivatives as 20S proteasome stimulators. Bioorgan. Med. Chem. Lett. 2019, 29, 420–423. [Google Scholar] [CrossRef]
- Coleman, R.A.; Trader, D.J. Development and Application of a Sensitive Peptide Reporter to Discover 20S Proteasome Stimulators. ACS Comb. Sci. 2018, 20, 269–276. [Google Scholar] [CrossRef]
- Coleman, R.A.; Trader, D.J. A Sensitive High-Throughput Screening Method for Identifying Small Molecule Stimulators of the Core Particle of the Proteasome. Curr. Protoc. Chem. Biol. 2018, 10, e52. [Google Scholar] [CrossRef]
- Jones, C.L.; Njomen, E.; Sjogren, B.; Dexheimer, T.S.; Tepe, J.J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol. 2017, 12, 2240–2247. [Google Scholar] [CrossRef]
- Njomen, E.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.; Tepe, J.J. Small Molecule Modulation of Proteasome Assembly. Biochemistry 2018, 57, 4214–4224. [Google Scholar] [CrossRef]
- Trippier, P.C.; Zhao, K.T.; Fox, S.G.; Schiefer, I.T.; Benmohamed, R.; Moran, J.; Kirsch, D.R.; Morimoto, R.I.; Silverman, R.B. Proteasome Activation is a Mechanism for Pyrazolone Small Molecules Displaying Therapeutic Potential in Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2014, 5, 823–829. [Google Scholar] [CrossRef]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. [Google Scholar] [CrossRef]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef]
- Shi, Y.; Long, M.J.; Rosenberg, M.M.; Li, S.; Kobjack, A.; Lessans, P.; Coffey, R.T.; Hedstrom, L. Boc3Arg-Linked Ligands Induce Degradation by Localizing Target Proteins to the 20S Proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef]
- Jacomin, A.C.; Taillebourg, E.; Fauvarque, M.O. Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities? Cells 2018, 7, 112. [Google Scholar] [CrossRef]
- Pal, A.; Young, M.A.; Donato, N.J. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014, 74, 4955–4966. [Google Scholar] [CrossRef]
- Shanmugham, A.; Ovaa, H. DUBs and disease: Activity assays for inhibitor development. Curr. Opin. Drug Discov. Dev. 2008, 11, 688–696. [Google Scholar]
- Farshi, P.; Deshmukh, R.R.; Nwankwo, J.O.; Arkwright, R.T.; Cvek, B.; Liu, J.; Dou, Q.P. Deubiquitinases (DUBs) and DUB inhibitors: A patent review. Expert Opin. Ther. Pat. 2015, 25, 1191–1208. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terminal Sequence | Hydrolysis (%WT) |
|---|---|
| LYR (WT) | 100 |
| LY- | 4 |
| LYD | 5 |
| LYA | 100 |
| LYW | 106 |
| LYL | 13 |
| LYG | 77 |
| YRA | 2 |
| No PA | 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, C.L.; Tepe, J.J. Proteasome Activation to Combat Proteotoxicity. Molecules 2019, 24, 2841. https://doi.org/10.3390/molecules24152841
Jones CL, Tepe JJ. Proteasome Activation to Combat Proteotoxicity. Molecules. 2019; 24(15):2841. https://doi.org/10.3390/molecules24152841
Chicago/Turabian StyleJones, Corey L., and Jetze J. Tepe. 2019. "Proteasome Activation to Combat Proteotoxicity" Molecules 24, no. 15: 2841. https://doi.org/10.3390/molecules24152841
APA StyleJones, C. L., & Tepe, J. J. (2019). Proteasome Activation to Combat Proteotoxicity. Molecules, 24(15), 2841. https://doi.org/10.3390/molecules24152841