
Hybrid Solid-Phase Extraction for Selective Determination of Methamphetamine and Amphetamine in Dyed Hair by Using Gas Chromatography–Mass Spectrometry


Abstract
1. Introduction
2. Results and Discussion
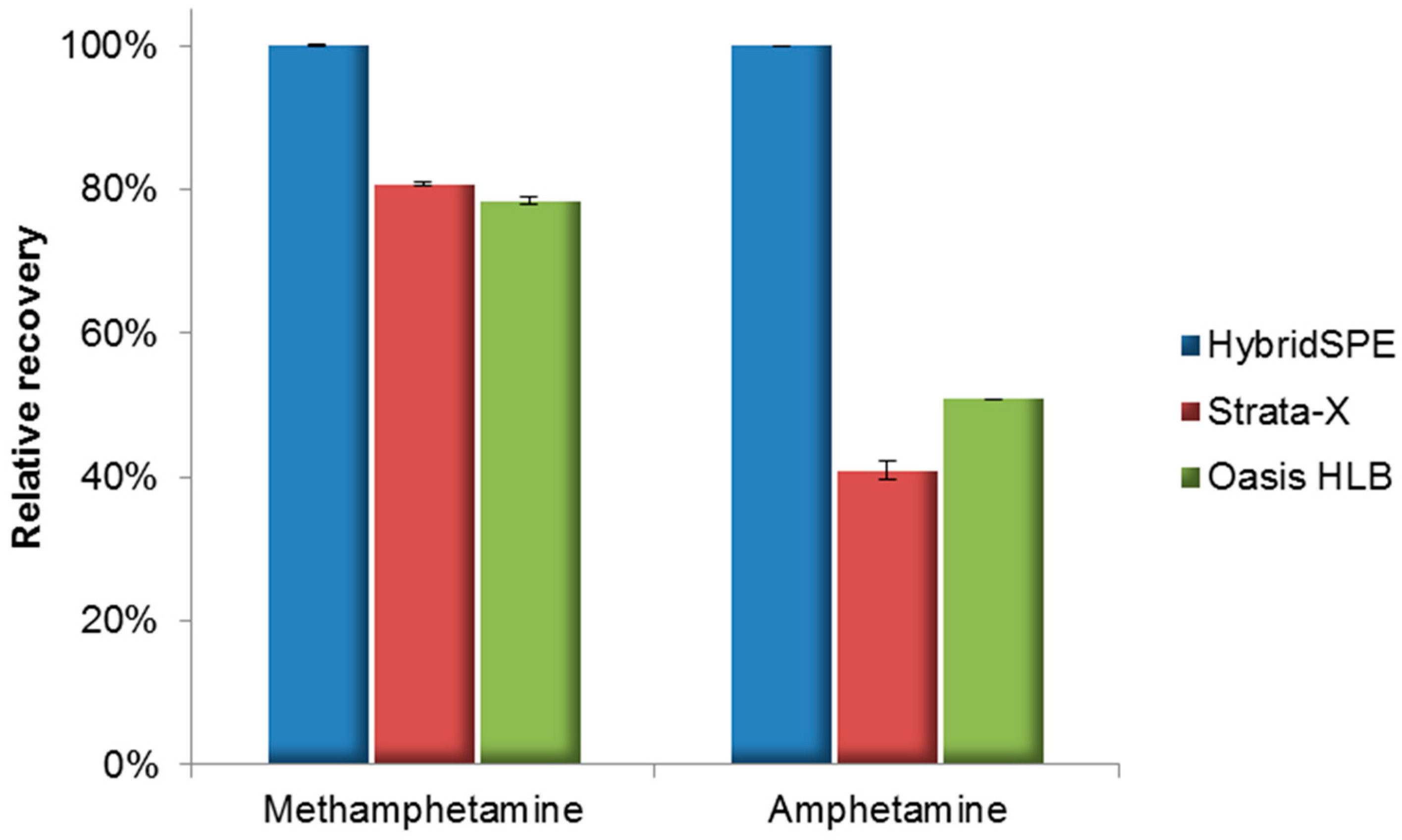
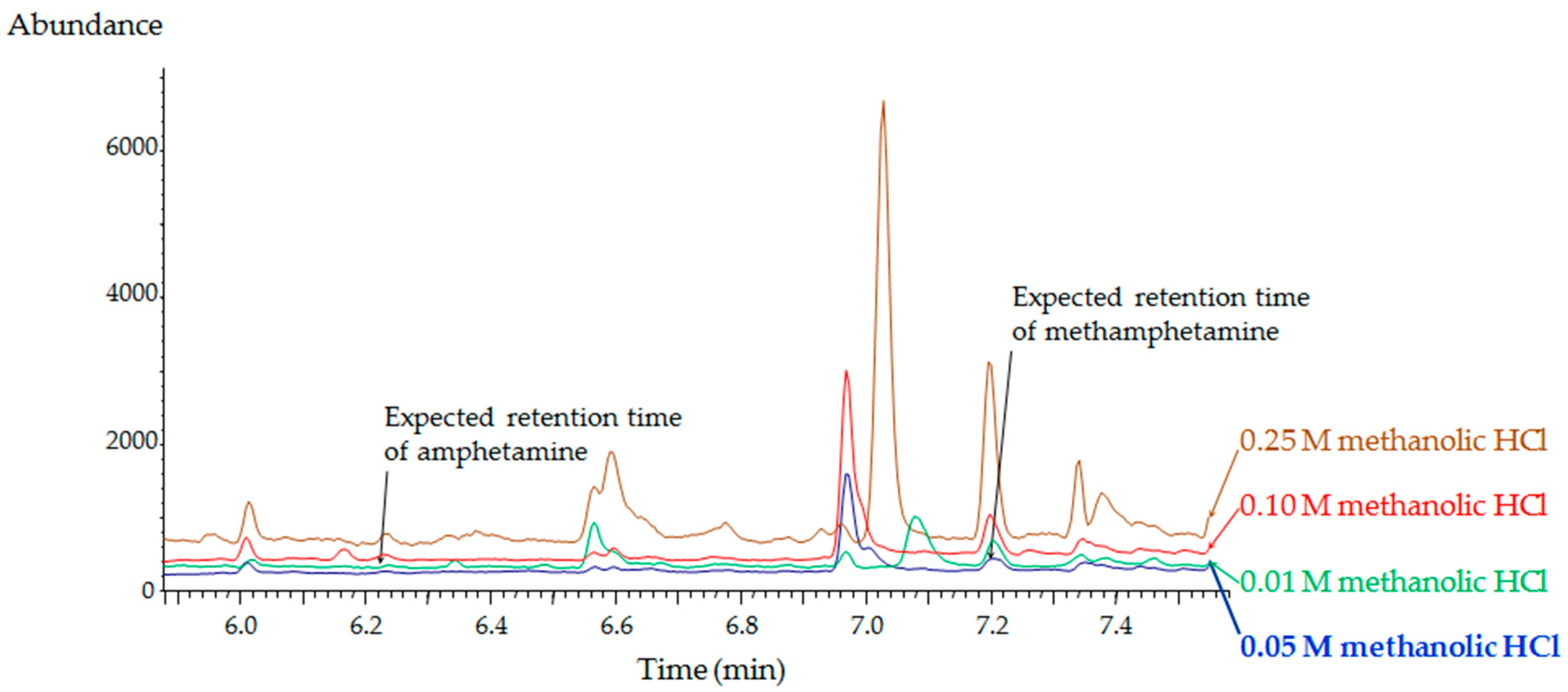
