
Dihydrogen Bond in the Aminoborane Complex of a Nicergoline Intermediate


Abstract
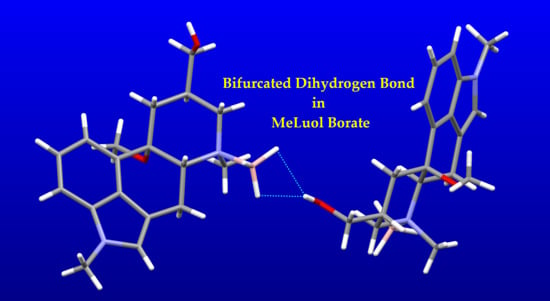
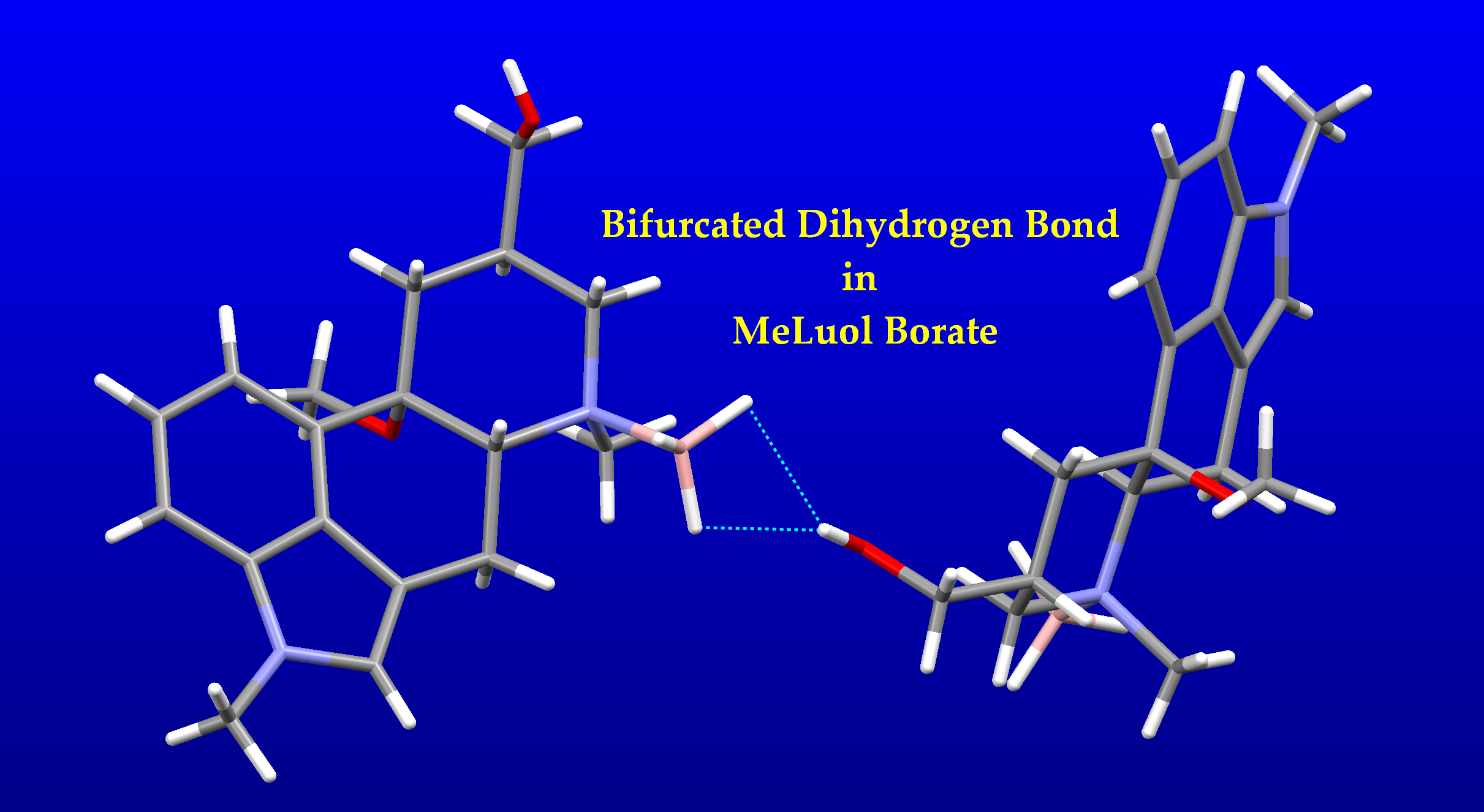
1. Introduction
Dihydrogen Bond
2. Results and Discussion
2.1. Preparation of Single Crystals
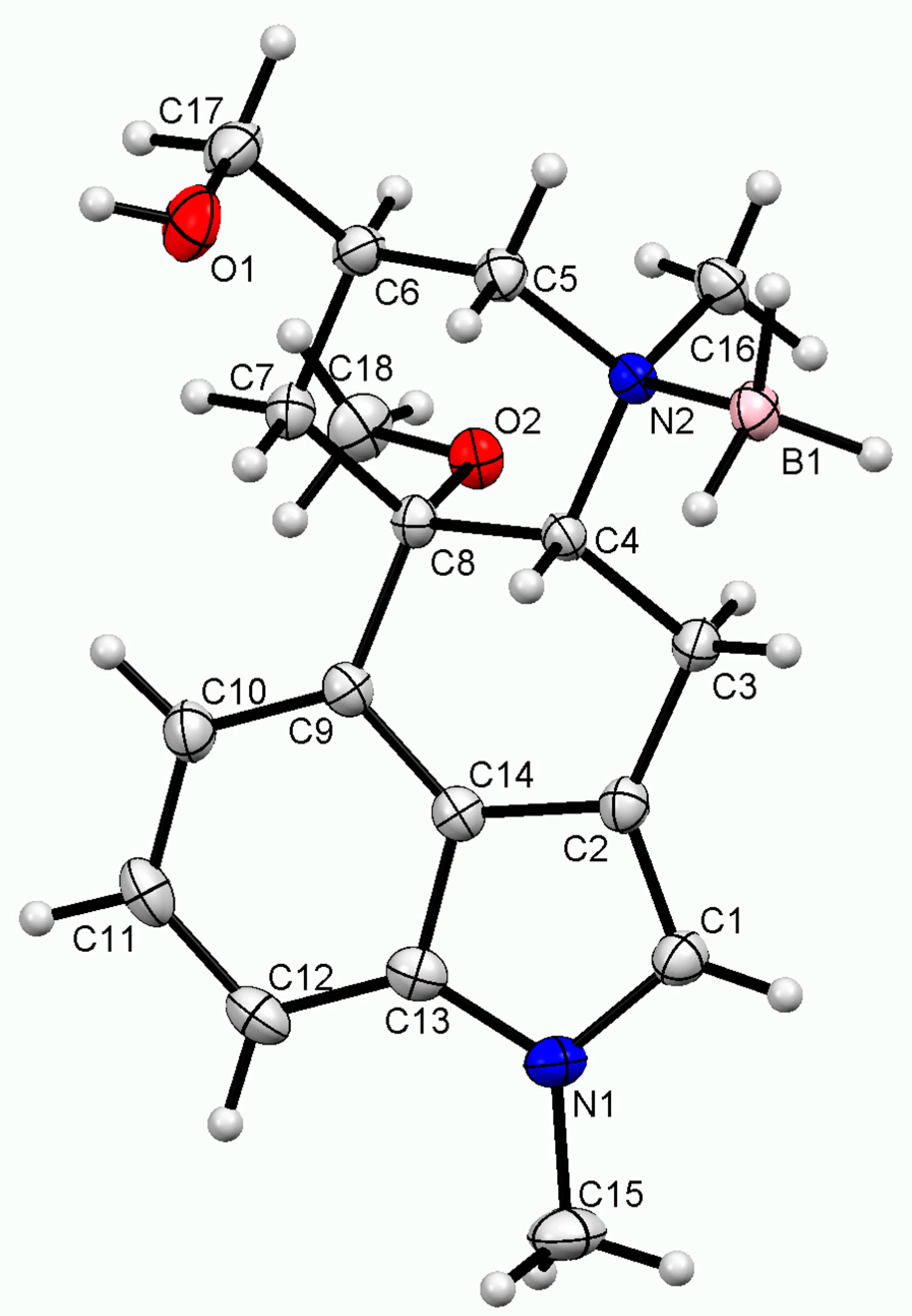
2.2. Single-Crystal X-ray Diffraction Structure
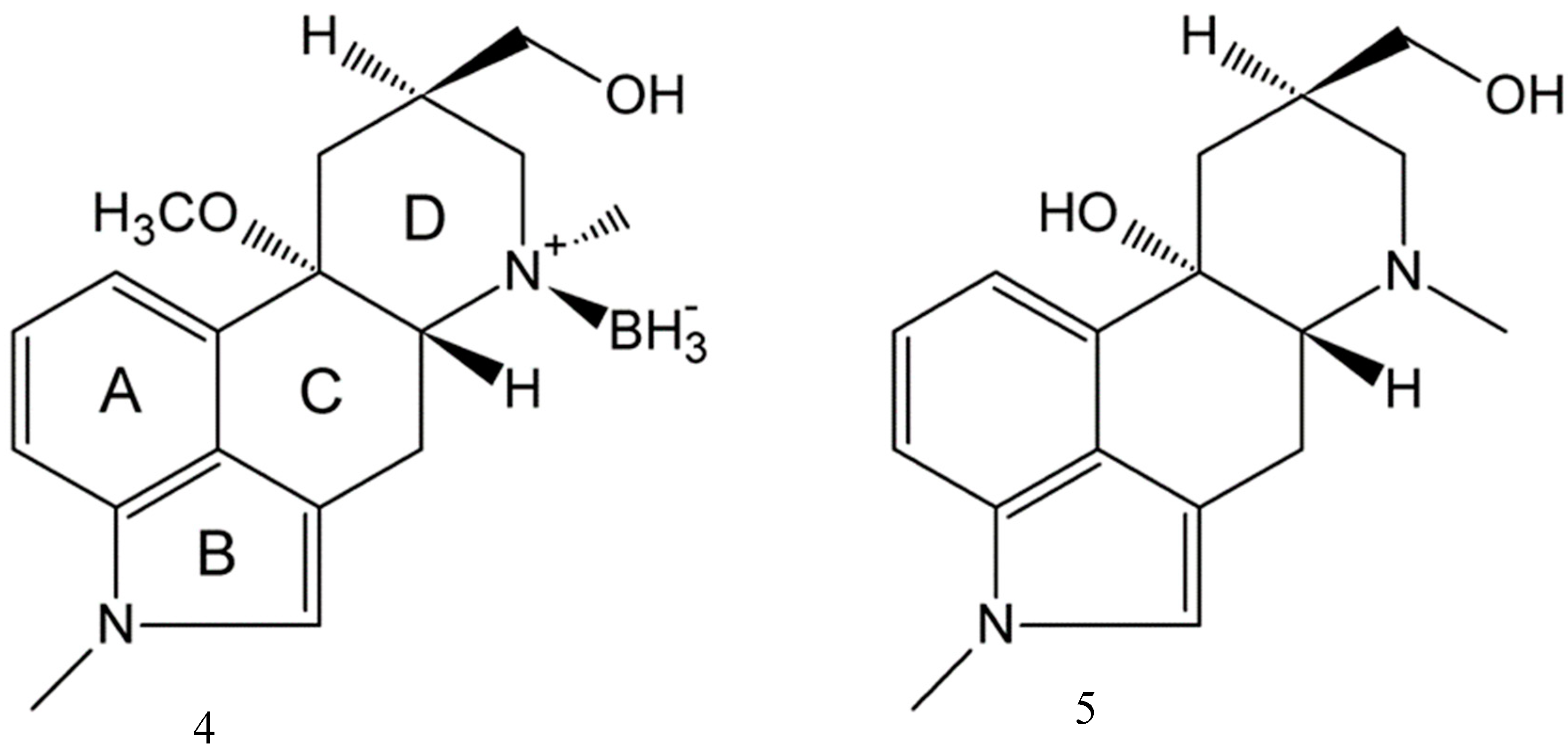
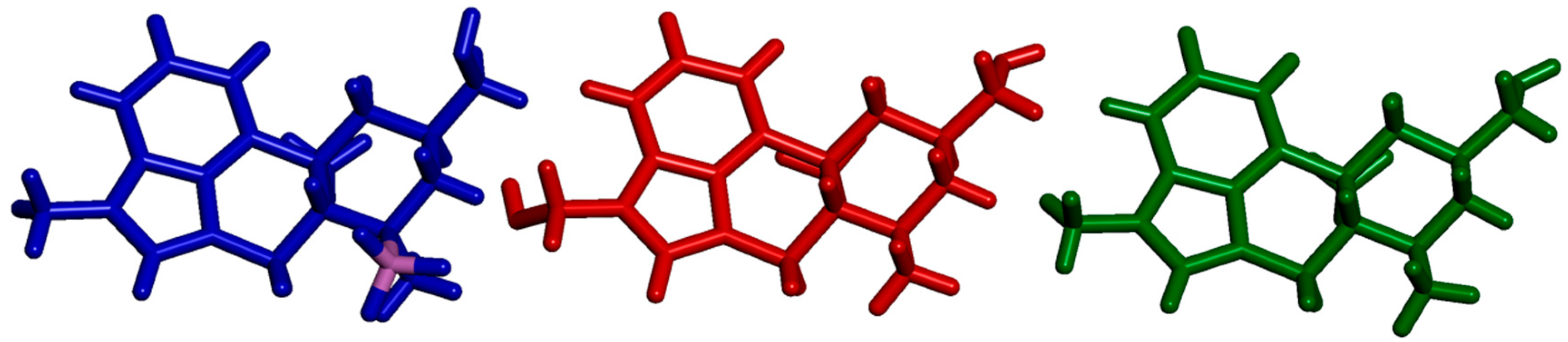
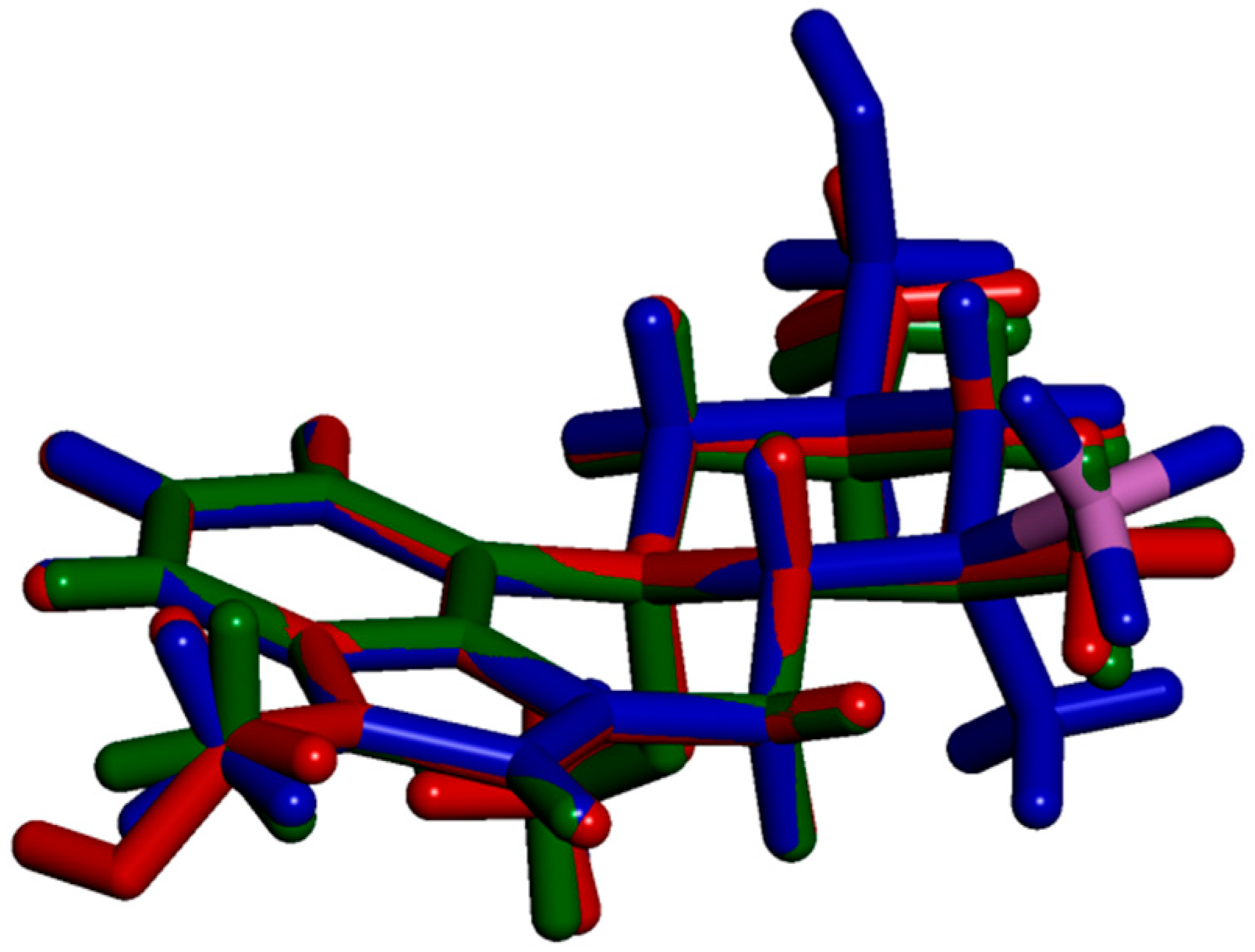
2.3. Impact of BH3 on the MeLuol Conformation
2.4. Structural Features
2.4.1. Molecular Packing Compared to Related Derivatives
