
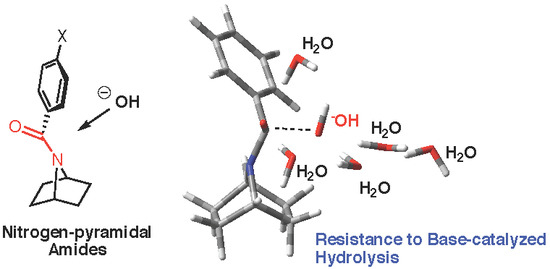
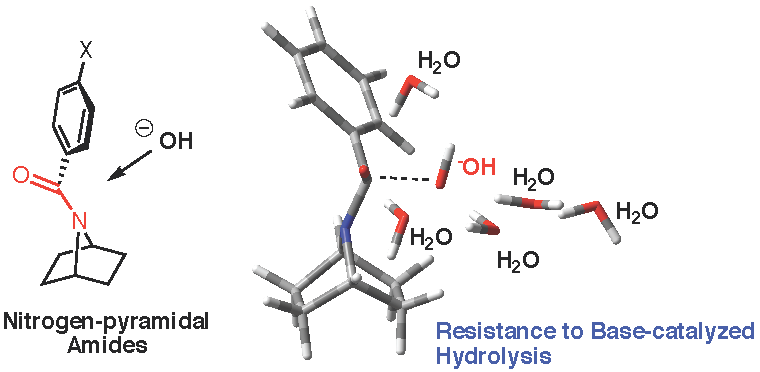
Unexpected Resistance to Base-Catalyzed