2.1. Method Development
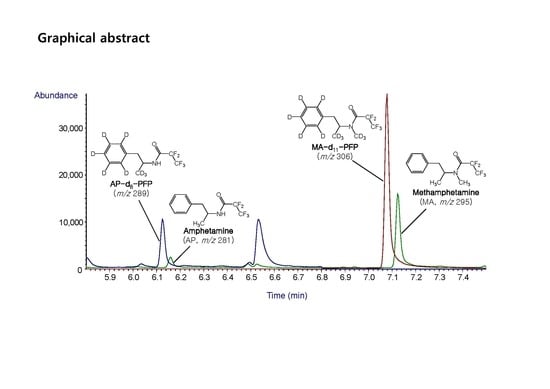
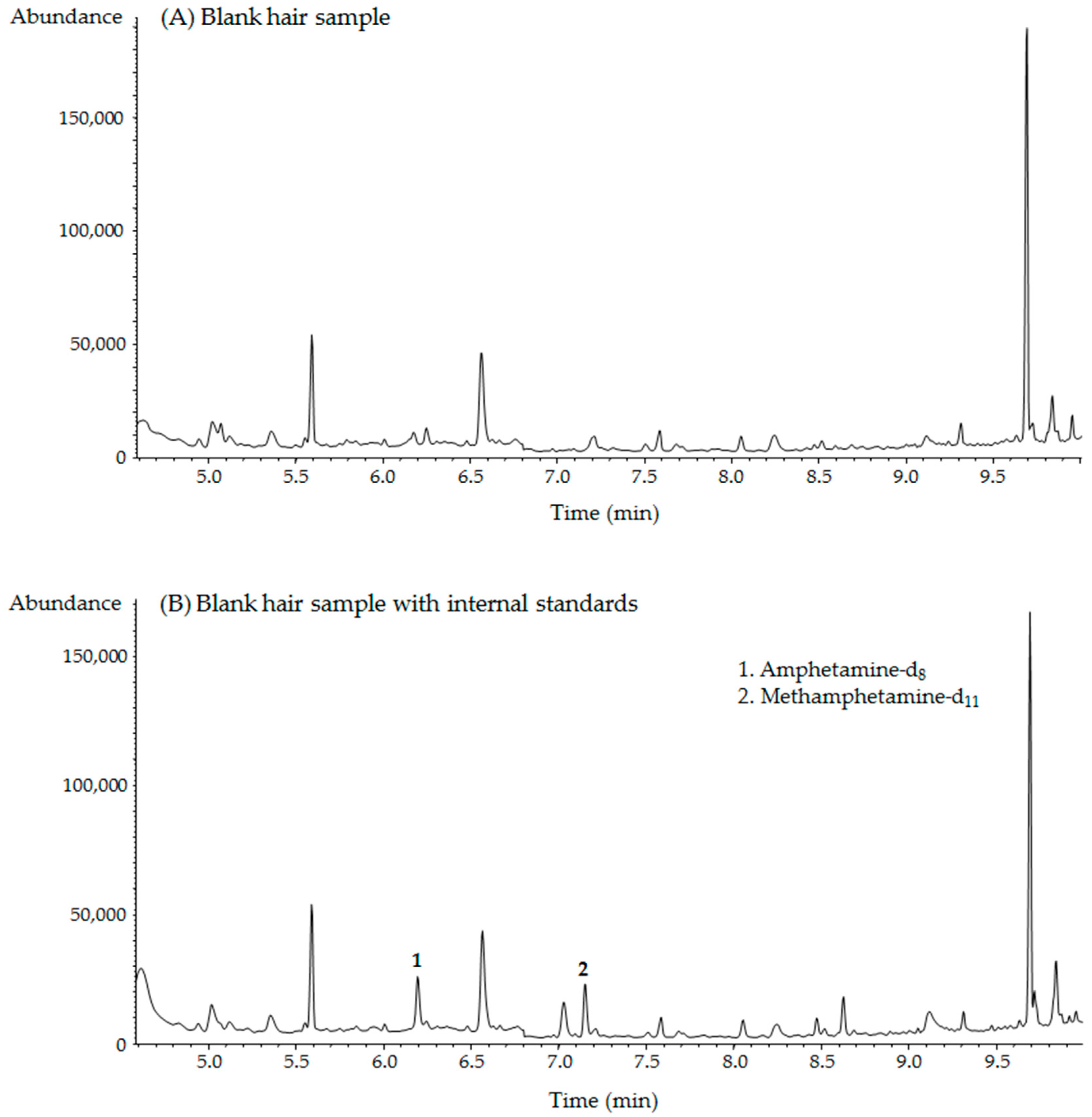
2.2. GC–MS Analysis
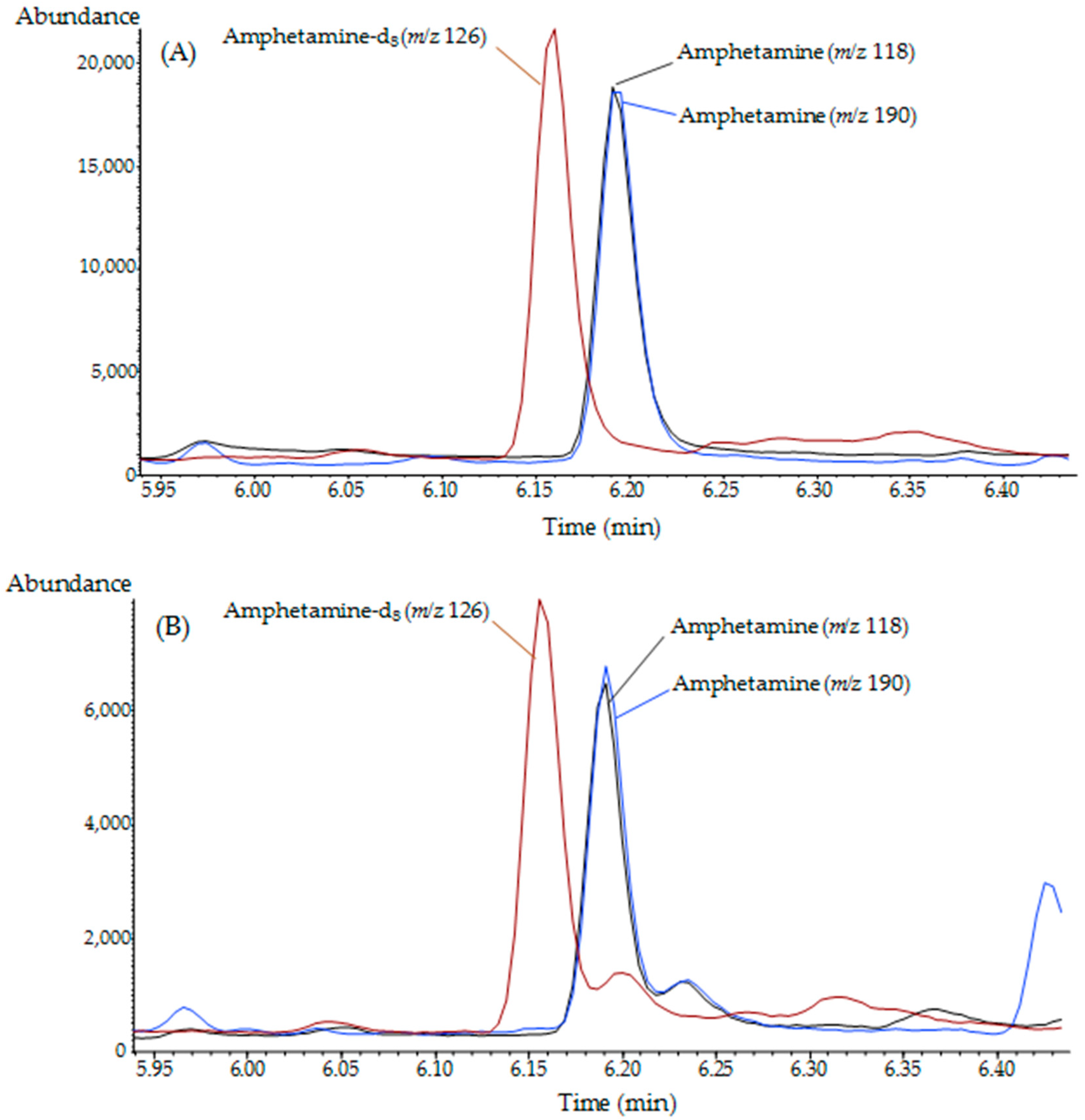
2.3. Method Validation
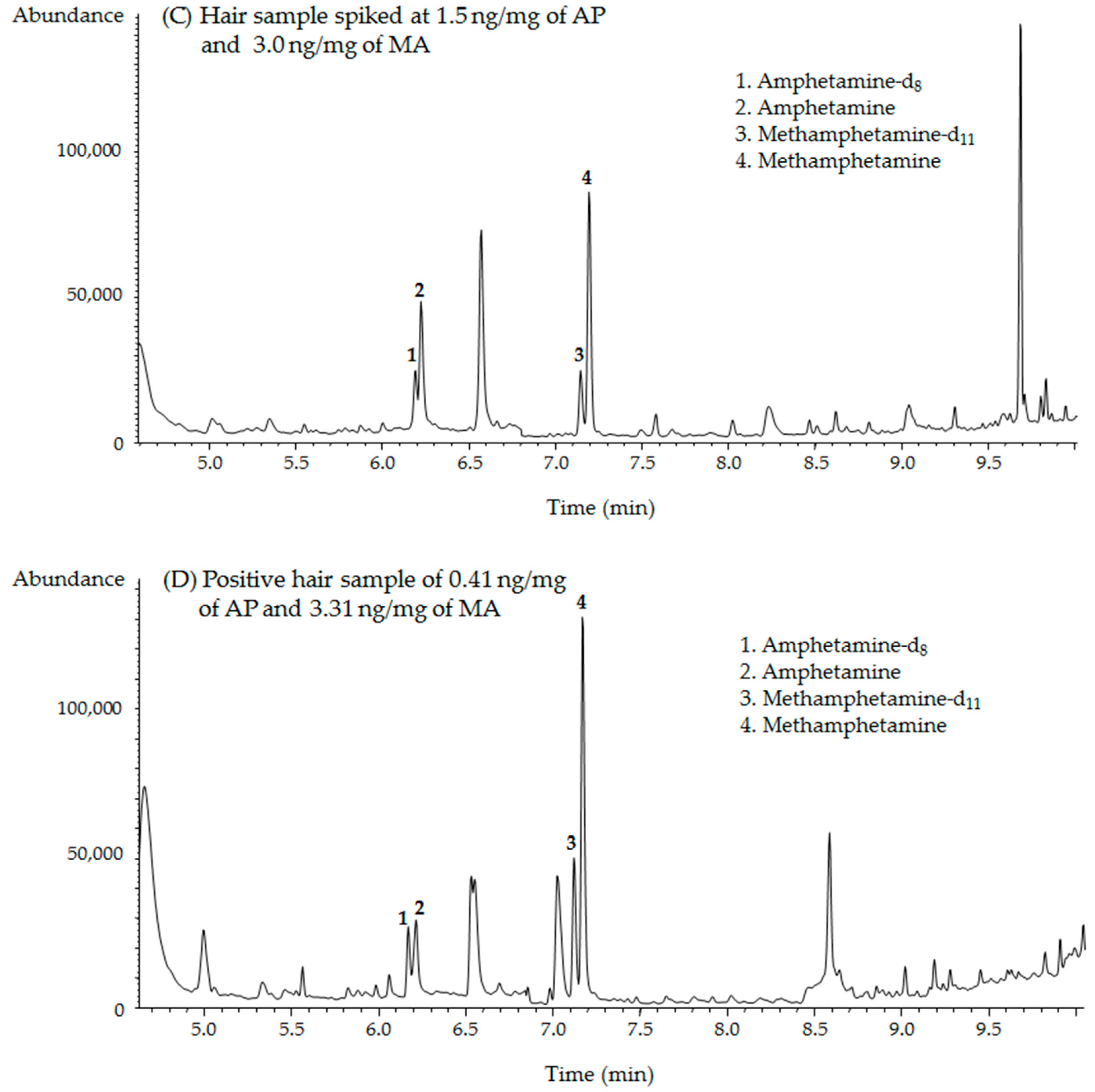
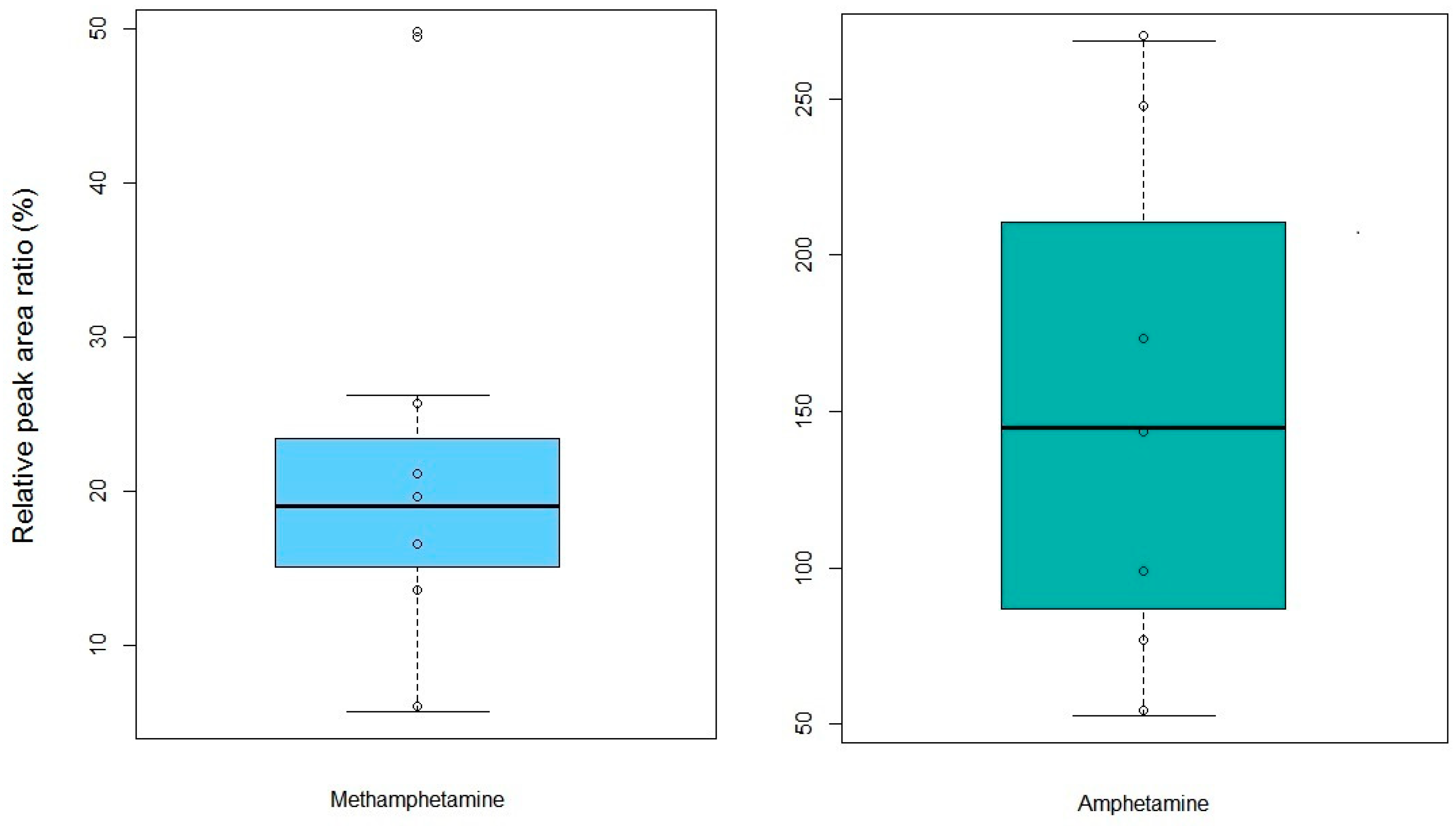
2.4. Forensic Applications
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Hair Specimens
4.3. Sample Preparation
4.4. GC–MS analysis
4.5. Method validation
Author Contributions
Funding
Conflicts of Interest
References
- United Nations Office on Drugs and Crime: “Methamphetamine Continues to Dominate Synthetic Drug Markets”, Global Smart Update Volume 20, August 2018. Available online: https://www.unodc.org/documents/scientific/Global_Smart_Update_20_web.pdf (accessed on 15 May 2019).
- Ando, E.; Hayashida, M.; Nihira, M.; Yamada, T.; Ohno, Y. GC-MS analysis of methamphetamine and amphetamine in hair of Thai drug addicts. J. Alcohol Stud. Drug Depend. 2004, 39, 168–179. [Google Scholar]
- Supreme Prosecutors’ Office: 2017 White Paper on Drug-Related Crimes. Available online: http://antidrug.drugfree.or.kr/page/?mIdx=190&mode=view&idx=12433&retUrl=mIdx%3D190 (accessed on 15 May 2019).