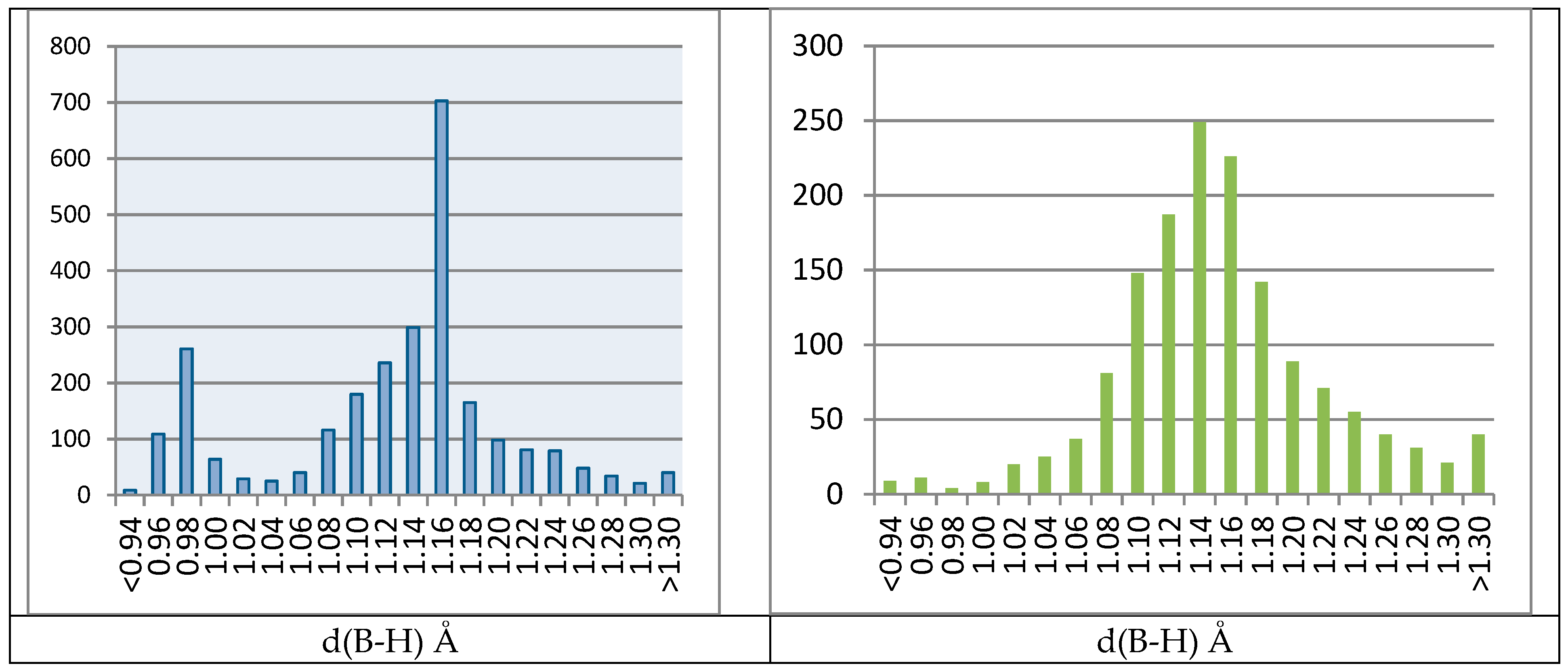
2.4.2. Biased B–H Bonds in CSD
2.4.3. Impact of BH3 on the Molecular Packing
3. Materials and Methods
3.1. Single-Crystal X-ray Diffraction
3.2. Materials Used
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schweitzer, B.A.; Egholm, M.; Koch, T.H. Mechanistic studies of the reduction of daunomycin with sodium borohydride. Formation and reaction of borate esters. J. Am. Chem. Soc. 1992, 114, 242–248. [Google Scholar] [CrossRef]
- Liu, Q.; Xiong, F.-J.; He, Q.-Q.; Chen, F.-E. Development of an Efficient Process for the Decomposition of the Borate Complexes Formed during the Large-Scale