
Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—A Brief Look at Progress on Small Molecules, Peptides and Macrocycles

 , , , ,
, , , ,
Abstract
1. Introduction
2. Inhibitors of the PD-1/PD-L1 Interaction
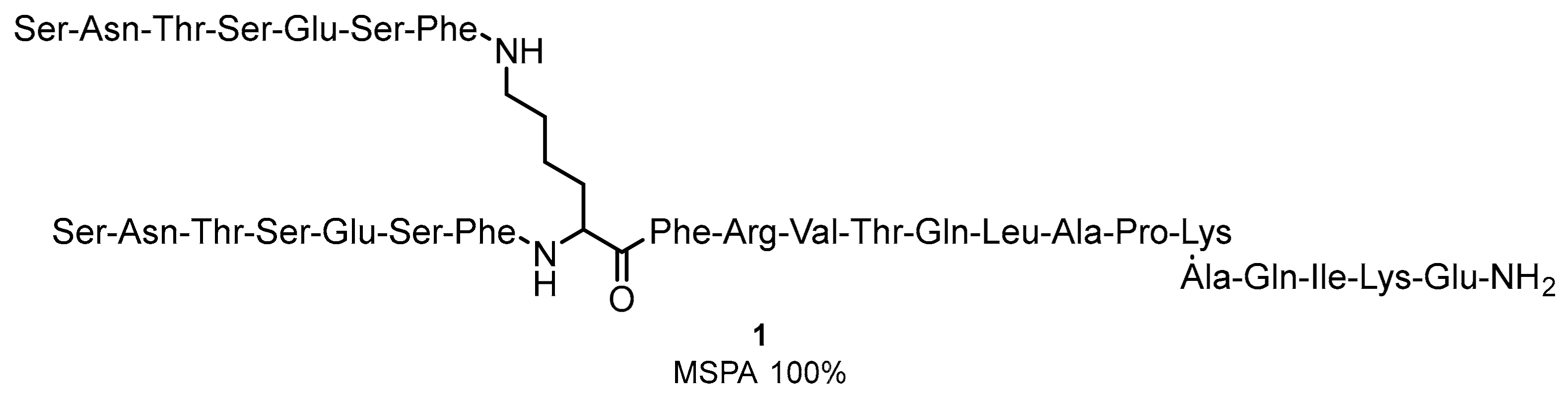
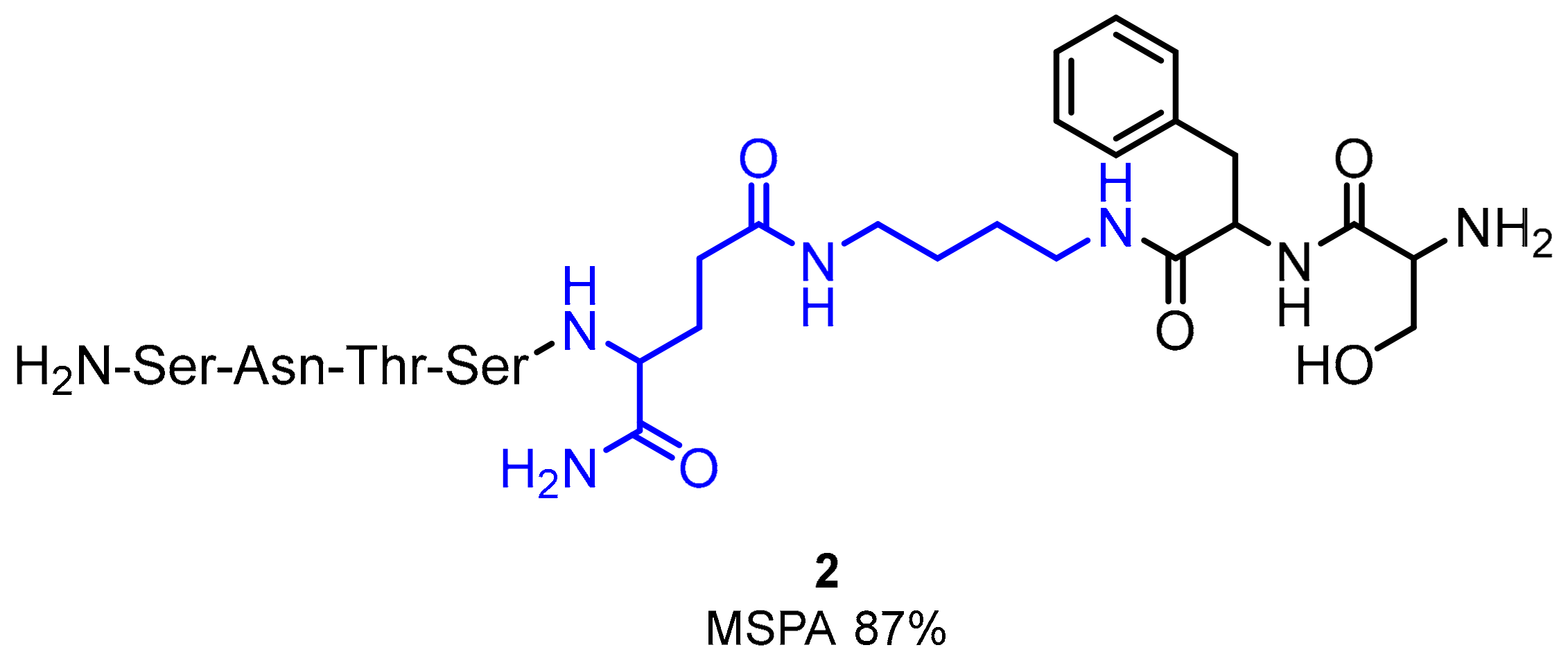
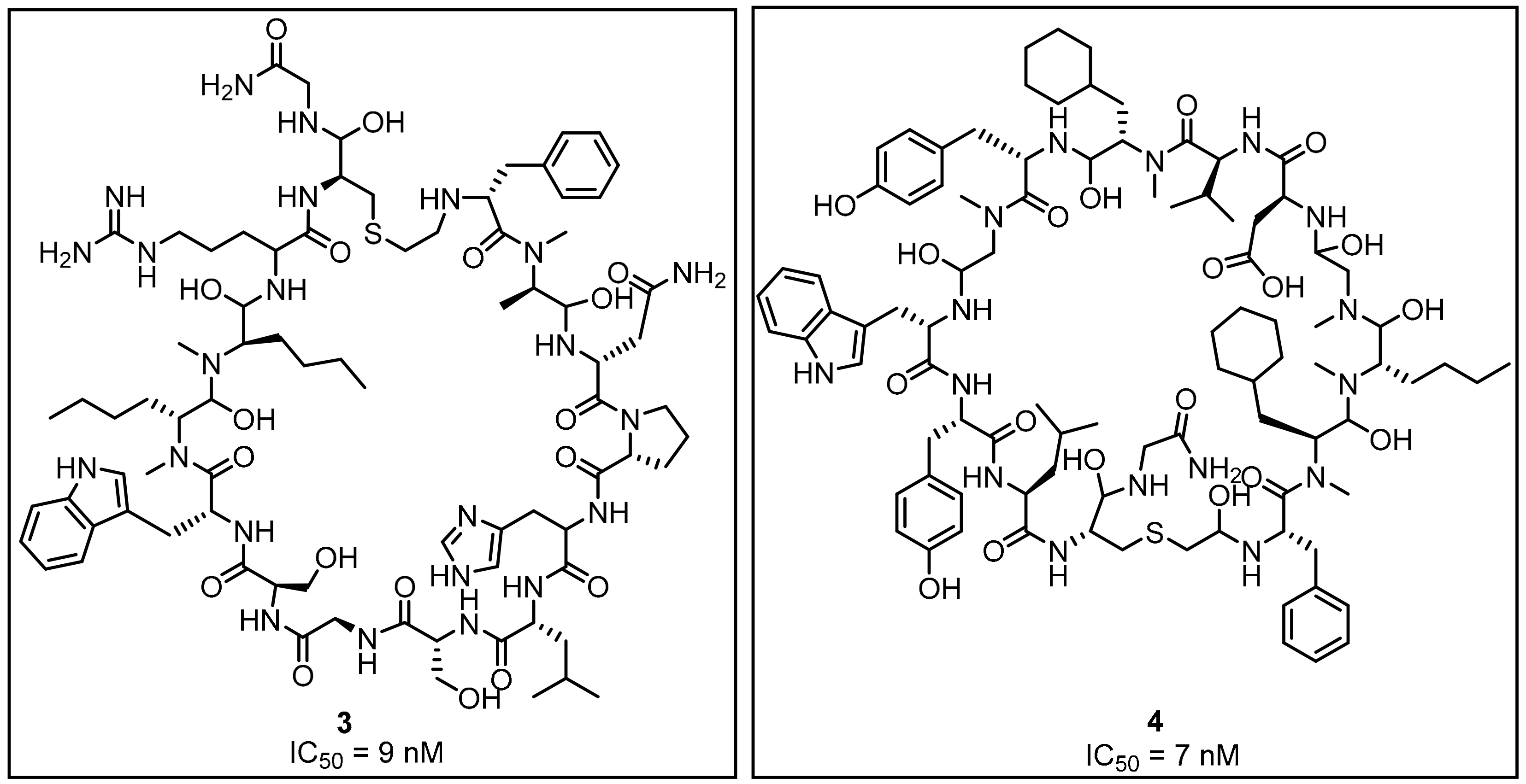
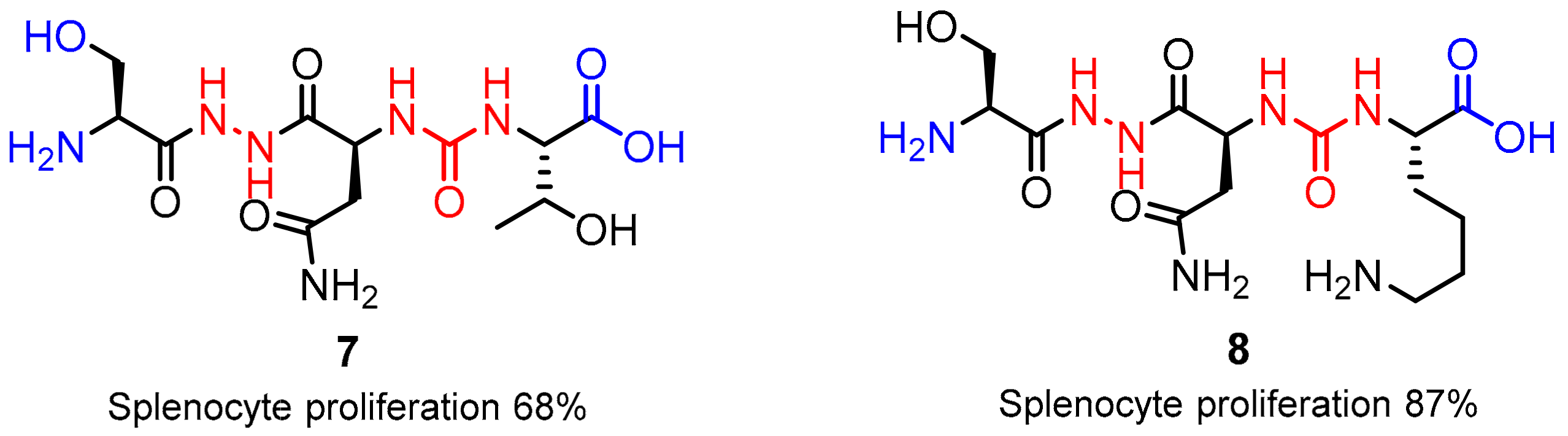
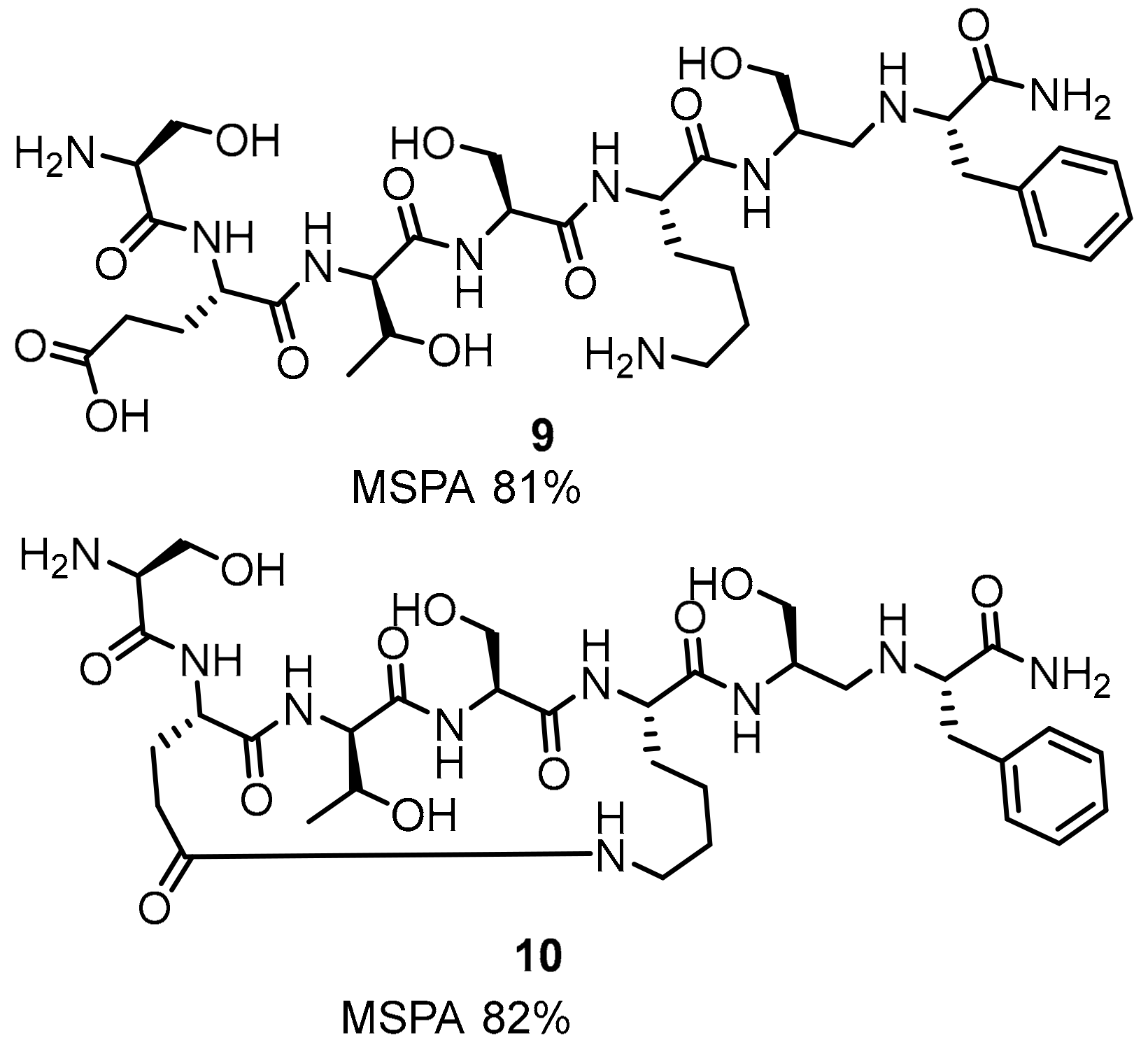
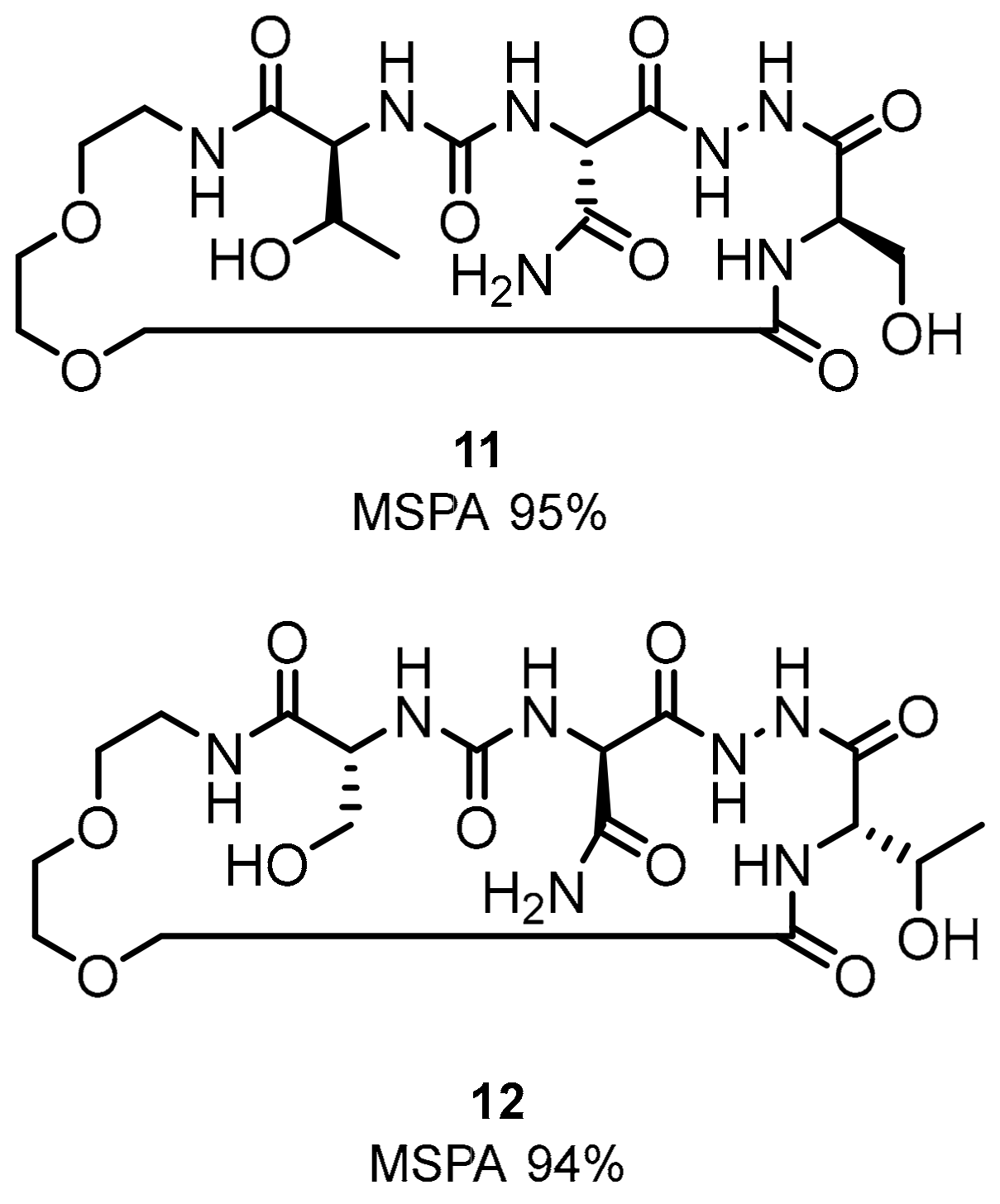
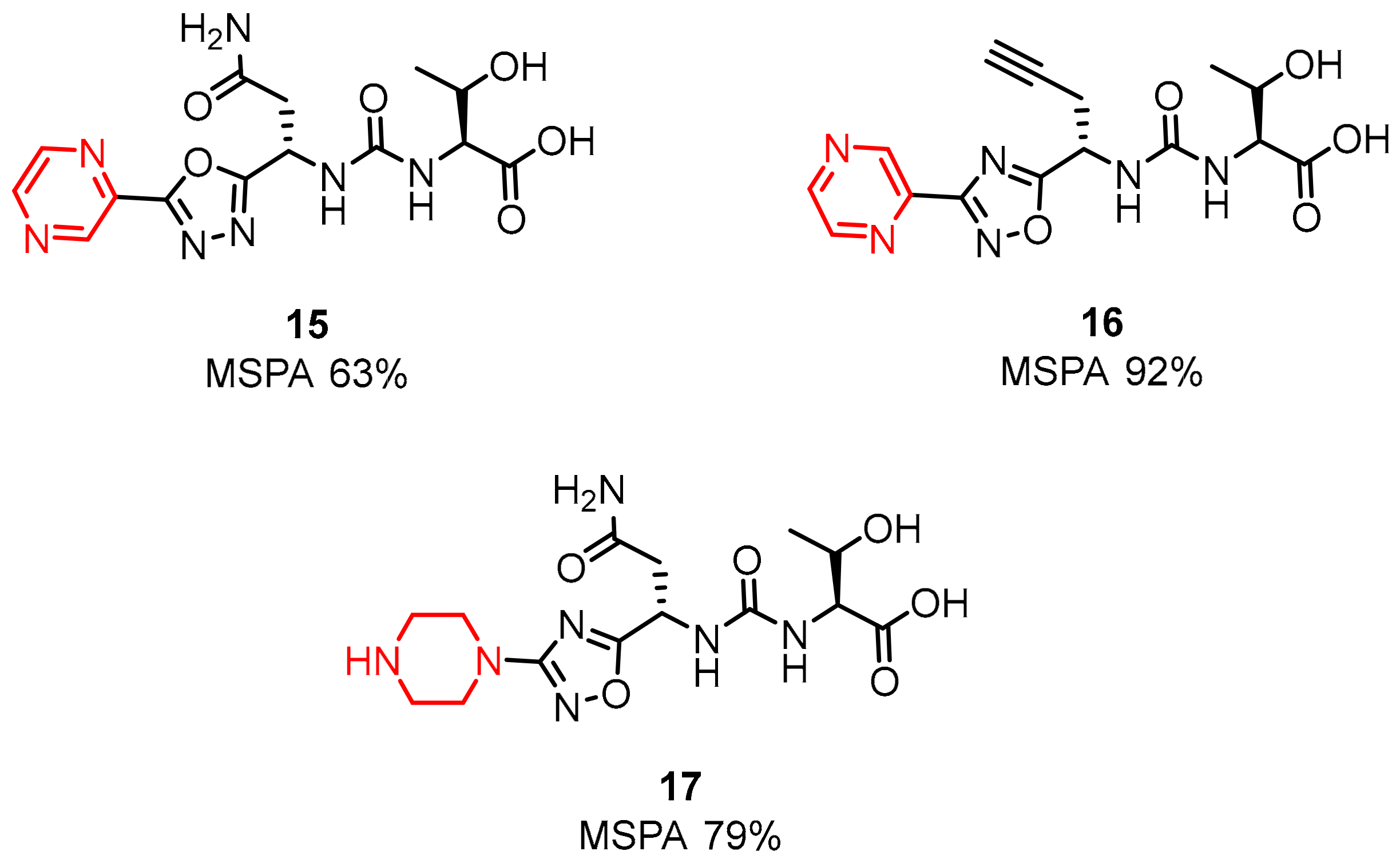
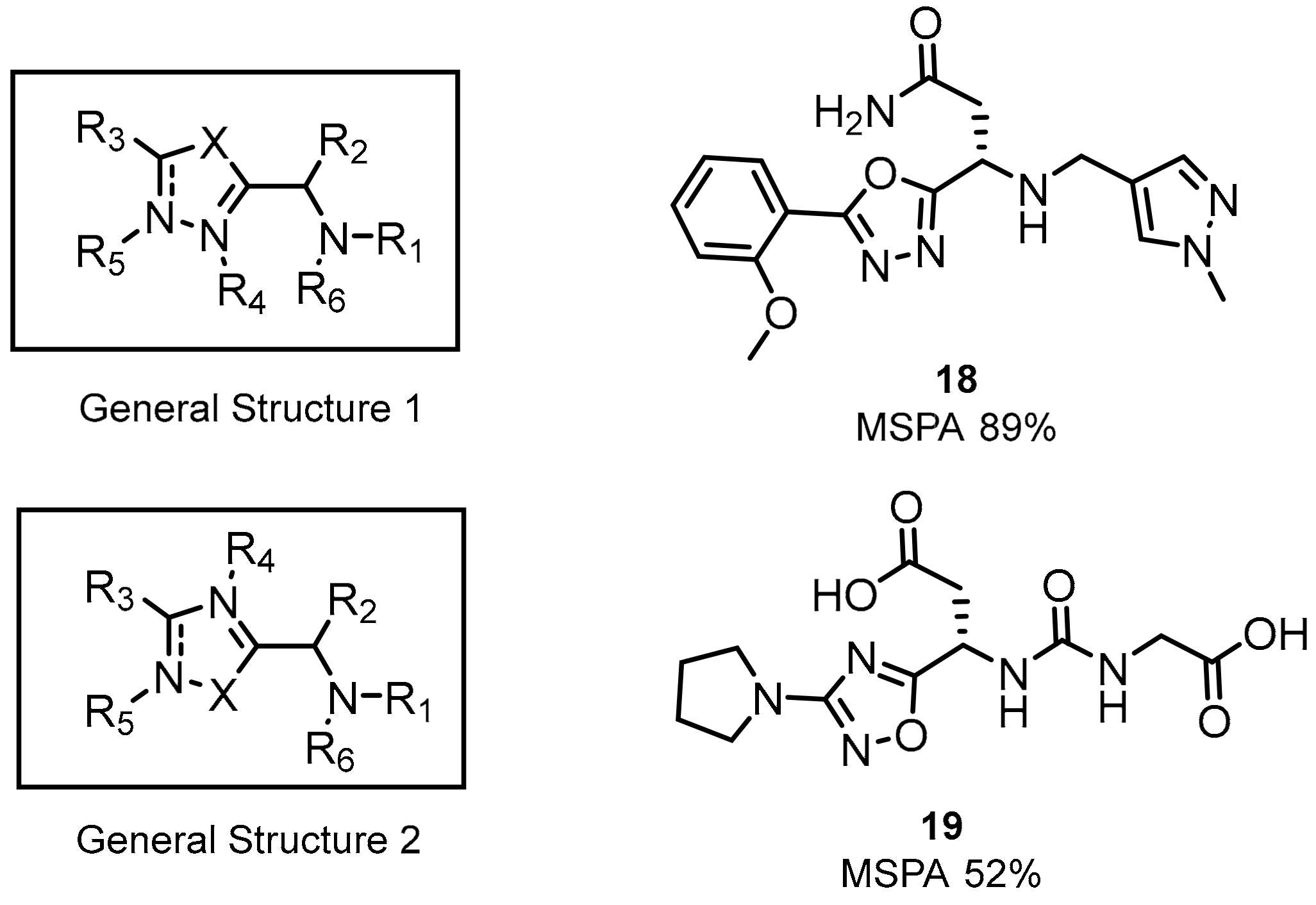
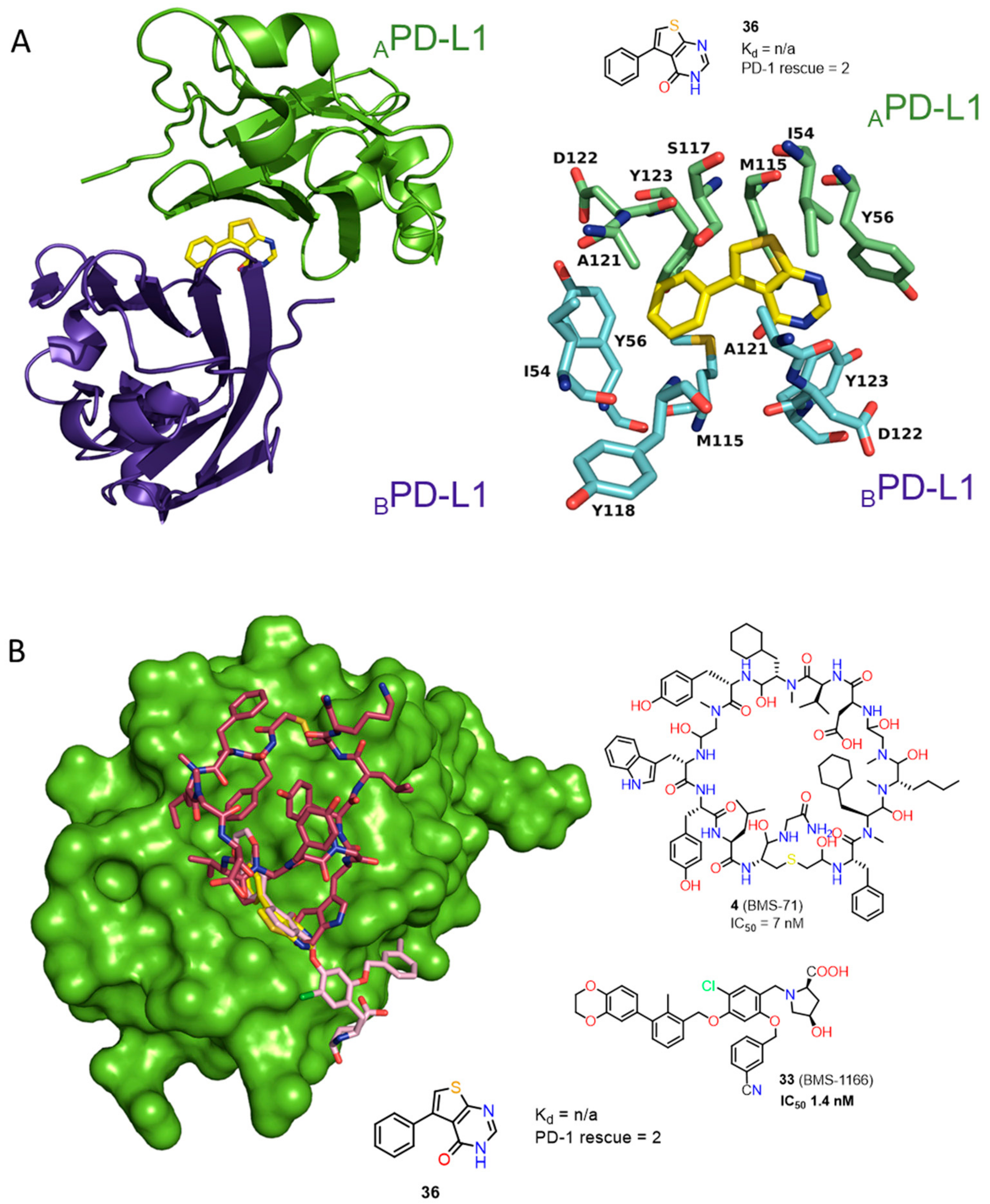
2.1. Peptides and Peptidomimetics as Inhibitors of the PD-1/PD-L1 Pathway
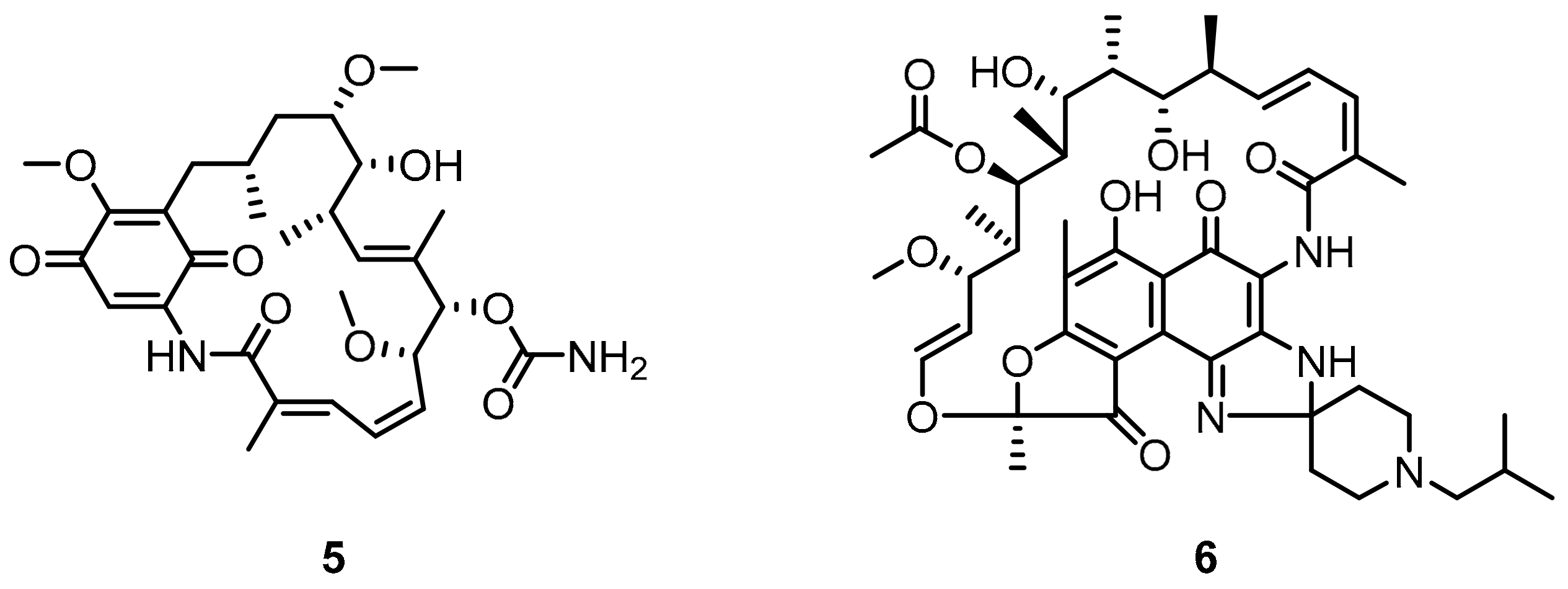
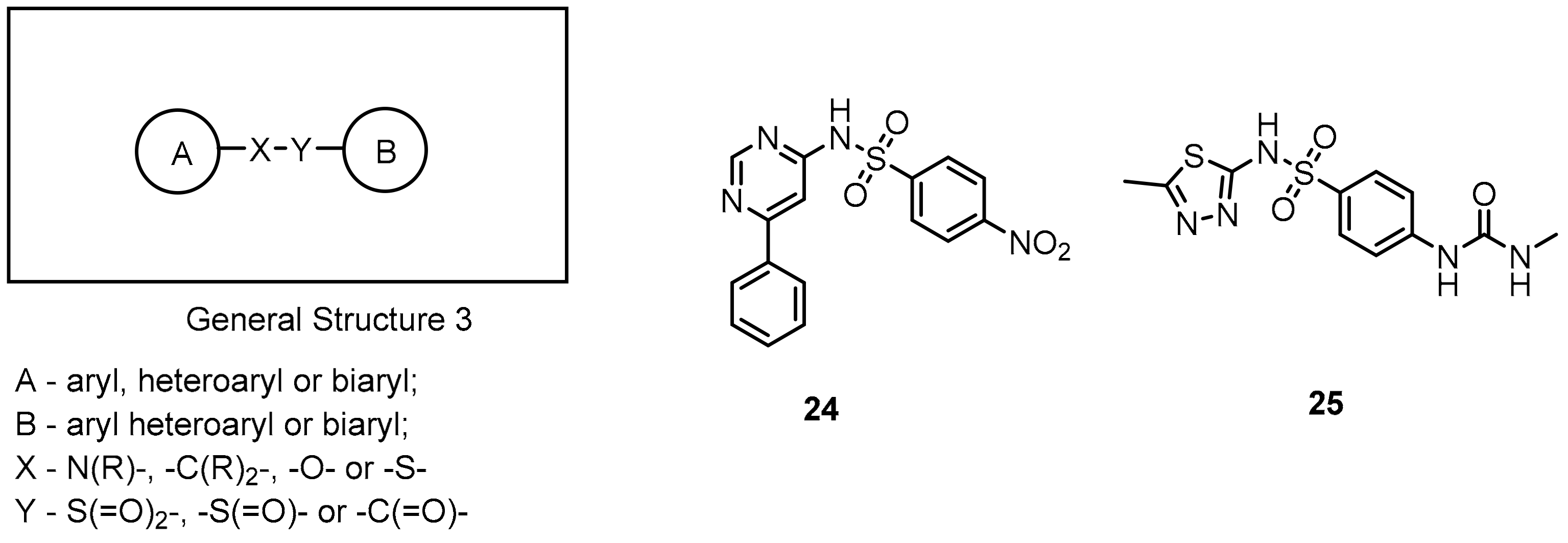
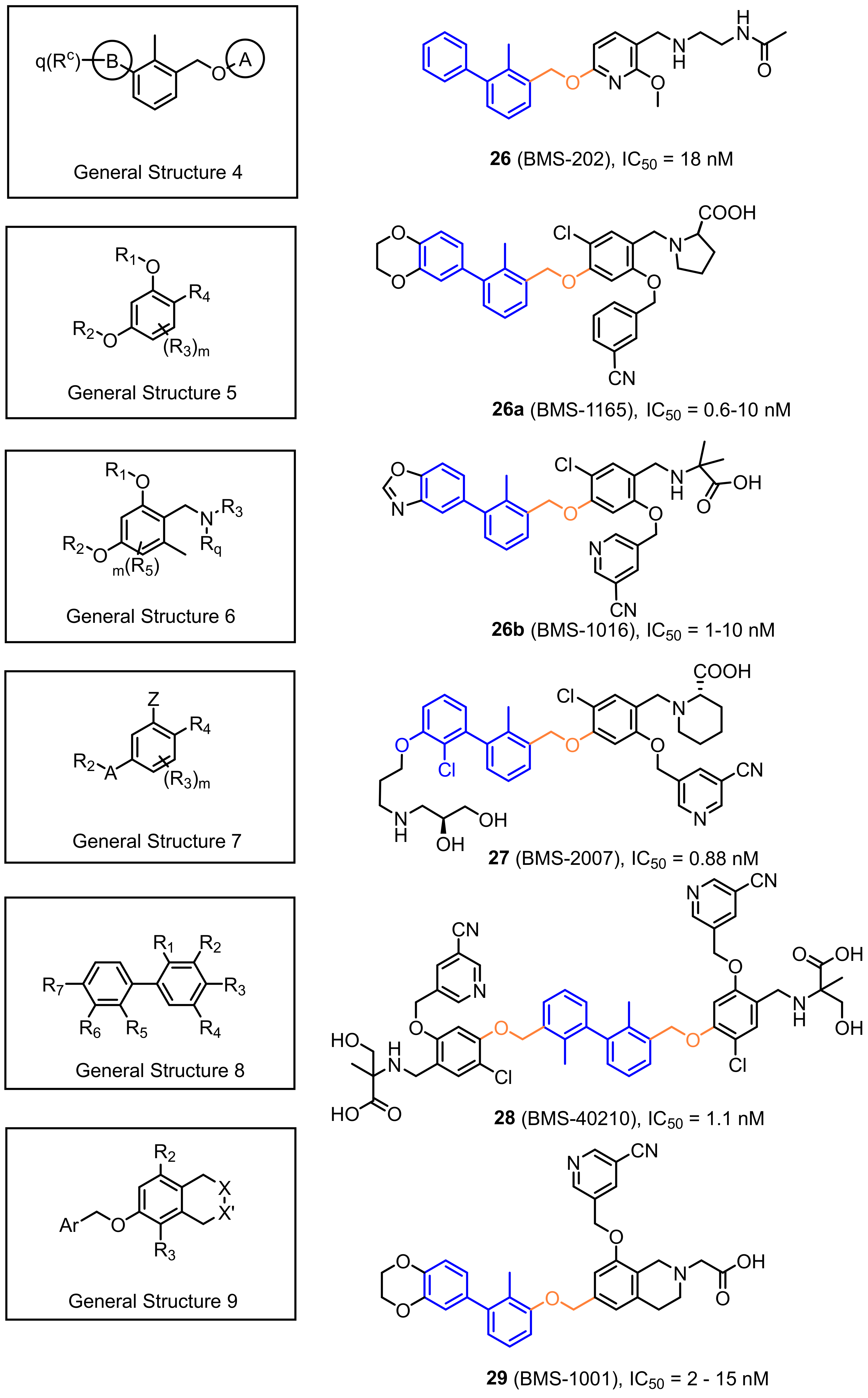
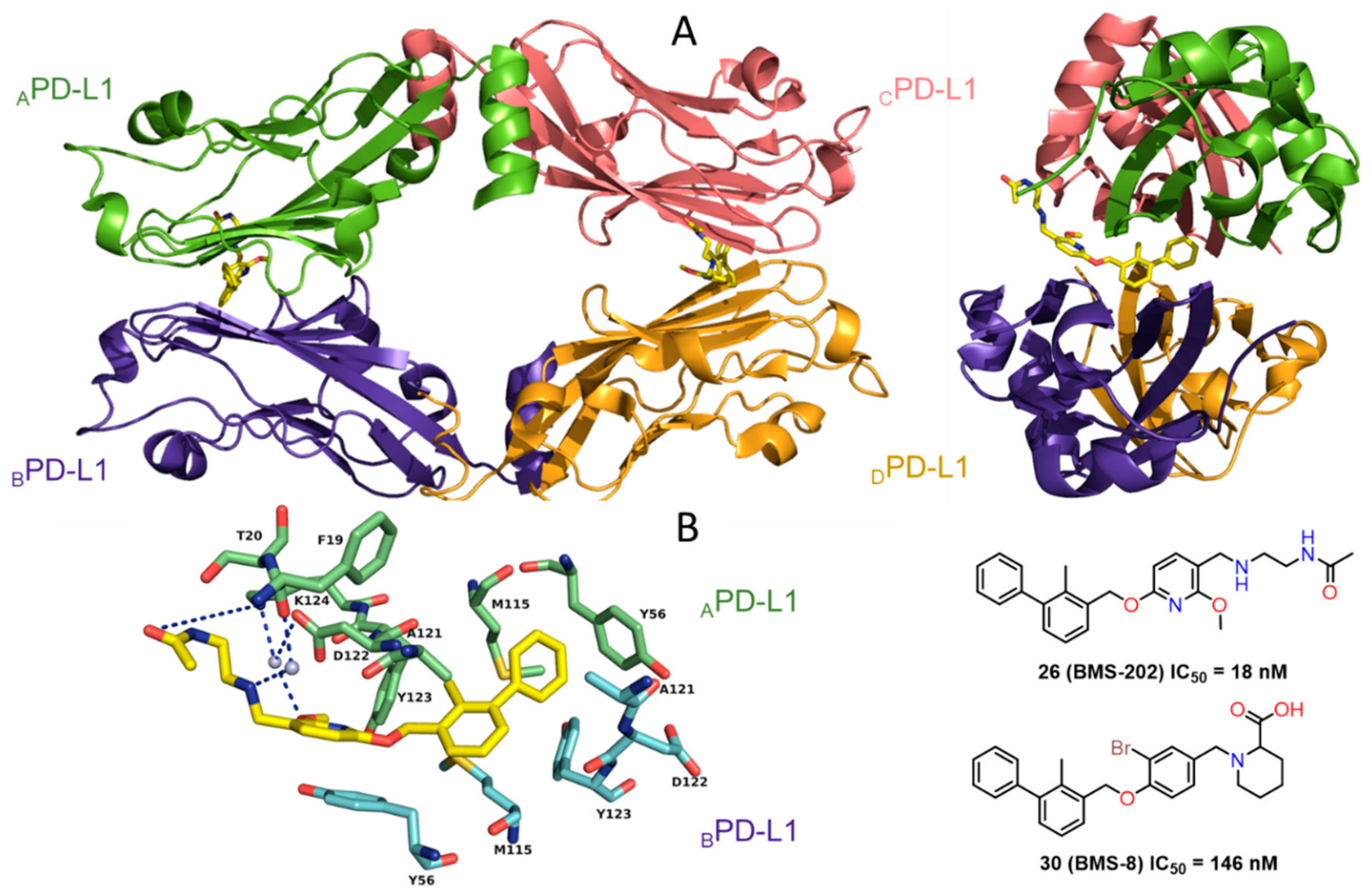
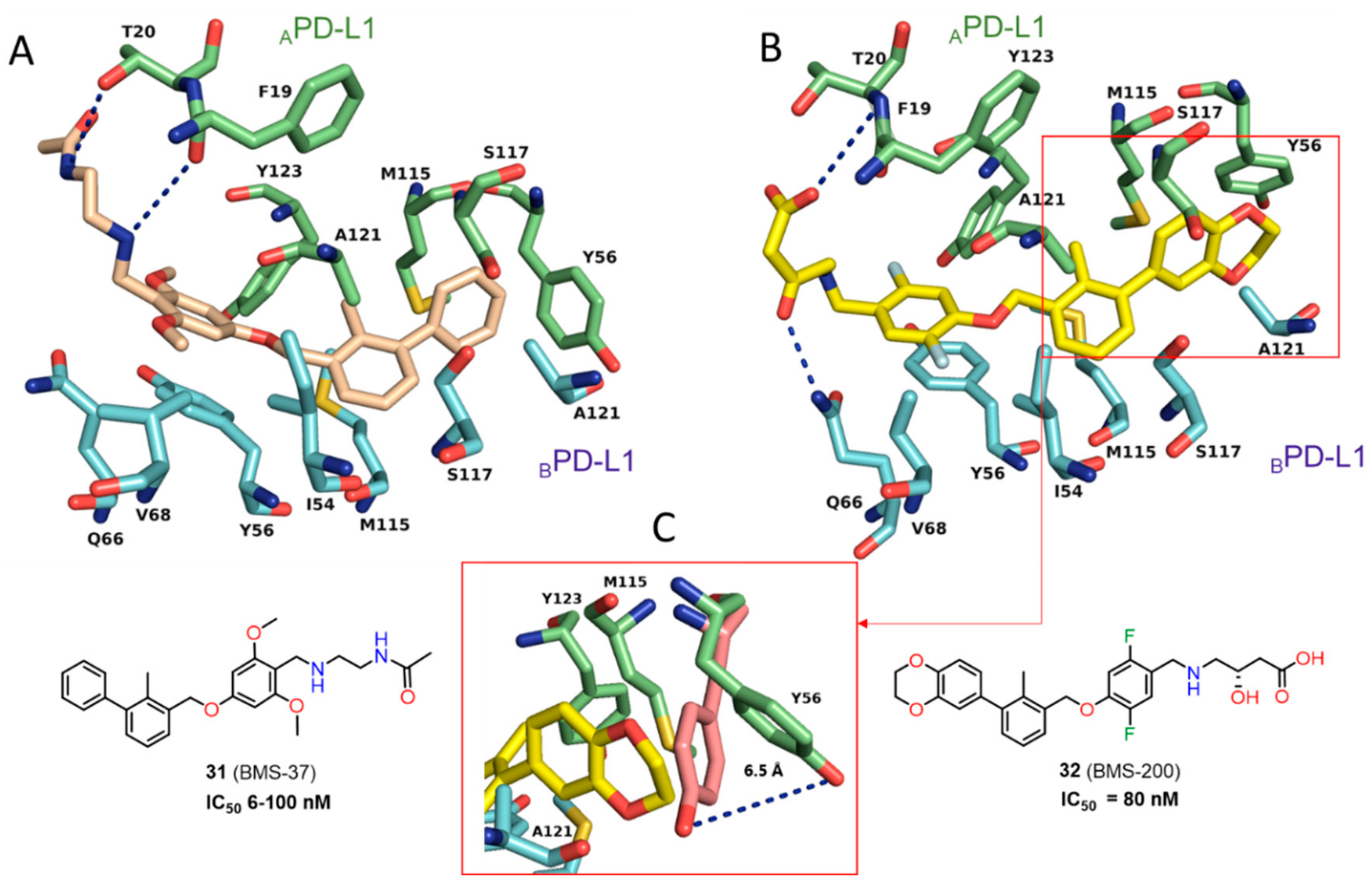
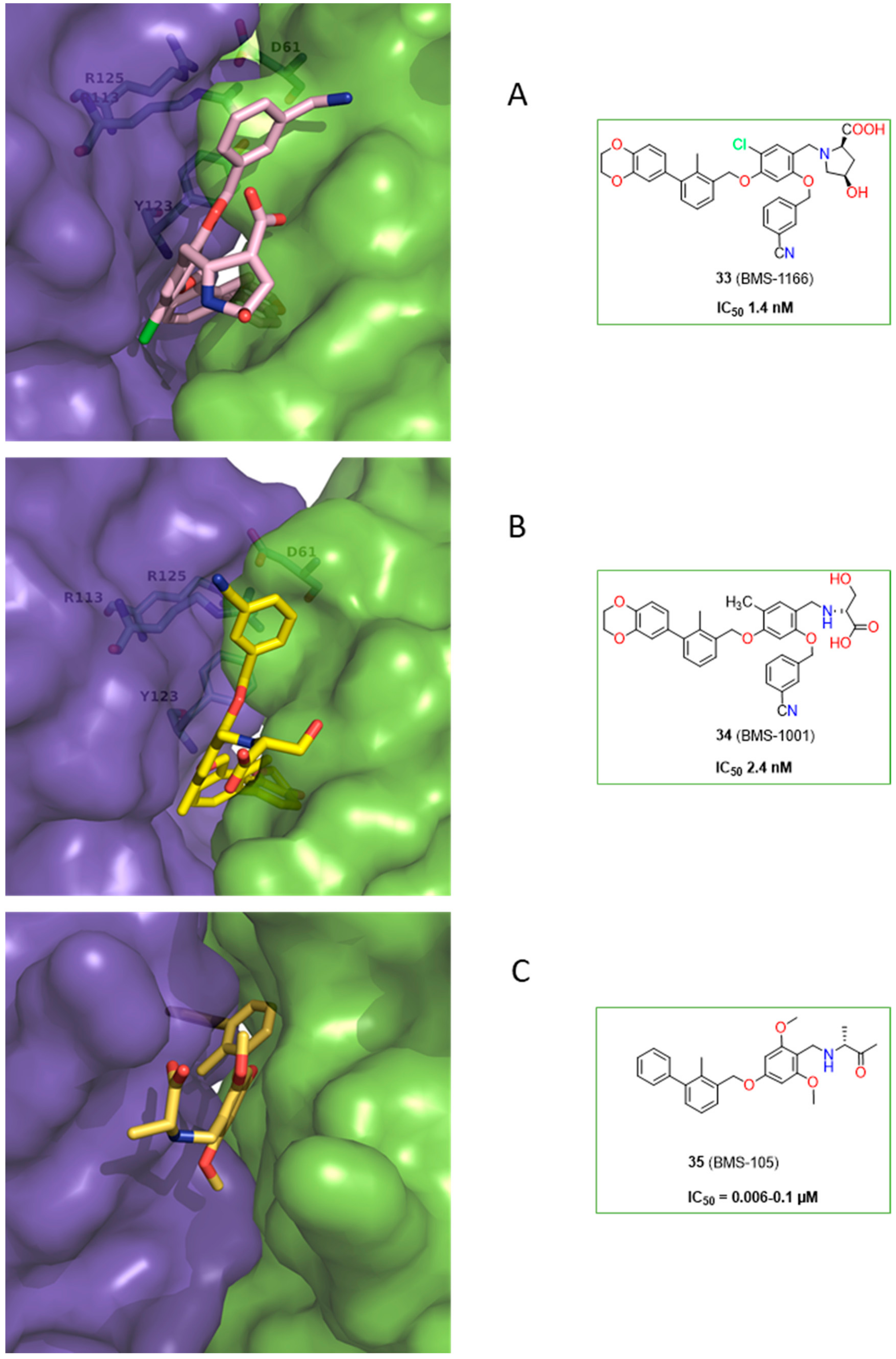
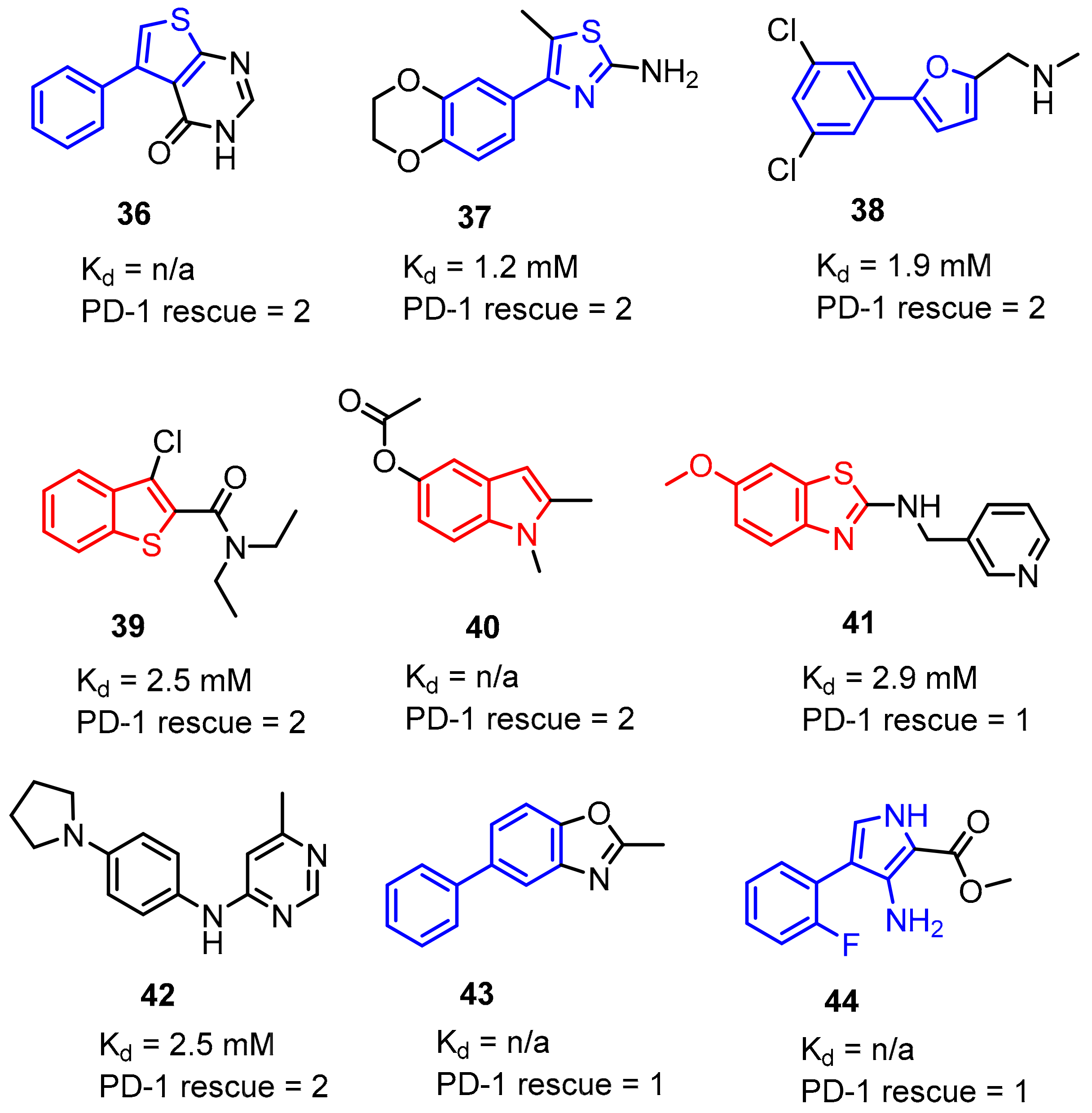
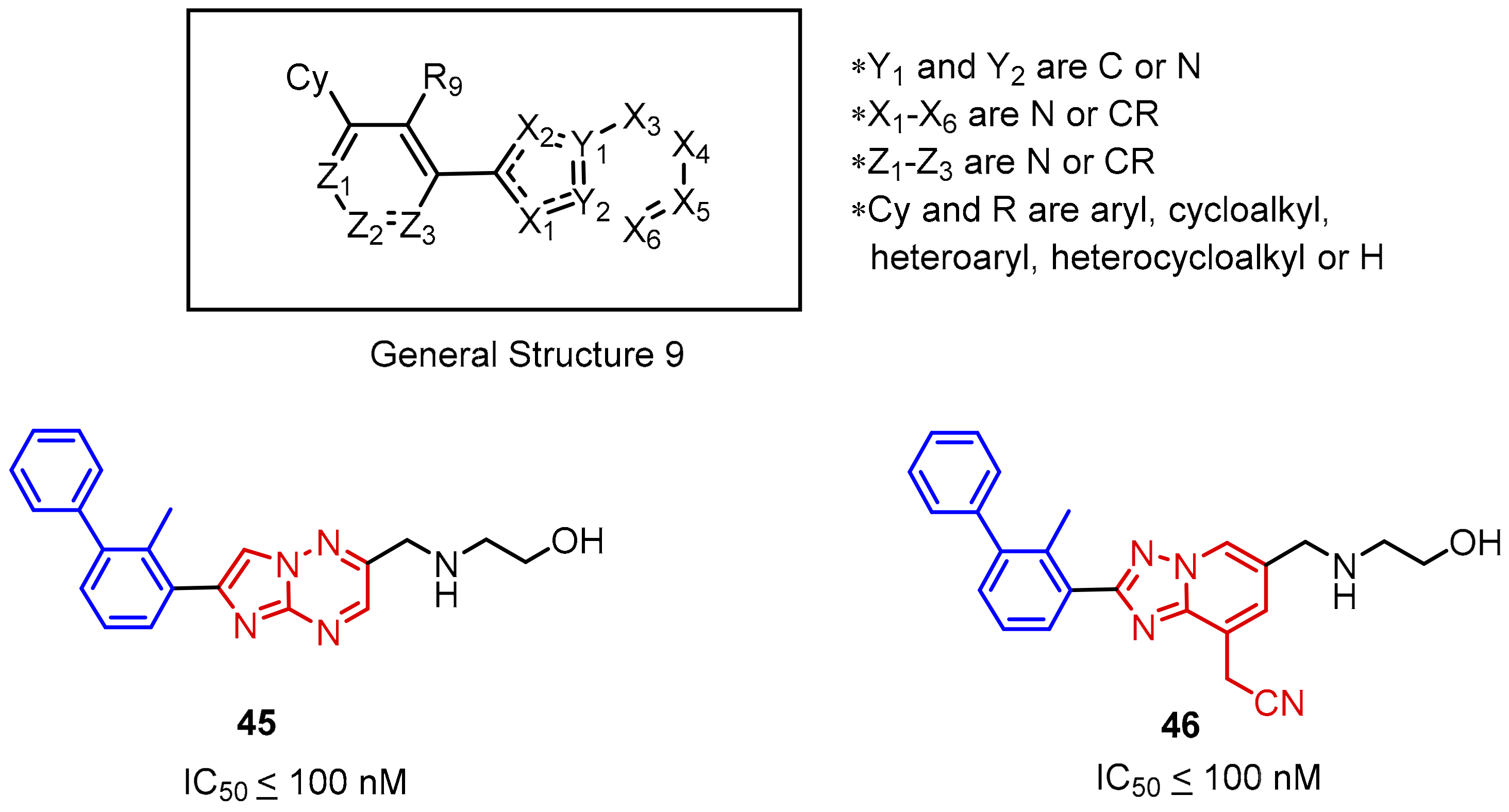
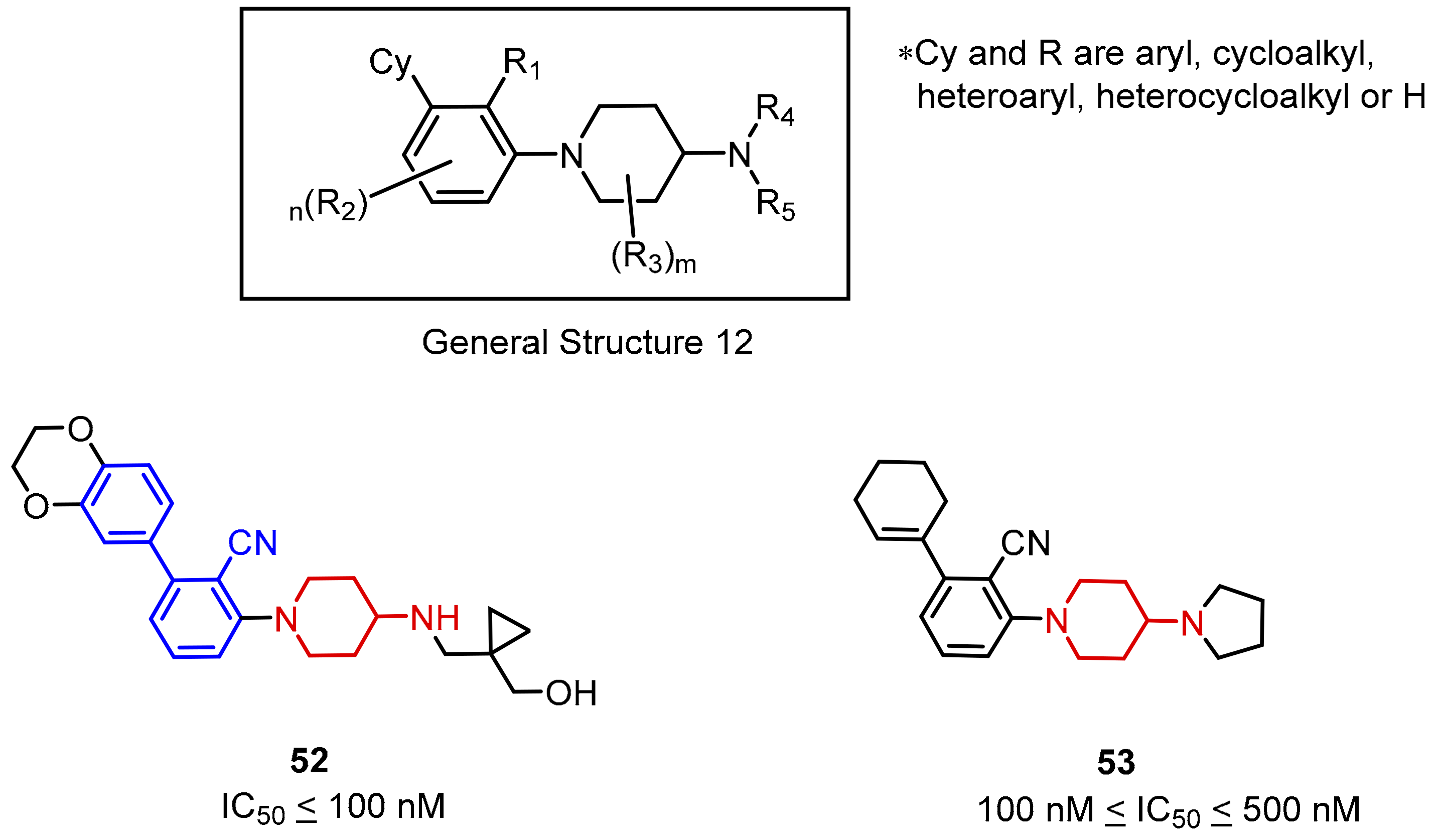
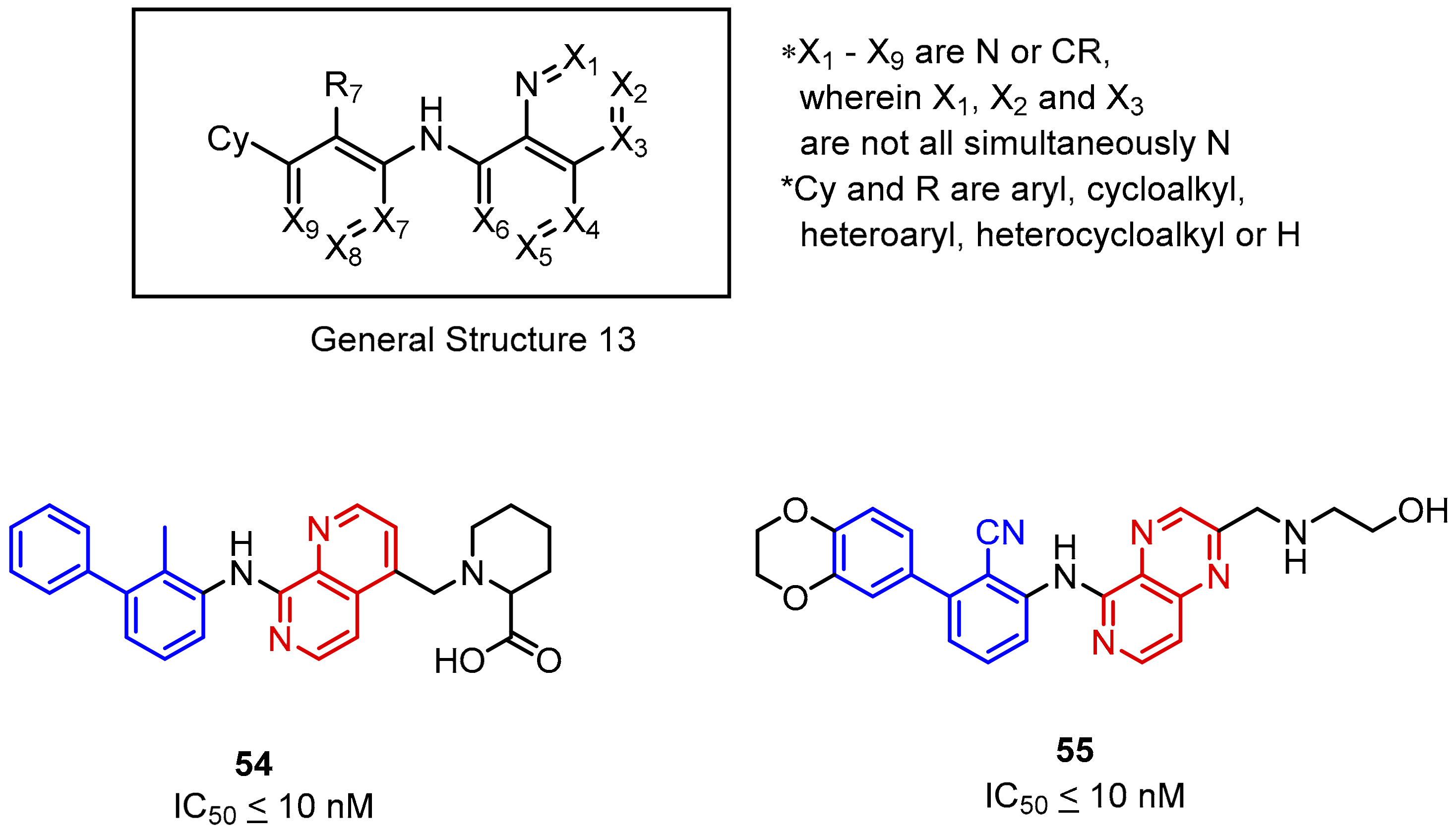
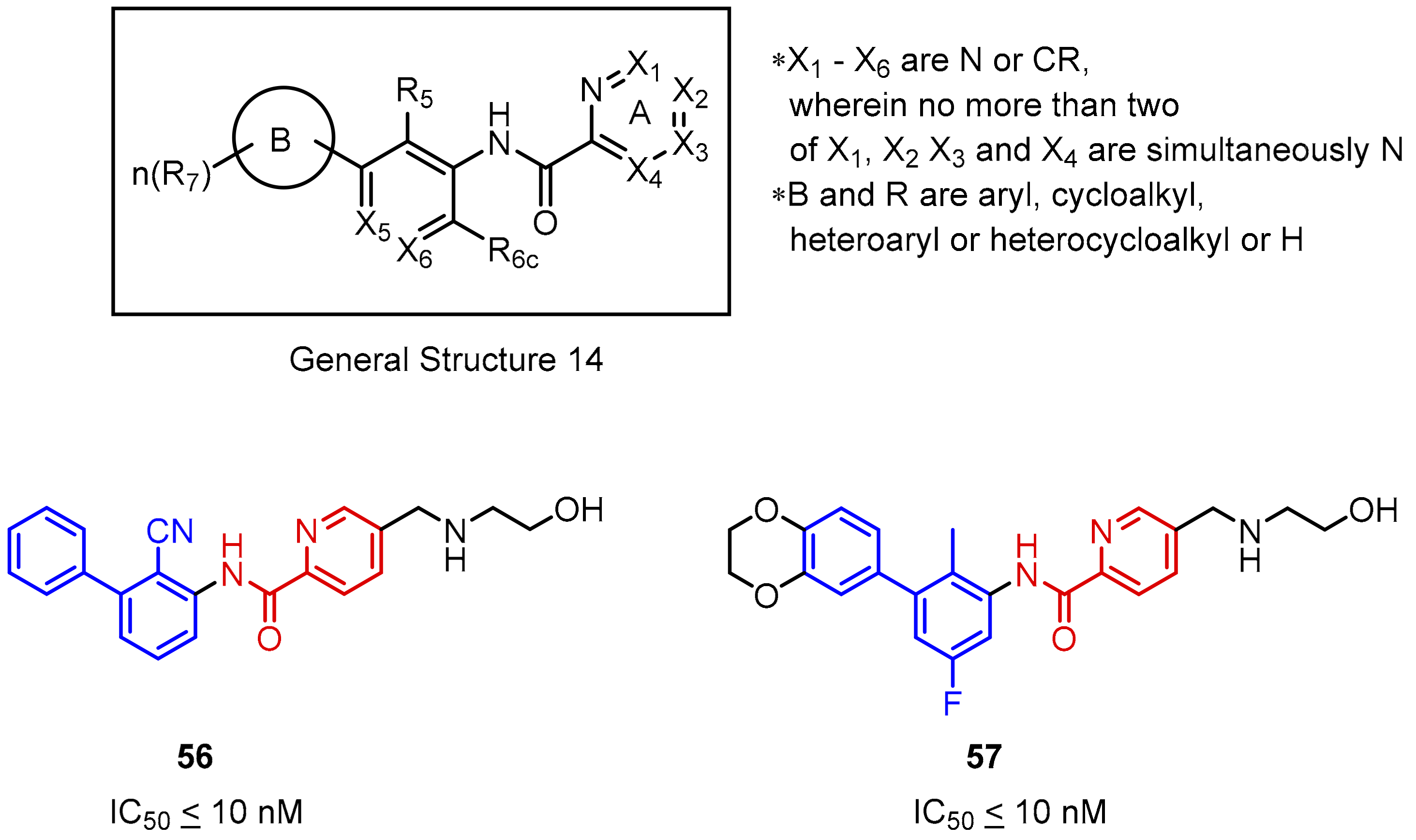
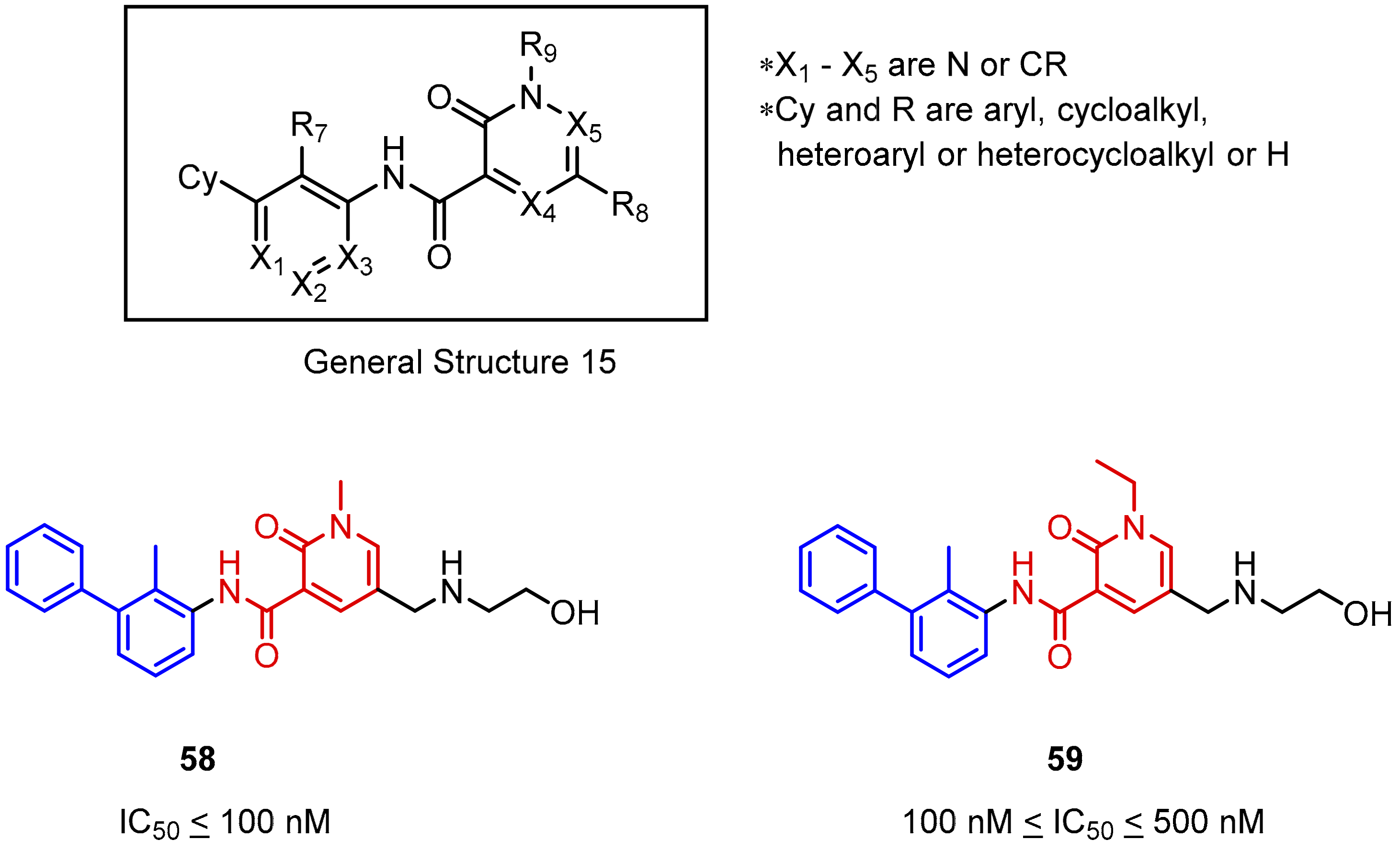
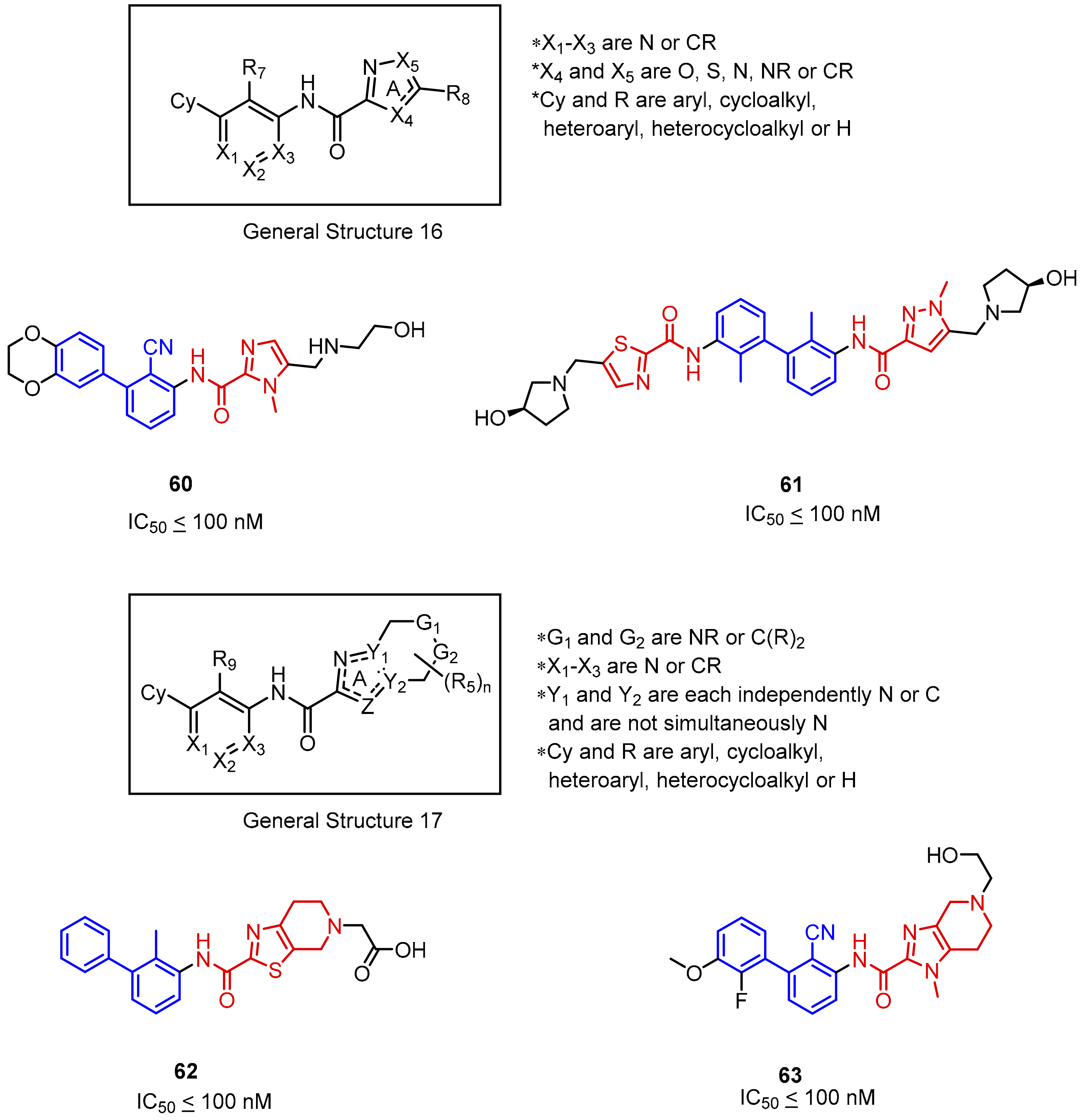
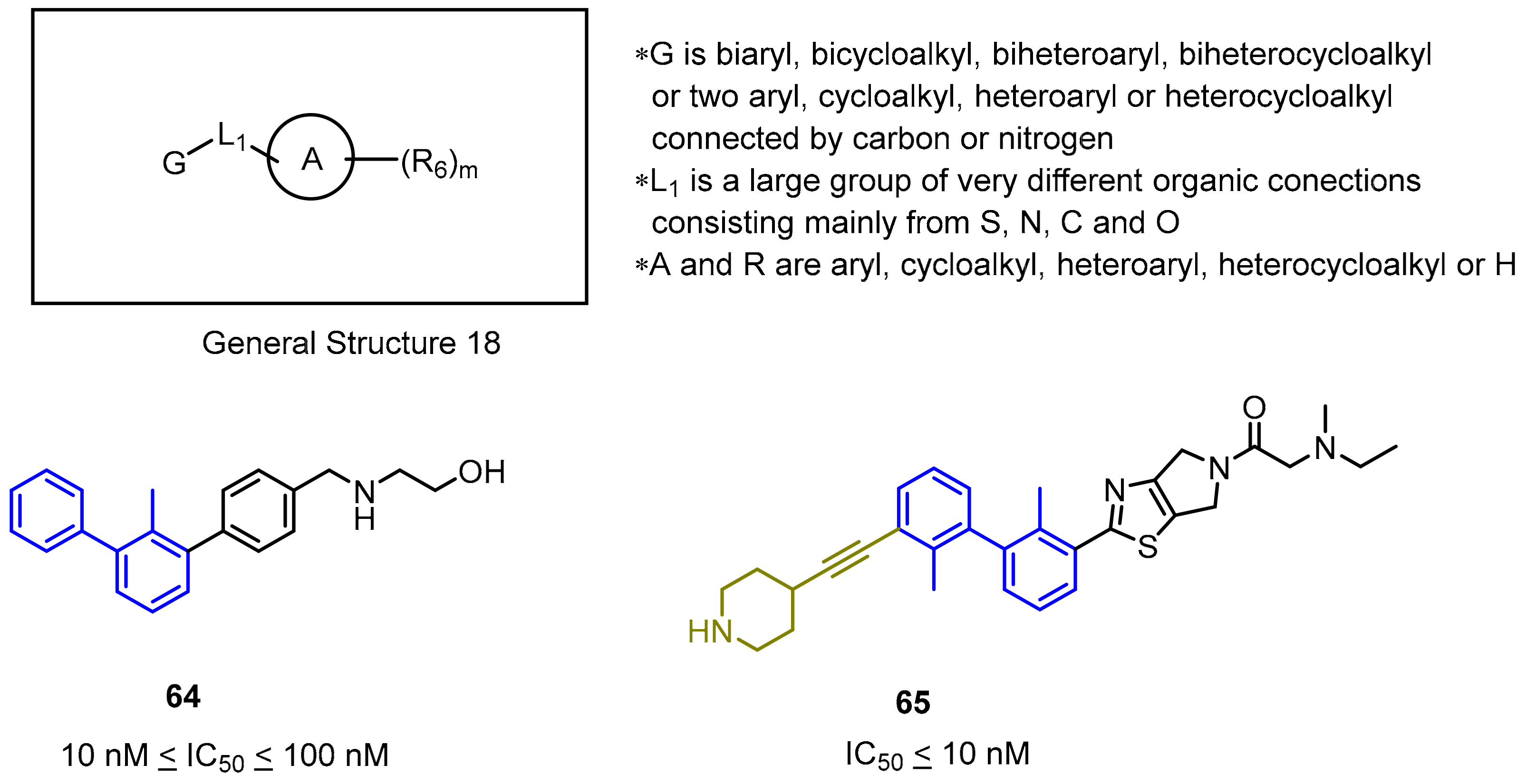
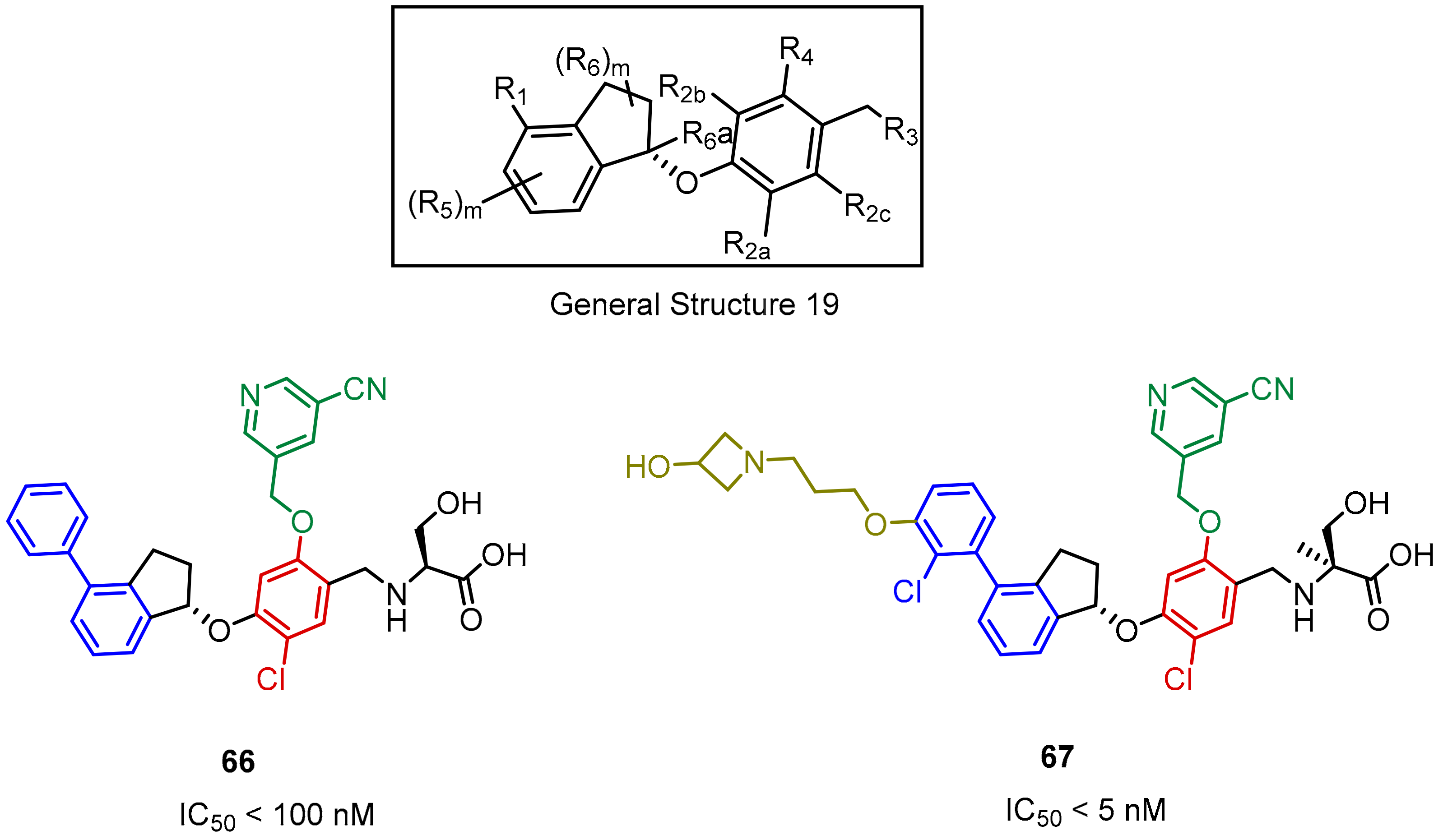
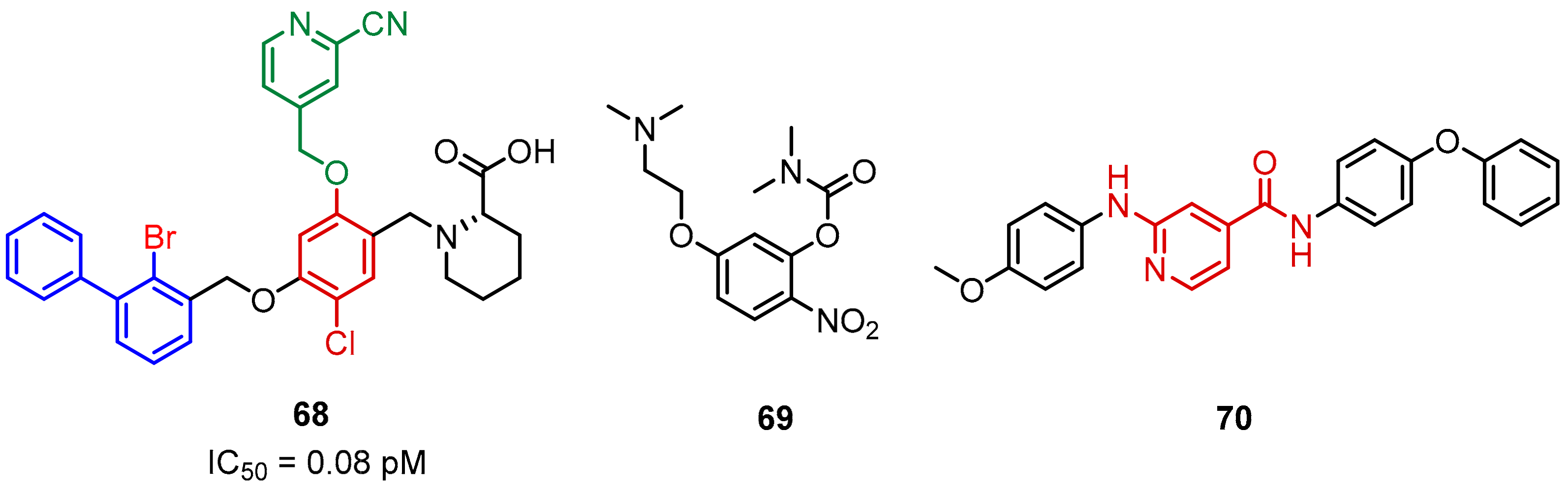
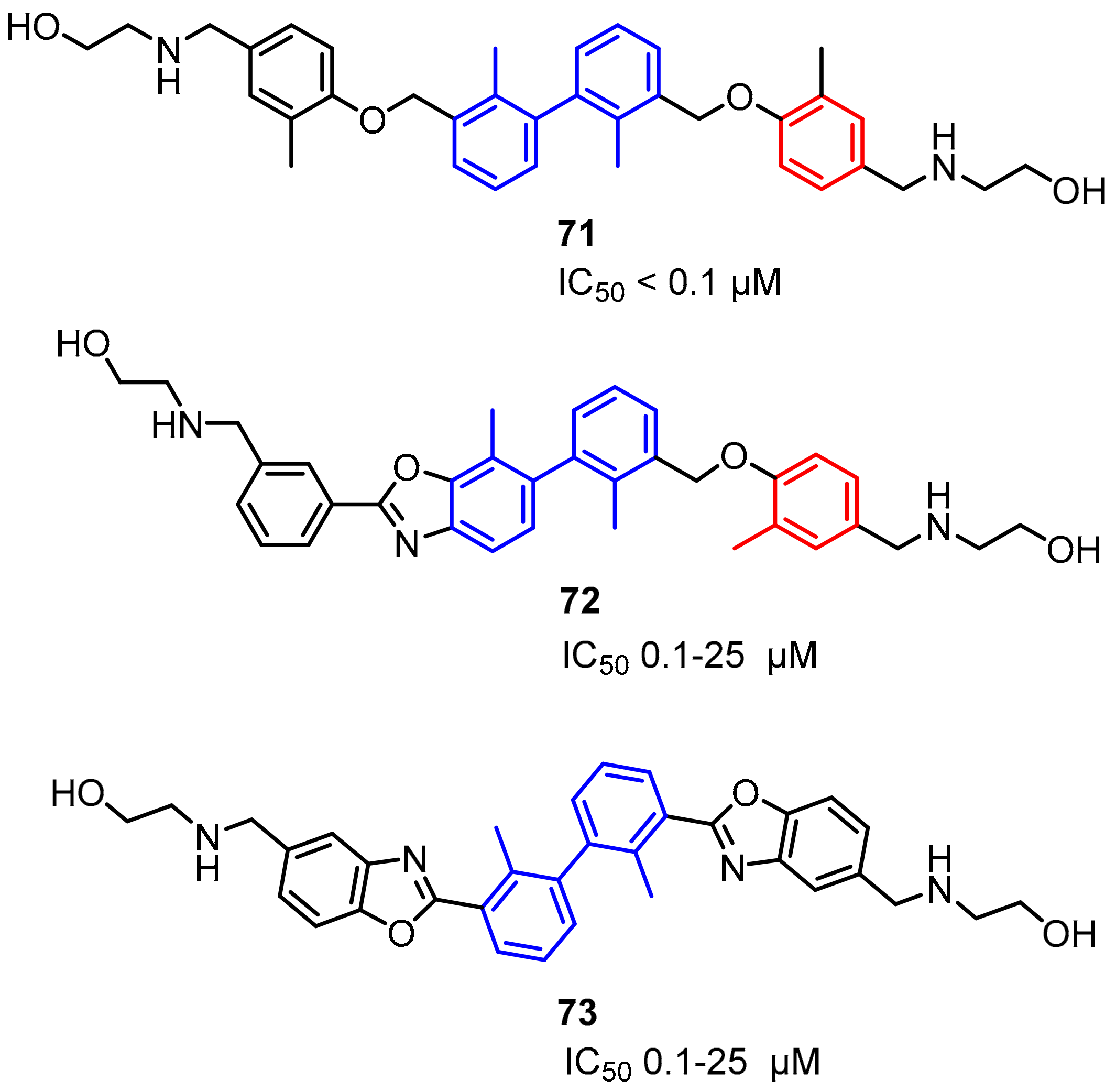
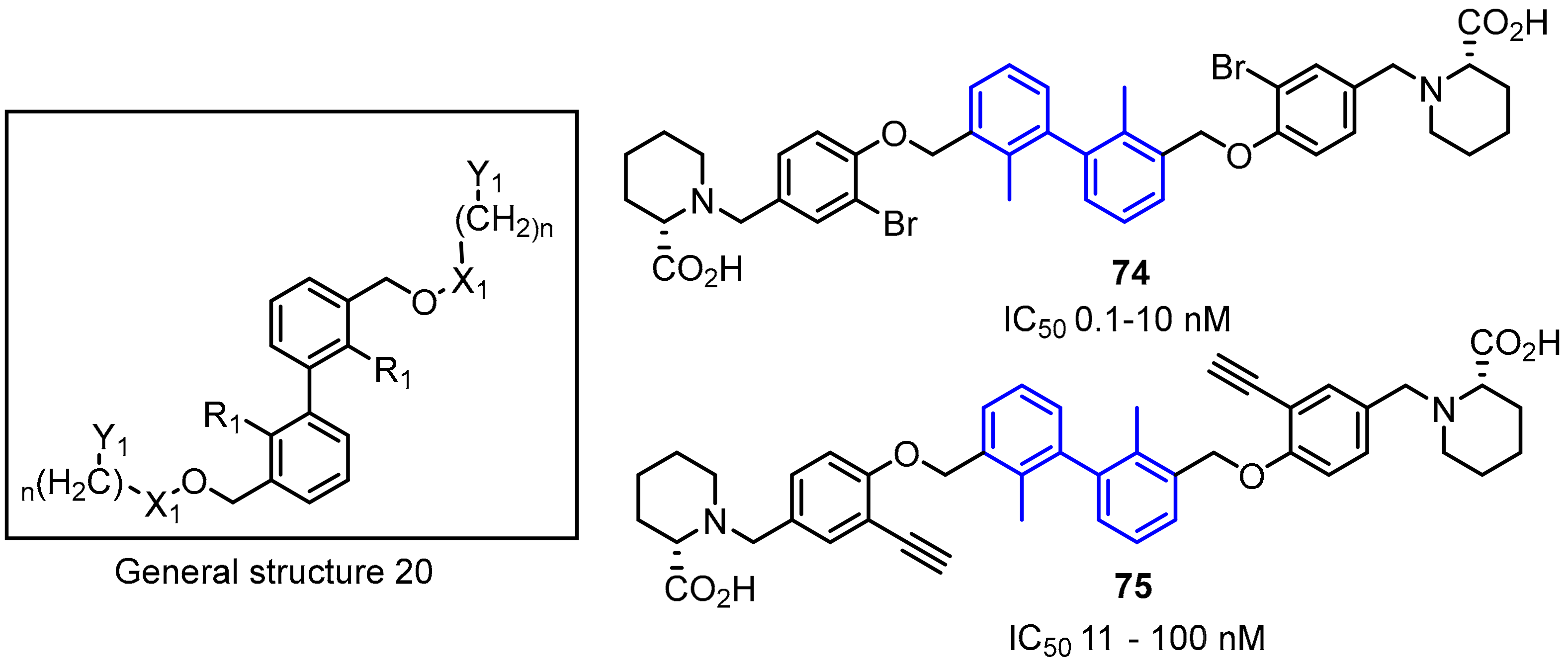
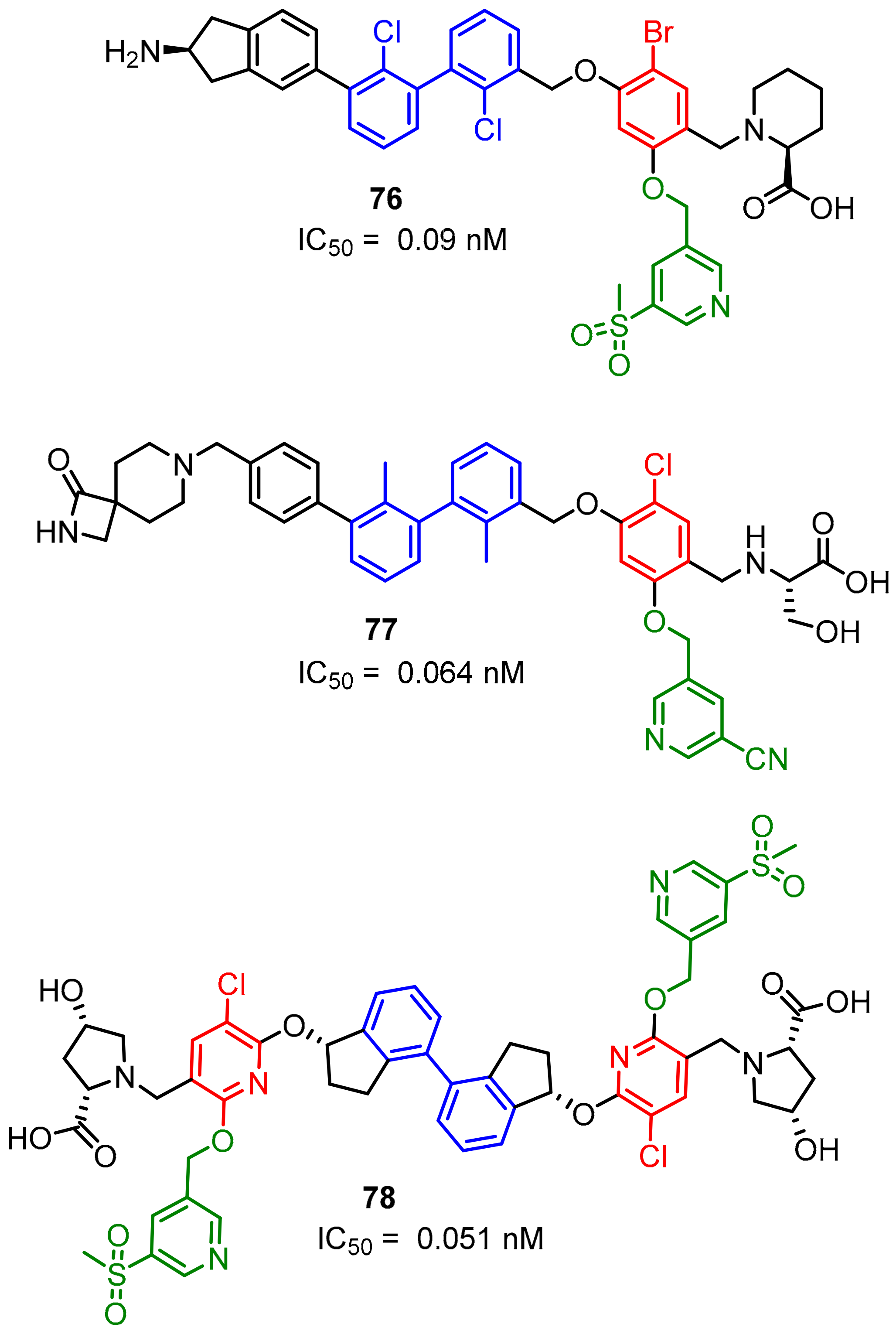
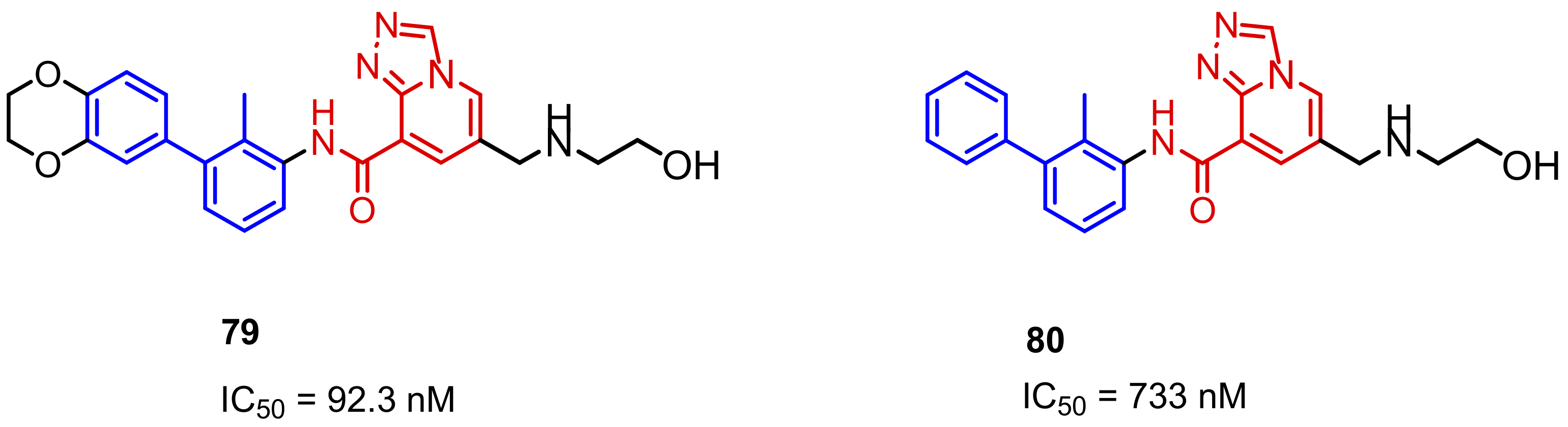
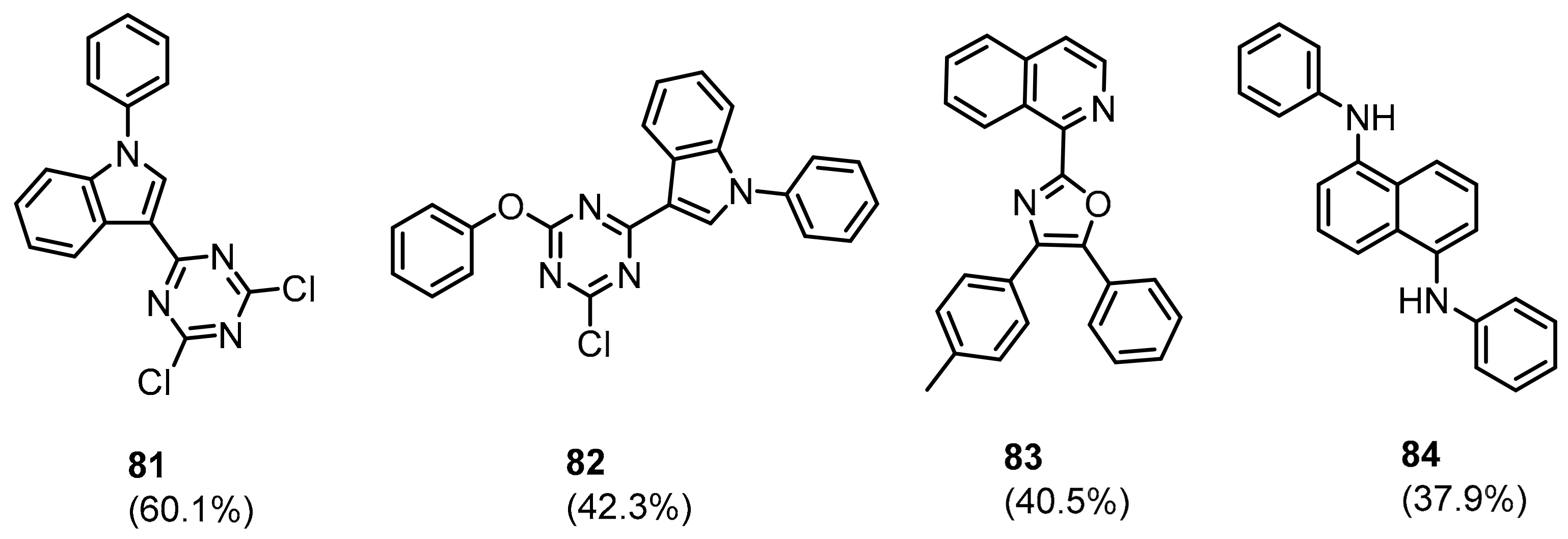
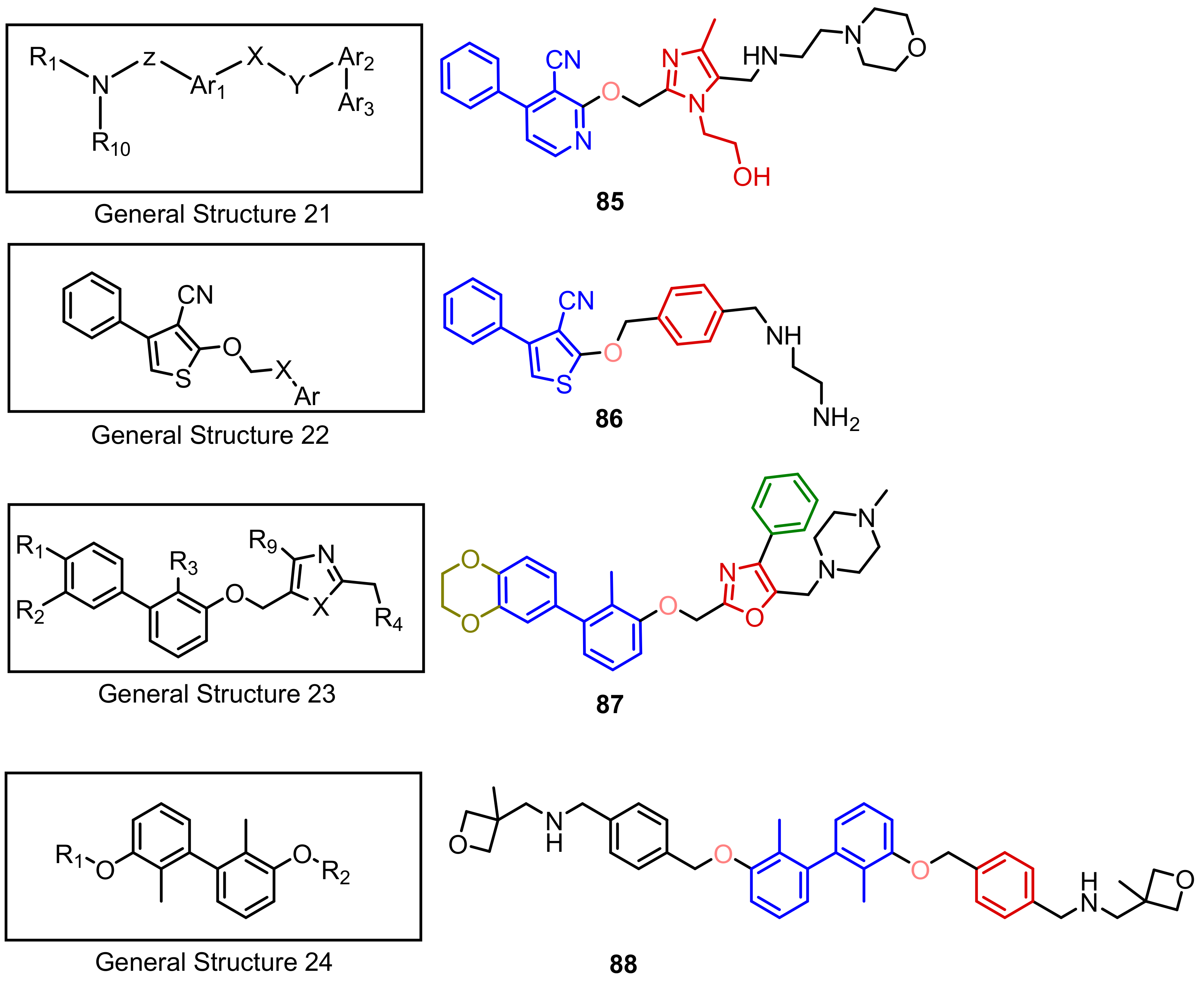
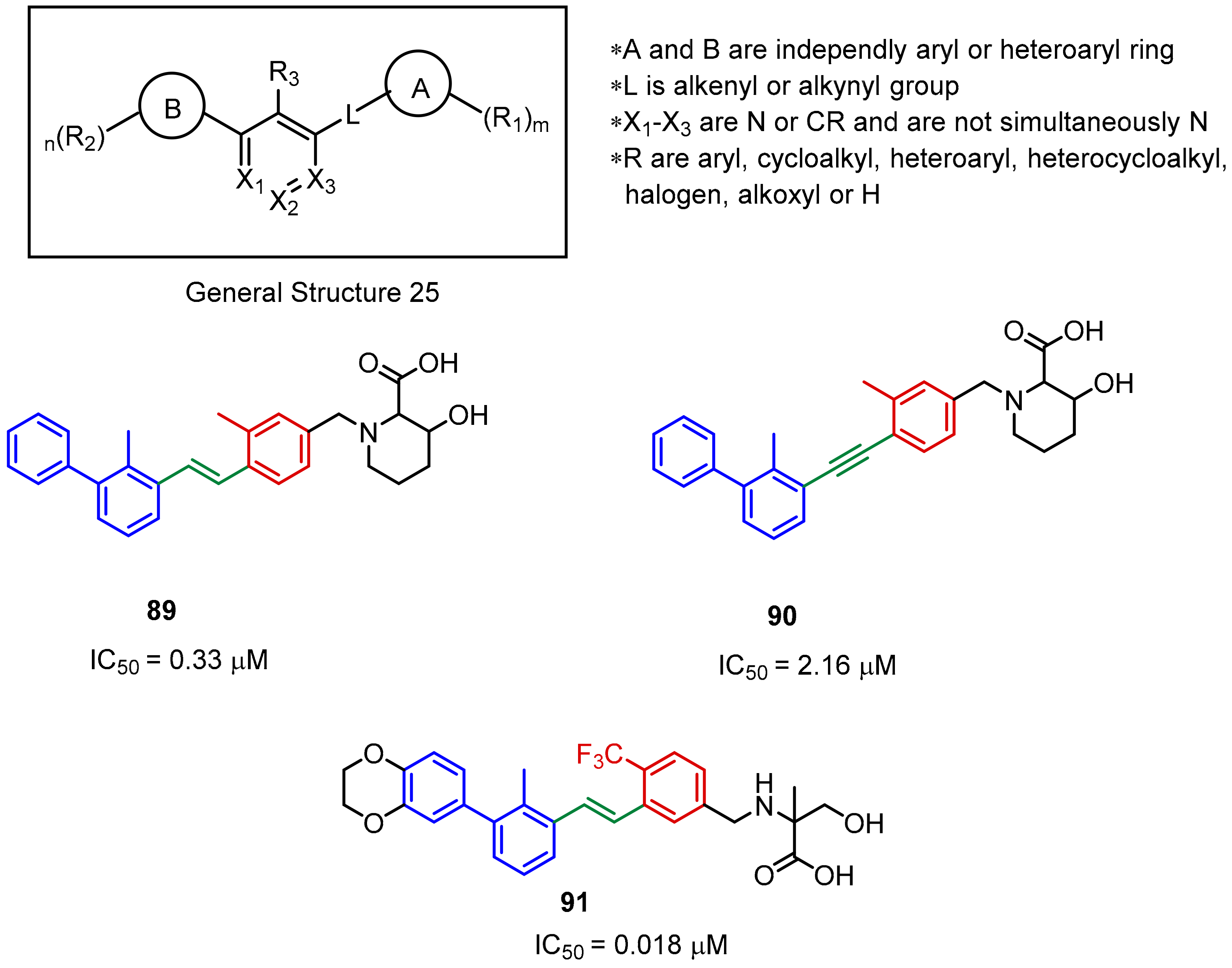
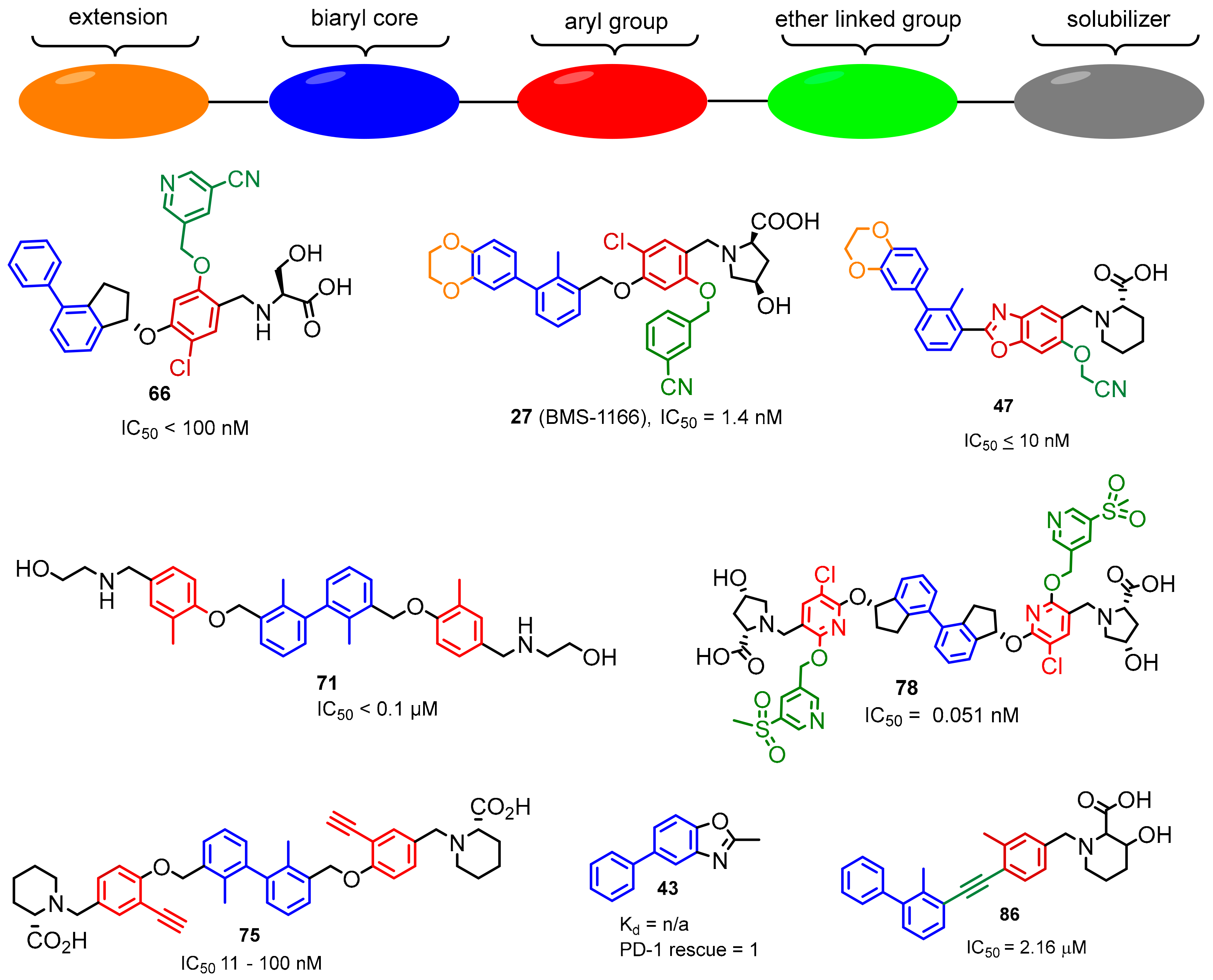
2.2. Nonpeptidic Small-Molecule Inhibitors
3. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Nishimura, H.; Agata, Y.; Kawasaki, A.; Sato, M.; Imamura, S.; Minato, N.; Yagita, H.; Nakano, T.; Honjo, T. Developmentally regulated expression of the PD-1 protein on the surface of double-negative(CD4(−)CD8(−)) thymocytes. Int. Immunol. 1996, 8, 773–780. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2018, 7, e1364828. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Magiera, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, S.H.; Heo, Y.-S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Shinohara, T.; Taniwaki, M.; Ishida, Y.; Kawaichi, M.; Honjo, T. Structure and Chromosomal Localization of the Human PD-1 Gene (PDCD1). Genomics 1994, 23, 704–706. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Schwartz, J.-C.D.; Guo, X.; Bhatia, S.; Cao, E.; Chen, L.; Zhang, Z.-Y.; Edidin, M.A.; Nathenson, S.G.; Almo, S.C. Structural and Functional Analysis of the Costimulatory Receptor Programmed Death-1. Immunity 2004, 20, 337–347. [Google Scholar] [CrossRef]