- Supreme Prosecutors’ Office: 2016 White Paper on Drug-Related Crimes. Available online: http://antidrug.drugfree.or.kr/page/?mIdx=190&mode=view&idx=10839&retUrl=mIdx%3D190%26pIdx%26field%26keyword%3D%25EB%25A7%2588%25EC%2595%25BD%25EB%25A5%2598%2B%25EB%25B2%2594%25EC%25A3%2584%26page%3D3 (accessed on 15 May 2019).
- Clauwaert, K.M.; Van Bocxlaer, J.F.; Lambert, W.E.; De Leenheer, A.P. Segmental analysis for cocaine and metabolites by HPLC in hair of suspected drug overdose cases. Forensic Sci. Int. 2000, 110, 157–166. [Google Scholar] [CrossRef]
- Wu, Y.H.; Lin, K.L.; Chen, S.C.; Chang, Y.Z. Simultaneous quantitative determination of amphetamines, ketamine, opiates and metabolites in human hair by gas chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Lin, K.L.; Chen, S.C.; Chang, Y.Z. Integration of GC/EI-MS and GC/NCI-MS for simultaneous quantitative determination of opiates, amphetamines, MDMA, ketamine, and metabolites in human hair. J. Chromatogr. B 2008, 870, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Meng, P.; Zhu, D.; He, H.; Wang, Y.; Guo, F.; Zhang, L. Dtermination of amphetamines in hair by GC/MS after small-volume liquid extraction and microwave derivatization. Anal. Sci. 2009, 25, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Johansen, S.S.; Jornil, J. Determination of amphetamine, methamphetamine, MDA and MDMA in human hair by GC-EI-MS after derivatization with perfluorooctanoyl chloride. Scand. J. Clin. Lab. Invest. 2009, 69, 113–120. [Google Scholar] [CrossRef]
- Kintz, P. Analytical and Practical Aspects of Drug Testing in Hair; CRC Press: Boca Raton, FL, USA, 2006; pp. 143–162. [Google Scholar]
- Cooper, G.A.; Kronstrand, R.; Kintz, P. Society of Hair Testing guidelines for drug testing in hair. Forensic Sci. Int. 2012, 218, 20–24. [Google Scholar] [CrossRef]
- Stout, P.R. Hair Testing for Drugs—Challenges for Interpretation. Forensic Sci. Rev. 2007, 19, 69–84. [Google Scholar]
- Skopp, G.; Pötsch, L.; Moeller, M.R. On cosmetically treated hair—Aspects and pitfalls of interpretation. Forensic Sci. Int. 1997, 84, 43–52. [Google Scholar] [CrossRef]
- Holme, D.J.; Peck, H. Analytical Biochemistry, 3rd ed.; Prentice Hall: Essex, UK, 1998; pp. 91–92. [Google Scholar]
- Kim, J.Y.; Shin, S.H.; Lee, J.I.; In, M.K. Rapid and simple determination of psychotropic phenylalkylamine derivatives in human hair by gas chromatography–mass spectrometry using micro-pulverized extraction. Forensic Sci. Int. 2010, 196, 43–50. [Google Scholar] [CrossRef]
- Van Elsué, N.; Yegles, M. Influence of cosmetic hair treatments on cannabinoids in hair: Bleaching, perming and permanent coloring. Forensic Sci. Int. 2019, 297, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Baeck, S.; Han, E.; Chung, H.; Pyo, M. Effects of repeated hair washing and a single hair dyeing on concentrations of methamphetamine and amphetamine in human hairs. Forensic Sci. Int. 2011, 206, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Hyötyläinen, T. Critical evaluation of sample pretreatment techniques. Anal. Bioanal. Chem. 2009, 394, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Plotka-Wasylka, J.; Szczepańska, N.; de la Guardia, M.; Namieśnik, J. Miniaturized solid-phase extraction techniques. Trends Anal. Chem. 2015, 73, 19–38. [Google Scholar] [CrossRef]