Synthesis of (S)-1,2,4-Butanetriol. Org. Process Res. Dev. 2013, 17, 1540–1542. [Google Scholar] [CrossRef]
- Niedenzu, K.; Dawson, J.W. Boron-Nitrogen Compounds. III.1,2 Aminoboranes, Part 2: The B-N Bond Character in Substituted Aminoboranes. J. Am. Chem. Soc. 1960, 82, 4223–4228. [Google Scholar] [CrossRef]
- Narayana, C.; Periasamy, M. Hydroboration of prochiral olefins with chiral Lewis base–borane complexes: Relationship to the mechanism of hydroboration. J. Chem. Soc. Chem. Commun. 1987, 1857–1859. [Google Scholar] [CrossRef]
- Le Toumelin, J.-B.; Baboulène, M. Chiral intramolecular amine-borane complexes as reducing agents for prochiral ketones. Tetrahedron Asymmetry 1997, 8, 1259–1265. [Google Scholar] [CrossRef]
- Burkhardt, E.R.; Matos, K. Boron Reagents in Process Chemistry: Excellent Tools for Selective Reductions. Chem. Rev. 2006, 106, 2617–2650. [Google Scholar] [CrossRef] [PubMed]
- Hisaki, I.; Shizuki, N.; Sasaki, T.; Ito, Y.; Tohnai, N.; Miyata, M. Handedness Determination of 21 Helical Motifs and Hierarchical Analysis of Crystal Structures Based on the Motifs: The Case of Cinchona Alkaloid Derivatives. Cryst. Growth Des. 2010, 10, 5262–5269. [Google Scholar] [CrossRef]