- Flies, D.B.; Sandler, B.J.; Sznol, M.; Chen, L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J. Biol. Med. 2011, 84, 409–421. [Google Scholar]
- Zhan, M.M.; Hu, X.Q.; Liu, X.X.; Ruan, B.F.; Xu, J.; Liao, C. From monoclonal antibodies to small molecules: The development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov. Today 2016, 21, 1027–1036. [Google Scholar] [CrossRef]
- Ghiotto, M.; Gauthier, L.; Serriari, N.; Pastor, S.; Truneh, A.; Nunès, J.A.; Olive, D. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int. Immunol. 2010, 22, 651–660. [Google Scholar] [CrossRef]
- Carter, L.L.; Fouser, L.A.; Jussif, J.; Fitz, L.; Deng, B.; Wood, C.R.; Collins, M.; Honjo, T.; Freeman, G.J.; Carreno, B.M. PD-1:PD-L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL-2. Eur. J. Immunol. 2002, 32, 634–643. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor- infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.B.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.G.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274(PD-L1, B7- H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Seo, S.H.; Kim, B.S.; Kim, C.D.; Lee, J.H.; Kang, J.S.; Maeng, P.J.; Lim, J.S. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J. Dermatol. Sci. 2005, 40, 95–103. [Google Scholar] [CrossRef]
- Kelly, P.N. The Cancer Immunotherapy Revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Nisbet, I. Cancer immunotherapy comes of age (Finally!). Australas. Biotechnol. 2016, 26, 38–40. [Google Scholar]
- Ledford, H.; Else, H.; Warren, M. Cancer immunologists scoop medicine Nobel prize. Nature 2018, 562, 20–21. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2202. [Google Scholar] [CrossRef]
- Hoos, A. Development of immuno-oncology drugs—From CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Tan, S.; Zhang, C.W.-H.; Gao, G.F. Seeing is believing: Anti-PD-1/PD-L1 monoclonal antibodies in action for checkpoint blockade tumor immunotherapy. Signal Transduct. Target. Ther. 2016, 1, 16029. [Google Scholar] [CrossRef]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Domling, A.; Dubin, G.; Holak, T.A. Structure of the complex of human programmed death 1, PD-1, and Its ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef]