- Hennion, M.C. Solid-phase extraction: method development, sorbents, and coupling with liquid chromatography. J. Chromatogr. A 1999, 856, 3–54. [Google Scholar] [CrossRef]
- Bylda, C.; Thiele, R.; Kobold, U.; Volmer, D.A. Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC-MS/MS. Analyst 2014, 139, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Ariffin, M.M.; Miller, E.I.; Cormack, P.A.; Anderson, R.A. Molecularly imprinted solid-phase extraction of diazepam and its metabolites from hair samples. Anal. Chem. 2007, 79, 256–262. [Google Scholar] [CrossRef]
- Thibert, V.; Legeay, P.; Chapuis-Hugon, F.; Pichon, V. Synthesis and characterization of molecularly imprinted polymers for the selective extraction of cocaine and its metabolite benzoylecgonine from hair extract before LC–MS analysis. Talanta 2012, 88, 412–419. [Google Scholar] [CrossRef]
- Pujadas, M.; Pichini, S.; Poudevida, S.; Menoyo, E.; Zuccaro, P.; Farré, M.; de la Torre, R. Development and validation of a gas chromatography-mass spectrometry assay for hair analysis of amphetamine, methamphetamine and methylenedioxy derivatives. J. Chromatogr. B 2003, 798, 249–255. [Google Scholar] [CrossRef]
- Míguez-Framil, M.; Moreda-Piñeiro, A.; Bermejo-Barrera, P.; Álvarez-Freire, I.; Tabernero, M.J.; Bermejo, A.M. Matrix solid-phase dispersion on column clean-up/pre-concentration as a novel approach for fast isolation of abuse drugs from human hair. J. Chromatogr. A 2010, 1217, 6342–6349. [Google Scholar] [CrossRef]
- Lendoiro, E.; Quintela, Ó.; de Castro, A.; Cruz, A.; López-Rivadulla, M.; Elena, M.C. Target screening and confirmation of 35 licit and illicit drugs and metabolites in hair by LC–MSMS. Forensic Sci. Int. 2012, 217, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Miyaguchi, H.; Kakuta, M.; Iwata, Y.T.; Matsuda, H.; Tazawa, H.; Kimura, H.; Inoue, H. Development of a micropulverized extraction method for rapid toxicological analysis of methamphetamine in hair. J. Chromatogr. A 2007, 1163, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Tzatzarakis, M.N.; Alegakis, A.K.; Kavvalakis, M.P.; Vakonaki, E.; Stivaktakis, P.D.; Kanaki, K.; Vardavas, A.I.; Barbounis, E.G.; Tsatsakis, A.M. Comparative evaluation of drug deposition in hair samples collected from different anatomical body sites. J. Anal. Toxicol. 2017, 41, 214–223. [Google Scholar] [CrossRef][Green Version]
- Gentili, S.; Torresi, A.; Marsili, R.; Chiarotti, M.; Macchia, T. Simultaneous detection of amphetamine-like drugs with headspace solid-phase microextraction and gas chromatography–mass spectrometry. J. Chromatogr. B 2002, 780, 183–192. [Google Scholar] [CrossRef]