- Hodgkin, D.C. The X-ray Analysis of Complicated Molecules. Science 1965, 150, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H.; Siegbahn, P.E.M.; Eisenstein, O.; Rheingold, A.L.; Koetzle, T.F. A New Intermolecular Interaction: UnconventionalHydrogen Bonds with Element−Hydride Bonds as ProtonAcceptor. Acc. Chem. Res. 1996, 29, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology; International Union of Crystallography monographs on crystallography; first publ. in paperback; Oxford University Press: Oxford, UK, 2001; ISBN 978-0-19-850970-7. [Google Scholar]
- Bakhmutov, V.I. Dihydrogen Bonds: Principles, Experiments, and Applications; Wiley-Interscience: Hoboken, NJ, USA, 2008; ISBN 978-0-470-18096-9. [Google Scholar]
- Čejka, J.; Kratochvíl, B.; Cvak, L.; Jegorov, A. Crystal structure of 1-hydroxymethyl-10α-methoxy-9,10-dihydrolysergol, C18H24N2O3. Z. Krist. New Cryst. Struct. 2005, 220, 371–372. [Google Scholar] [CrossRef][Green Version]
- Čejka, J.; Kratochvíl, B.; Cvak, L.; Jegorov, A. Crystal structure of 1-methyl-10α-methoxy-9,10-dihydrolysergol, C18H24N2O2. Z. Krist. New Cryst. Struct. 2005, 220, 217–218. [Google Scholar] [CrossRef]
- Discovery Studio Modeling Environment; Dassault Systèmes BIOVIA; Dassault Systèmes: San Diego, CA, USA, 2016.