- Weinmann, H. Cancer Immunotherapy: Selected Targets and Small-Molecule Modulators. ChemMedChem 2016, 11, 450–466. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Konstantinidou, M.; Gao, Y.; Krzemien, D.; Zak, K.; Dubin, G.; Holak, T.A.; Krzemien, D.; Zak, K.; Dubin, G.; et al. Inhibitors of programmed cell death 1 (PD-1): A patent review (2010–2015). Expert Opin. Ther. Pat. 2016, 3776, 1–6. [Google Scholar] [CrossRef]
- Toogood, P.L. Small molecule immuno-oncology therapeutic agents. Bioorg. Med. Chem. Lett. 2018, 28, 319–329. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, H.T.; Shin, W.; Chae, J.; Choi, J.; Kim, S.H.; Lim, H.; Won Heo, T.; Park, K.Y.; Lee, Y.J.; et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 2016, 7, 13354. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Zarganes-Tzitzikas, T.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies? Angew. Chem. Int. Ed. 2018, 57, 4840–4848. [Google Scholar] [CrossRef]
- Wang, T.; Wu, X.; Guo, C.; Zhang, K.; Xu, J.; Li, Z.; Jiang, S. Development of Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Signaling Pathway. J. Med. Chem. 2019, 62, 1715–1730. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, Q.; Liu, Z.; Chen, Y.; Feng, F.; Sun, H. Peptide-based and small synthetic molecule inhibitors on PD-1/PD-L1 pathway: A new choice for immunotherapy? Eur. J. Med. Chem. 2019, 161, 378–398. [Google Scholar] [CrossRef]
- Sasikumar, P.G.N.; Ramachandra, M.; Vadlamani, S.K.; Vemula, K.R.; Satyam, L.K.; Subbarao, K.; Shrimali, K.R.; Kandepudu, S. Immunosuppression Modulating Compounds. US20110318373A1, 29 December 2011. [Google Scholar]
- Sasikumar, P.G.; Satyam, L.K.; Shrimali, R.K.; Subbarao, K.; Ramachandra, R.; Vadlamani, S.; Reddy, A.; Kumar, A.; Srinivas, A.; Reddy, S.; et al. Abstract 2850: Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway. Cancer Res. 2012, 72. [Google Scholar] [CrossRef]
- Sasikumar, P.G.N.; Ramachandra, M.; Vadlamani, S.K.; Shrimali, K.; Subbarao, K. Therapeutic Compounds for Immunomodulation. WO2012168944, 13 December 2012. [Google Scholar]