- Puccia, V.; Di Palma, S.; Alfieri, A.; Bonelli, F.; Monteagudo, E. A novel strategy for reducing phospholipids-based matrix effect in LC–ESI-MS bioanalysis by means of HybridSPE. J. Pharm. Biomed. Anal. 2009, 50, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Kalra, H.; Gupta, A.; Raut, B.; Hussain, A.; Rahman, A. HybridSPE: A novel technique to reduce phospholipid-based matrix effect in LC-ESI-MS bioanalysis. J. Pharm. Bioallied. Sci. 2012, 4, 267–275. [Google Scholar]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef]
- Society of Forensic Toxicologists/American Academy of Forensic Sciences: The Forensic Toxicology Laboratory Guidelines. Available online: http://soft-tox.org/files/Guidelines_2006_Final.pdf (accessed on 15 May 2019).
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
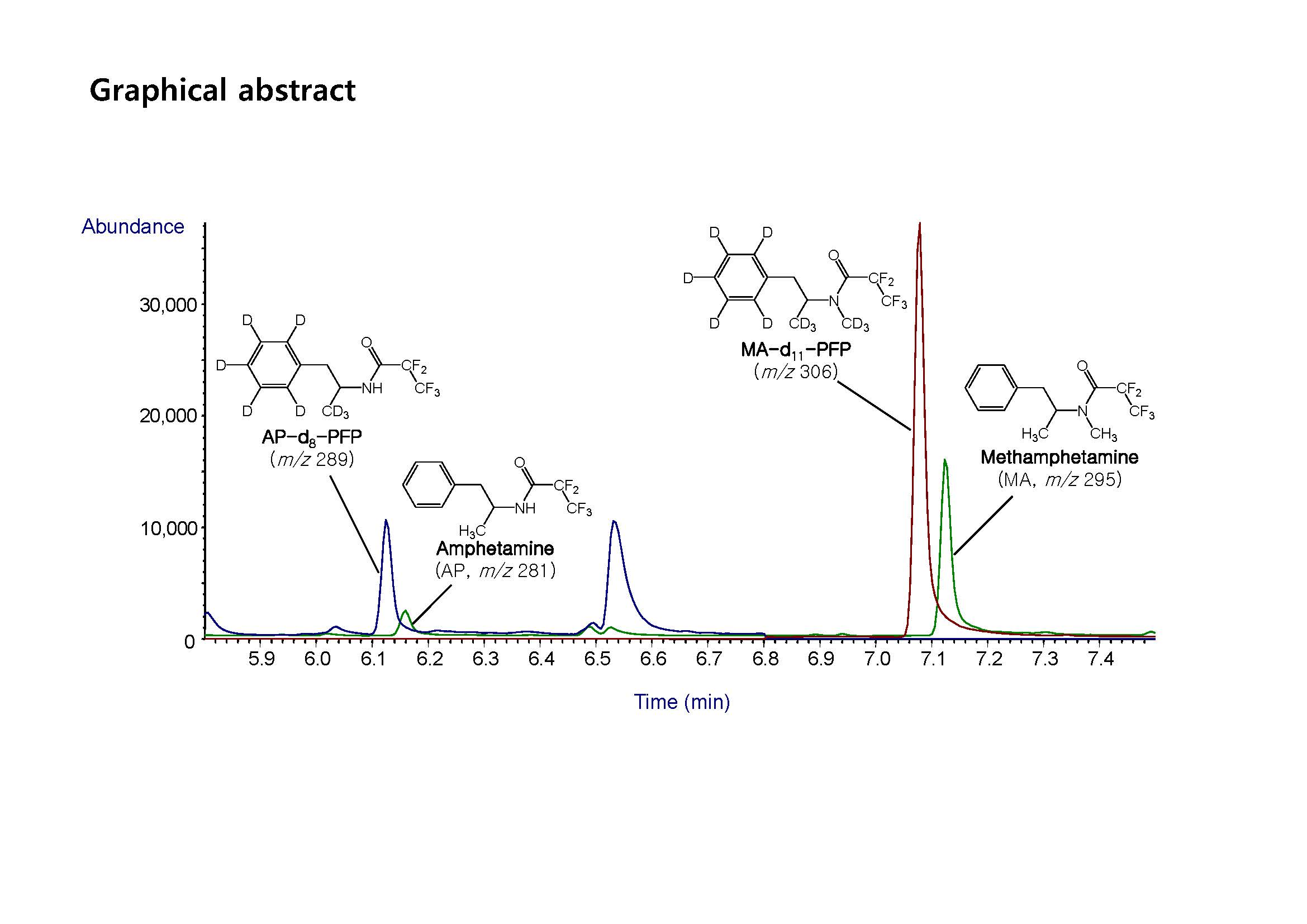
| Compound | Retention Time (min) | Molecular Weight | Ions Monitored (m/z) | ||
|---|---|---|---|---|---|
| Quantifier Ion | Qualifier Ions | ||||
| AP-d8 | 6.19 | 289 | 126 | - | - |
| AP | 6.22 | 281 | 118 | 190 | 91 |
| MA-d11 | 7.15 | 306 | 210 | - | - |
| MA | 7.20 | 295 | 118 | 204 | 160 |
| Compound | Concentration Range (ng/mg) | Slope (Mean ± SD) | y-Intercept (mean) | Linearity 1 (R2) | LOD 2 (ng/mg) | LLOQ 3 (ng/mg) |
|---|---|---|---|---|---|---|
| AP | 0.05–5.0 | 1.1968 ± 0.0553 | 0.0178 | 0.9998 | 0.016 | 0.05 |
| MA | 0.1–10.0 | 0.2826 ± 0.0205 | 0.0321 | 0.9999 | 0.031 | 0.10 |
| Compound | QC (ng/mg) | Recovery (%) | Intra-Day (n = 7) | Inter-Day (n = 28) | ||
|---|---|---|---|---|---|---|
| Precision 1 (% CV) | Accuracy 2 (% bias) | Precision (% CV) | Accuracy (% bias) | |||
| AP | 0.15 | 87.1 | 2.2 | −9.4 | 6.7 | 0.3 |
| 1.5 | 96.8 | 1.4 | −0.8 | 3.1 | −2.2 | |
| 3 | 91.8 | 0.4 | −4.2 | 1.8 | −5.1 | |
| MA | 0.3 | 83.4 | 8.3 | 5.5 | 5.9 | 3.1 |
| 3 | 94.8 | 1.4 | −1.0 | 4.8 | −1.0 | |
| 6 | 92.0 | 0.7 | −5.8 | 3.9 | −4.7 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, N.H.; Lee, Y.R.; Kim, H.S.; Cheong, J.C.; Kim, J.Y. Hybrid Solid-Phase Extraction for Selective Determination of Methamphetamine and Amphetamine in Dyed Hair by Using Gas Chromatography–Mass Spectrometry. Molecules 2019, 24, 2501. https://doi.org/10.3390/molecules24132501
Kwon NH, Lee YR, Kim HS, Cheong JC, Kim JY. Hybrid Solid-Phase Extraction for Selective Determination of Methamphetamine and Amphetamine in Dyed Hair by Using Gas Chromatography–Mass Spectrometry. Molecules. 2019; 24(13):2501. https://doi.org/10.3390/molecules24132501
Chicago/Turabian StyleKwon, Nam Hee, Yu Rim Lee, Hee Seung Kim, Jae Chul Cheong, and Jin Young Kim. 2019. "Hybrid Solid-Phase Extraction for Selective Determination of Methamphetamine and Amphetamine in Dyed Hair by Using Gas Chromatography–Mass Spectrometry" Molecules 24, no. 13: 2501. https://doi.org/10.3390/molecules24132501
APA StyleKwon, N. H., Lee, Y. R., Kim, H. S., Cheong, J. C., & Kim, J. Y. (2019). Hybrid Solid-Phase Extraction for Selective Determination of Methamphetamine and Amphetamine in Dyed Hair by Using Gas Chromatography–Mass Spectrometry. Molecules, 24(13), 2501. https://doi.org/10.3390/molecules24132501