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Flores-Parra, A.; Sánchez-Ruiz, S.A.; Guadarrama, C.; Nöth, H.; Contreras, R. BHδ—δ+HC Interactions in N-Borane and N-Chloroborane Adducts Derived from 1,3,5-Heterocyclohexanes. Eur. J. Inorg. Chem. 1999, 1999, 2069–2073. [Google Scholar] [CrossRef]
- Peters, K.; Peters, E.-M.; Drinkuth, S.; Groetsch, S.; Christi, M. Crystal structure of (la,5β,8β,8aβ)-1-methyl-1,2,3,5,8,8a-hexahydro-5,8- epoxychinolin(N-B)boran, C9H10NO(CH3)(BH3). Z. Krist. New Cryst. Struct. 2000, 215, 600–602. [Google Scholar] [CrossRef]
- Peters, E.-M.; Peters, K.; Groetsch, S.; Christi, M. Crystal structure of (1α,2ß,8α)-2-methyl-8-phenyl-2-azabicyclo[4.2.0]oct- 5-ene(N-B)boran, (C6H5)C7H9N(CH3)(BH3). Z. Krist. New Cryst. Struct. 2001, 216, 121–122. [Google Scholar] [CrossRef]
- Kawaguchi, K. Fourier transform infrared spectroscopy of the BH3 ν3 band. J. Chem. Phys. 1992, 96, 3411–3415. [Google Scholar] [CrossRef]
- Jones, D.S.; Lipscomb, W.N. Analysis of diborane X-ray diffraction data utilizing structure factors calculated from molecular wave functions. Acta Crystallogr. Sect. A 1970, 26, 196–207. [Google Scholar] [CrossRef]
- Milovanović, M.M.; Andrić, J.M.; Medaković, V.B.; Djukic, J.-P.; Zarić, S.D. Investigation of interactions in Lewis pairs between phosphines and boranes by analyzing crystal structures from the Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2018, 74, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, P.R.; Kim, S.-K.; Schulze, V.K.; White, J.D.; Yokochi, A.F.T. Asymmetric synthesis of (+)-loline, a pyrrolizidine alkaloid from rye grass and tall fescue. J. Chem. Soc. Perkin Trans. 1 2001, 1831–1847. [Google Scholar] [CrossRef]
- Groselj, U.; Bevk, D.; Jakse, R.; Meden, A.; Stanovnik, B.; Svete, J. CCDC 286431: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2006. [Google Scholar]
- White, J.M.; McClure, M.S. CCDC 1026489: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2014. [Google Scholar]
- Gainsford, G.J.; Luxenburger, A.; Woolhouse, A.D. CCDC 790649: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2010. [Google Scholar]
- Pinaka, A.; Vougioukalakis, G.; Dimotikali, D.; Psyharis, V.; Papadopoulos, K. A Convenient One-Step Synthesis of Stable β-Amino Alcohol N-Boranes from α-Amino Acids. Synthesis 2012, 44, 1057–1062. [Google Scholar] [CrossRef]
- Apex 3, Saint; Bruker AXS Inc.: Madison, WI, USA, 2016.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.D.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Cryst. 2008, 41, 96–103. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C18H27BN2O2 |
| Formula weight | 314.24 g/mol |
| Temperature | 180 K |
| Wavelength | 1.54178 Å |
| Crystal system | Orthorhombic |
| Space group | P 21 21 21 |
| Unit cell dimensions | a = 9.6759(14) Å |
| b = 11.8394(16) Å | |
| c = 14.752(3) Å | |
| V | 1690.0(7) Å3 |
| Z | 4 |
| Density (calculated) | 1.235 g/cm3 |
| Absorption coefficient | 0.623 mm−1 |