- Miller, M.M.; Mapelli, C.; Allen, M.P.; Bowsher, M.S.; Boy, K.M.; Gillis, E.P.; Langley, D.R.; Mull, E.; Poirier, M.A.; Sanghvi, N.; et al. Macrocyclic Inhibitors of the PD-1/PD-L1 and CD80(B7-1)/PD- L1 Protein/Protein Interactions. WO2014151634, 25 September 2014. [Google Scholar]
- Miller, M.M.; Mapelli, C.; Allen, M.P.; Bowsher, M.S.; Gillis, E.P.; Langley, D.R.; Mull, E.; Poirier, M.A.; Sanghvi, N.; Sun, L.Q.; et al. Macrocyclic Inhibitors of the PD-1/PD-L1 and CD80(B7-1)/PD-LI Protein/Protein Interactions. WO2016039749, 17 March 2016. [Google Scholar]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, Ł.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew. Chem. Int. Ed. Engl. 2017, 56, 13732–13735. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; Krzanik, S.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef]
- Perry, E.; Mills, J.J.; Zhao, B.; Wang, F.; Sun, Q.; Christov, P.P.; Tarr, J.C.; Rietz, T.A.; Olejniczak, E.T.; Lee, T.; et al. Fragment-based screening of programmed death ligand 1 (PD-L1). Bioorg. Med. Chem. Lett. 2019, 29, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P.; Yoon, S.-C.; Aradhya, A.G.; Hofer, J.; Fink, M.A.; Enley, E.S.; Fisher, J.E.; Herb, M.C.; Klingos, A.; Proulx, J.T.; et al. Macrocyclic Compounds from Ansamycin Antibiotic Class as Inhibitors of PD1–PDL1 Protein–Protein Interaction. Chem. Pharm. Bull. 2018, 66, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. Peptidomimetic Compounds as Immunomdulators. US20130237580, 12 September 2013. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. Immunomodulating Peptidomimetic Derivatives. WO2015036927A1, 19 March 2015. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. Therapeutic Immunomodulating Compounds. WO2015044900A1, 2 April 2015. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S.; Prasad, A. 3-Substituted-1,2,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators. US20180044329A1, 15 February 2018. [Google Scholar]
- Böger, C.; Behrens, H.M.; Krüger, S.; Röcken, C. The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? OncoImmunology 2017, 6, e1293215. [Google Scholar] [CrossRef]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. Dual Inhibitors of VISTA and PD-1 Pathways. WO2018073754, 26 April 2018. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Derivatives as Immunomodulators. WO2015033301A1, 12 March 2015. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. Cyclic Substituted-1,3,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators. WO2018051255A1, 22 March 2018. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. 1,2,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators. WO2016142833, 15 September 2016. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S.; Prasad, A. 3-Substituted 1,3,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators. WO2016142894A1, 15 September 2016. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. 1,2,4-Oxadiazole Derivatives as Immunomodulators. US10173989B2, 8 January 2019. [Google Scholar]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremaddepalli, S.S.S. 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Derivatives as Immunomodulators. US10160736B2, 25 December 2018. [Google Scholar]
- Shaabani, S.; Huizinga, H.P.S.; Butera, R.; Kouchi, A.; Guzik, K.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. A patent review on PD-1/PD-L1 antagonists: Small molecules, peptides, and macrocycles (2015–2018). Expert Opin. Ther. Pat. 2018, 28, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Butte, M.J.; Oyama, S. Modulators of Immunoinhibitory Receptor PD-1, and Methods of Use Thereof. WO2011082400 A2, 7 July 2011. [Google Scholar]
- Chupak, L.S.; Zheng, X. Compounds useful as immunomodulators. WO2015034820 A1, 12 March 2015. [Google Scholar]
- Chupak, L.S.; Ding, M.; Martin, S.W.; Zheng, X.; Hewawasam, P.; Connolly, T.P.; Xu, N.; Yeung, K.-S.; Zhu, J.; Langley, D.R.; et al. Compounds Useful as Immunomodulators. WO2015160641, 22 October 2015. [Google Scholar]
- Yeung, K.S.; Connolly, T.P.; Frennesson, D.B.; Grant-Young, K.A.; Hewawasam, P.; Langley, D.R.; Meng, Z.; Mull, E.; Parcella, K.E.; Saulnier, M.G.; et al. Compounds Useful as Immunomodulators. WO2017/066227, 20 April 2017. [Google Scholar]
- Yeung, K.S.; Grant-Young, K.A.; Zhu, J.; Saulnier, M.G.; Frennesson, D.B.; Meng, Z.; Scola, P.M. 1,3-dihydroxy-phenyl derivatives useful as immunomodulators. WO2018/009505 A1, 11 January 2018. [Google Scholar]
- Yeung, K.S.; Grant-Young, K.A.; Zhu, J.; Saulnier, M.G.; Frennesson, D.B.; Langley, D.R.; Hewawasam, P.; Wang, A.X.; Zhang, Z.; Meng, Z.; et al. Biaryl Compounds Useful as Immunomodulators. WO2018/044963A1, 8 March 2018. [Google Scholar]
- Yeung, K.S.; St. Laurent, D.R.; Romine, J.L.; Scola, P.M. Substituted Isoquionline Derivatives as Immunomodulators. WO2018/183171 A1, 18 October 2018. [Google Scholar]