| F000 | 680.0 |
| Crystal size | 0.152 × 0.280 × 0.408 mm3 |
| Theta range for data collection | 4.79° to 72.147° |
| Index ranges | −11 ≤ h ≤ 11, −14 ≤ k ≤ 14, −17 ≤ l ≤ 18 |
| Reflections collected | 20314 |
| Independent reflections | 3301 [Rint = 0.036] |
| Completeness to θ = 72.147° | 99.999% |
| Max. and min. transmission | 0.75 and 0.91 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3301/5/226 |
| Goodness-of-fit on F2 | 1.001 |
| Final R indices [I > 2σ(I)] | R1 = 0.0311, wR2 = 0.0731 |
| R indices (all data) | R1 = 0.0296, wR2 = 0.0700 |
| Extinction coefficient | 189(4) |
| Largest diff. peak and hole | 0.19 and −0.15 e·Å−3 |
| Flack x | 0.06(18) |
| Compound | d(B–N) (Å) | d(B–H) (Å) |
|---|---|---|
| 4 | 1.6358(18) | 1.144–1.160(13) |
| QOPGOR | 1.608(4) | 0.96 * |
| XANFUN | 1.621(3) | 0.96 * |
| ABUKAJ | 1.600(3) | 1.154(16) |
| DAZPIE | 1.580(6), 1.574(5) | 1.057–1.155(8), 0.97–1.21(8) |
| DORHAV | 1.606(4) | 1.02–1.13(3) |
| DUZNUI | 1.627(3) | 1.10–1.21(3) |
| WANTAI | 1.610(4) | 1.14–1.18(2) |
| D–H … A* | D–H (Å) | D … A (Å) | H … A (Å) | D–H … A (°) | D … B (Å) | A…H…A′ (°) |
|---|---|---|---|---|---|---|
| 4 | ||||||
| O1–H12 … H14 | 0.826(15) | 2.983(17) | 2.27(2) | 145(2) | 3.3911(18) | |
| O1–H12 … H15 | “ | 2.809(17) | 2.04(2) | 156(2) | 3.3911(18) | 51.2(7) |
| C1–H11 … H13 | 0.938 | 3.184(17) | 2.398(17) | 141.3(3) | 3.605(2) | |
| ABUKAJ | ||||||
| O17–H17O … H18B | 0.91(3) | 2.881(15) | 2.07(4) | 148(3) | 3.305 | |
| O17–H17O … H18C | “ | 2.738(11) | 1.93(3) | 148(3) | “ | 56.1(10) |
| DAZPIE | ||||||
| O22–H2 … H1A | 0.97 | 3.00 | 2.14 | 147 | 3.394(9) | |
| O22–H2 … H1B | “ | 2.76 | 2.11 | 122 | “ | 52 |
| O12–H1 … H2A | 1.12 | 2.890 | 2.28 | 112 | 3.408(8) | |
| O12–H1 … N21 | “ | 3.460(4) | 2.35 | 171 | - | 76 |
| DORHAV | ||||||
| O1–H1 … H91 | 0.78(3) | 2.80(3) | 2.05(4) | 165(3) | 3.249(5) | |
| DUZNUI | ||||||
| O2–H1 … H1B1 | 0.89(3) | 2.62(2) | 1.76(4) | 162(3) | 3.498(3) | |
| WANTAI | ||||||
| O–H10 … H2B | 0.88(3) | 2.68(2) | 1.81(4) | 173(3) | 3.264(3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čejka, J.; Cvak, L.; Žižková, S.; Kratochvíl, B.; Jegorov, A. Dihydrogen Bond in the Aminoborane Complex of a Nicergoline Intermediate. Molecules 2019, 24, 2548. https://doi.org/10.3390/molecules24142548
Čejka J, Cvak L, Žižková S, Kratochvíl B, Jegorov A. Dihydrogen Bond in the Aminoborane Complex of a Nicergoline Intermediate. Molecules. 2019; 24(14):2548. https://doi.org/10.3390/molecules24142548
Chicago/Turabian StyleČejka, Jan, Ladislav Cvak, Simona Žižková, Bohumil Kratochvíl, and Alexandr Jegorov. 2019. "Dihydrogen Bond in the Aminoborane Complex of a Nicergoline Intermediate" Molecules 24, no. 14: 2548. https://doi.org/10.3390/molecules24142548
APA StyleČejka, J., Cvak, L., Žižková, S., Kratochvíl, B., & Jegorov, A. (2019). Dihydrogen Bond in the Aminoborane Complex of a Nicergoline Intermediate. Molecules, 24(14), 2548. https://doi.org/10.3390/molecules24142548