- Krajewski, M.; Rothweiler, U.; D’Silva, L.; Majumdar, S.; Klein, C.; Holak, T.A. An NMR-based antagonist induced dissociation assay for targeting the ligand-protein and protein-protein interactions in competition binding experiments. J. Med. Chem. 2007, 50, 4382–4387. [Google Scholar] [CrossRef] [PubMed]
- D-Silva, L.D.; Ozdowy, P.; Krajewski, M.; Rothweiler, U.; Singh, M.; Holak, T.A. Monitoring the Effects of Antagonists on Protein—Protein Interactions with NMR Spectroscopy. J. Am. Chem. Soc. 2005, 127, 13220–13226. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, M.; Ozdowy, P.; D’Silva, L.; Rothweiler, U.; Holak, T.A. NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro. Nat. Med. 2005, 11, 1135–1136. [Google Scholar] [CrossRef]
- Wu, L.; Shen, B.; Li, J.; Li, Z.; Liu, K.; Zhang, F.; Yao, W. Heterocyclic Compounds as Immunomodulators. US20170107216 A1, 20 April 2017. [Google Scholar]
- Li, J.; Wu, L.; Yao, W. Heterocyclic Compounds as Immunomodulators. WO2017087777 A1, 26 May 2017. [Google Scholar]
- Li, Z.; Wu, L.; Yao, W. Heterocyclic Compounds as Immunomodulators. WO2017192961 A1, 9 November 2017. [Google Scholar]
- Lu, L.; Qian, D.Q.; Wu, L.; Yao, W. Heterocyclic Compounds as Immunomodulators. WO2017205464 A1, 30 November 2017. [Google Scholar]
- Lajkiewicz, N.; Wu, L.; Yao, W. Heterocyclic compounds as immunomodulators. US 20170174679 A1, 22 June 2017. [Google Scholar]
- Wu, L.; Yu, Z.; Zhang, F.; Yao, W. N-Phenyl-Pyridine-2-Carboxamide Derivatives and Their Use as PD-1/PD-L1 Protein/Protein Interaction Modulators. WO2017106634 A1, 22 June 2017. [Google Scholar]
- Yu, Z.; Wu, L.; Yao, W. Heterocyclic Compounds as Immunomodulators. WO2018013789 A1, 18 January 2018. [Google Scholar]
- Wu, L.; Zhang, F.; Yao, W. Heterocyclic Compounds as Immunomodulators. WO2018044783 A1, 8 March 2018. [Google Scholar]
- Xiao, K.; Zhang, F.; Wu, L.; Yao, W. Heterocyclic compounds as immunomodulators. US20170362253 A1, 21 December 2017. [Google Scholar]
- Wu, L.; Qian, D.Q.; Lu, L.; Lajkiewicz, N.; Konkol, L.C.; Li, Z.; Zhang, F.; Li, J.; Wang, H.; Xu, M.; et al. Heterocyclic Compounds as Immunomodulators. US 20180177784 A1, 28 June 2018. [Google Scholar]
- Lange, C.; McMurtrie, D.J.; Malathong, V.; Punna, S.; Singh, R.; Yang, J.; Zhang, P. Immunomodulator Compounds. WO2018005374A1, 11 January 2018. [Google Scholar]
- Lange, C.; McMurtrie, D.J.; Malathong, V.; Mali, V.R.; Mcmahon, J.; Roth, H.S.; Singh, R.; Wang, Y.; Yang, J.; Zhang, P. Immunomodulator Compounds. WO2019023575A1, 31 January 2019. [Google Scholar]
- Vilalta Colomer, M.; Li, S.; Malathong, V.; Lange, C.; McMurtrie, D.; Yang, J.; Roth, H.; McMahon, J.; Campbell, J.J.; Ertl, L.S.; et al. A small molecule human PD-1/PD-L1 inhibitor promotes T cell immune activation and reduces tumor growth in a preclinical model. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Yang, Y.; Zhou, C.; Lai, F.; Ji, M.; Jing, X.; Xue, N.; Zheng, Y.; Chen, H.; et al. Phenylate Derivative, Preparation Method Therefor, and Pharmaceutical Composition and Uses Therof. WO2017202276 A1, 30 November 2017. [Google Scholar]
- Li, S.; Xiao, J.; Liu, A.; Wei, X.; Zhong, W.; Zheng, Z.; Wang, X.; Xie, Y.; Zhao, G.; Li, H. Resorcinol Compound and Medicinal Use Thereof. CN107286057 A, 24 October 2017. [Google Scholar]
- Sun, H.; Xin, T.; Wen, X.; Wu, Y.; Yuan, H. 2-Substituted Isonicotinic Acid Compound, Preparation Method and Application Thereof. CN106632021, 10 May 2017. [Google Scholar]
- Wang, M. Symmetric or Semi-symmetric Compounds Useful as Immunomodulators. WO2018/026971, 8 February 2019. [Google Scholar]
- Webber, S.; Almassy, R.J. Immune Checkpoint Inhibitors Compositions and Methods Thereof. WO2018/045142 A1, 8 March 2018. [Google Scholar]
- Aktoudianakis, E.; Appleby, T.; Aesop, C.; Zhimin, D.; Graupe, M.; Guerrero, J.; Jabri, S.; Lad, L.; Machicao Tello, P.A.; Medley, J.W.; et al. PD-1/PD-L1 INHIBITORS. US 2018/0305315 A1, 25 October 2018. [Google Scholar]
- Qin, M.; Cao, Q.; Zheng, S.; Tian, Y.; Zhang, H.; Xie, J.; Xie, H.; Liu, Y.; Zhao, Y.; Gong, P. Discovery of [1,2,4]Triazolo[4,3-a]pyridines as Potent Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2019, 62, 4703–4715. [Google Scholar] [CrossRef]
- Patil, S.P.; Fink, M.A.; Enley, E.S.; Fisher, J.E.; Herb, M.C.; Klingos, A.; Proulx, J.T.; Fedorky, M.T. Identification of Small-Molecule Inhibitors of PD-1/PD-L1 Protein-Protein Interaction. ChemistrySelect 2018, 3, 2185–2189. [Google Scholar] [CrossRef]
- Monga, M.; Sausville, E.A. Developmental therapeutics program at the NCI: Molecular target and drug discovery process. Leukemia 2002, 16, 520–526. [Google Scholar] [CrossRef]
- Dömling, A. Inhibitors of the PD-1/PD-L1 protein/protein interaction. WO2017/118762 A1, 13 July 2017. [Google Scholar]
- Dömling, A. 3-Cyanotiphene Derivatives as Inhibitors of the PD-1/PD-L1 Interaction. WO2019/008152 A1, 10 January 2019. [Google Scholar]
- Dömling, A. 3-(azolylmethoxy)biphenyl Derivatives as Inhibitors of the PD-1/PD-L1 Protein-Protein Interaction. WO2019/008154 A1, 10 January 2019. [Google Scholar]
- Dömling, A. Inhibitors of the PD-1/PD-L1 interaction. WO2019/008156 A1, 10 January 2019. [Google Scholar]
- Wang, Y.; Xu, Z.; Wu, T.; He, M.; Zhang, N. 2018, Aromatic Acetylene or Aromatic Ethylene Compound, Intermediate, Preparation Method, Pharmaceutical Composition and Use Thereof. WO2018006795, 11 January 2018. [Google Scholar]
- Wang, Y.; Zhou, H.; Zhang, N.; Wang, F.; Zhao, Q.; Wu, T.; Zhu, H.; Liu, Y. Abstract 3851: Novel small-molecule inhibitor of PD1/PDL1 pathway demonstrated single agent and drug combo effectiveness in cancer immunotherapy. In Proceedings of the American Association for Cancer Research Annual Meeting, Chicago, IL, USA, 14–18 April 2018; AACR Cancer Research: Philadelphia, PA, USA, 2018; Volume 78. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
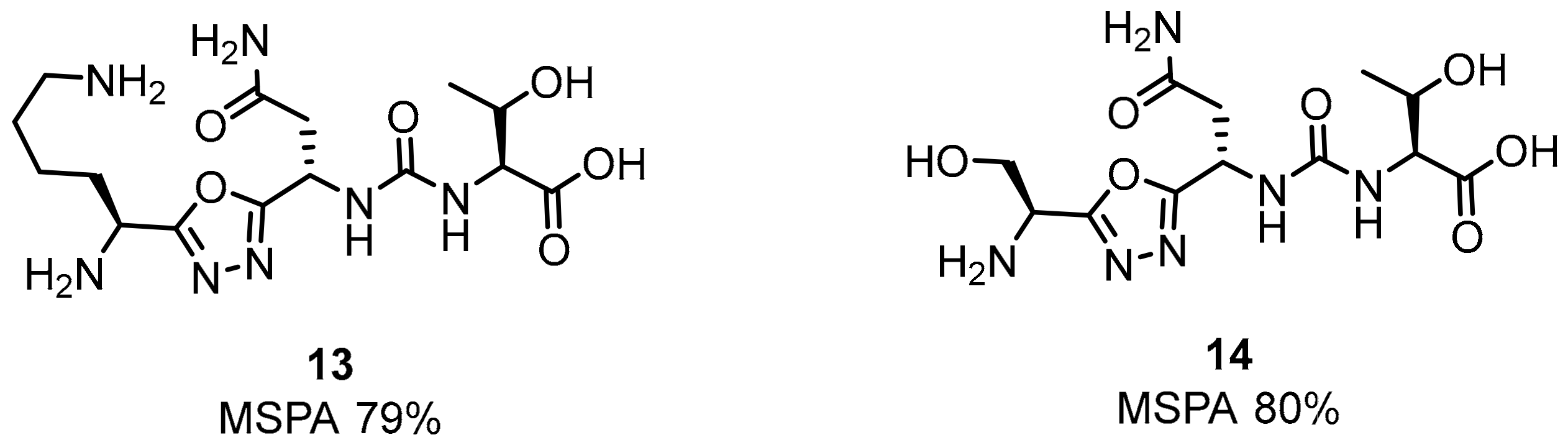{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


























© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Błaszkiewicz, U.; Pustuła, M.; Butera, R.; Dömling, A.; Holak, T.A. Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. https://doi.org/10.3390/molecules24112071
Guzik K, Tomala M, Muszak D, Konieczny M, Hec A, Błaszkiewicz U, Pustuła M, Butera R, Dömling A, Holak TA. Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules. 2019; 24(11):2071. https://doi.org/10.3390/molecules24112071
Chicago/Turabian StyleGuzik, Katarzyna, Marcin Tomala, Damian Muszak, Magdalena Konieczny, Aleksandra Hec, Urszula Błaszkiewicz, Marcin Pustuła, Roberto Butera, Alexander Dömling, and Tad A. Holak. 2019. "Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—A Brief Look at Progress on Small Molecules, Peptides and Macrocycles" Molecules 24, no. 11: 2071. https://doi.org/10.3390/molecules24112071
APA StyleGuzik, K., Tomala, M., Muszak, D., Konieczny, M., Hec, A., Błaszkiewicz, U., Pustuła, M., Butera, R., Dömling, A., & Holak, T. A. (2019). Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules, 24(11), 2071. https://doi.org/10.3390/molecules24112071