Hydrolysis of Nitrogen Pyramidal Amides Based on the 7-Azabicyclic[2.2.1]heptane Scaffold


Abstract
:
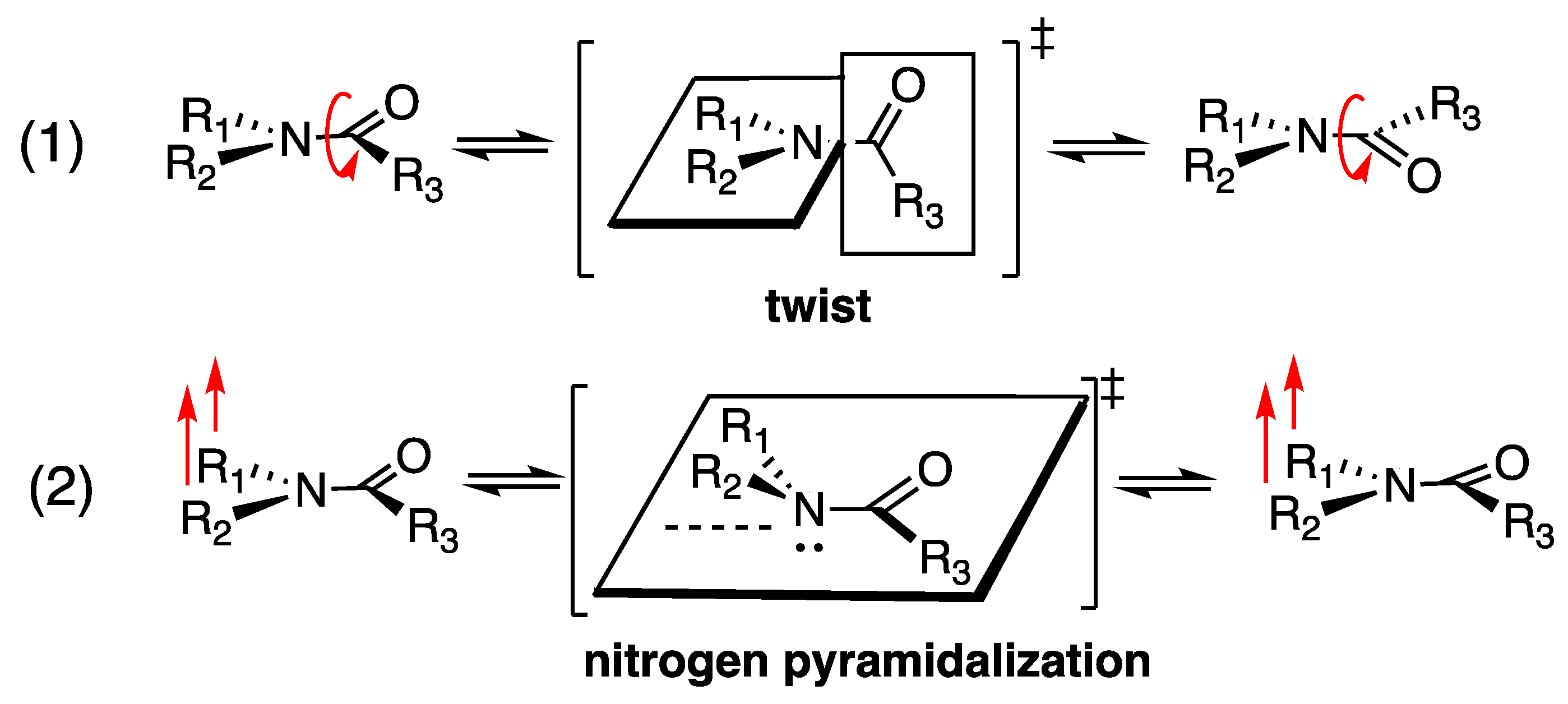
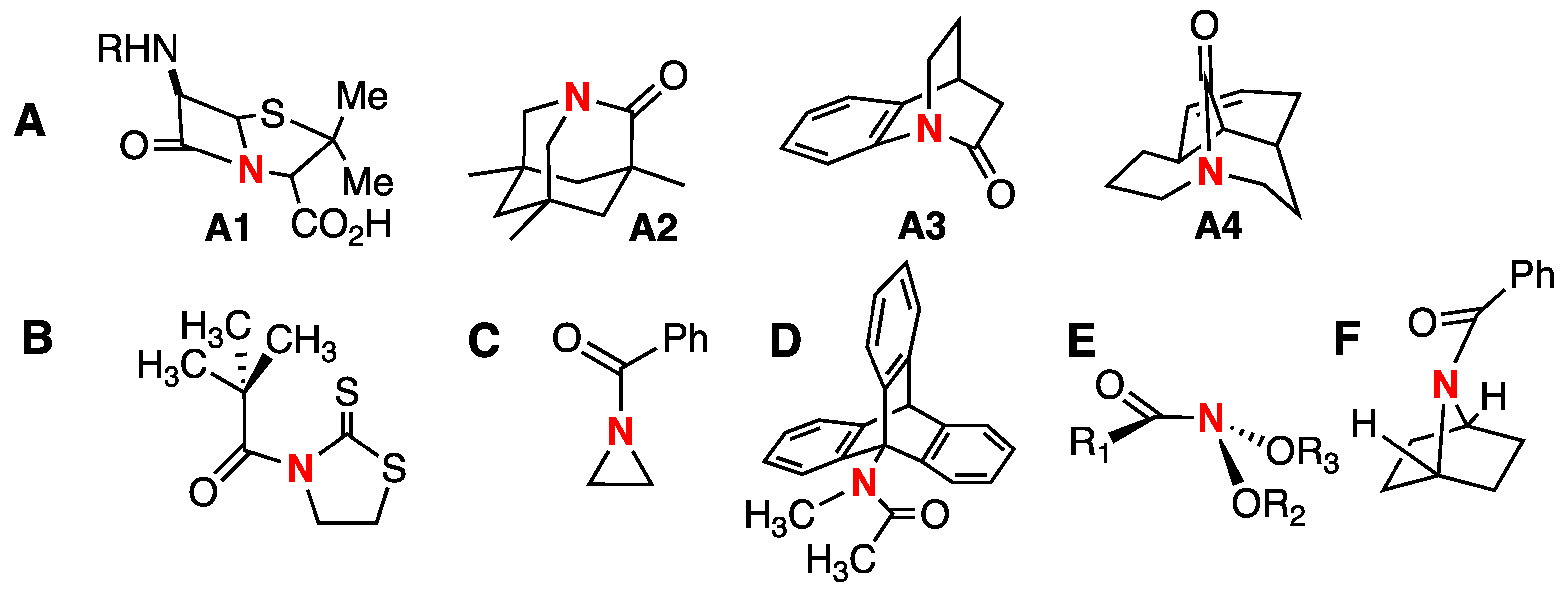
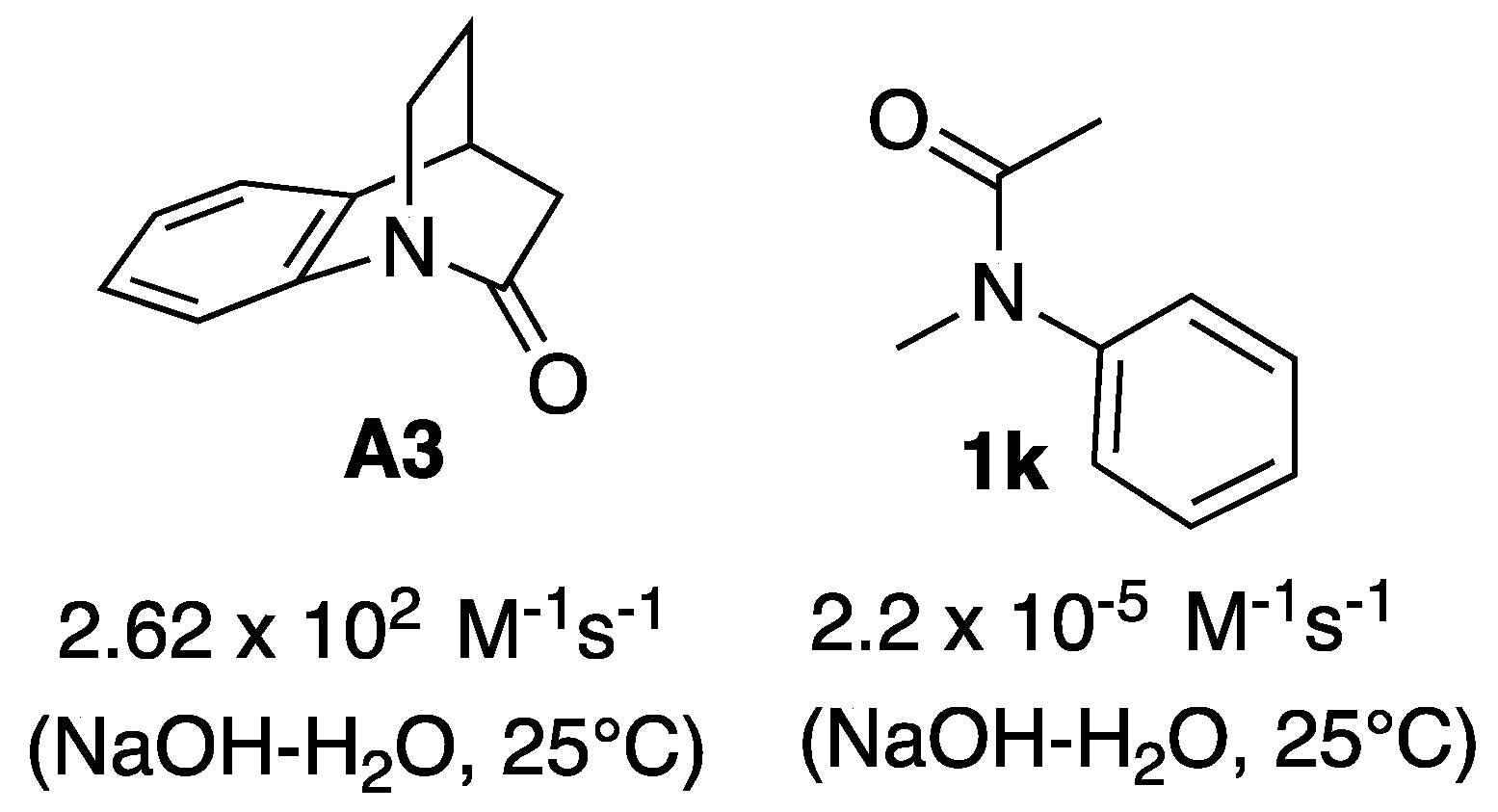
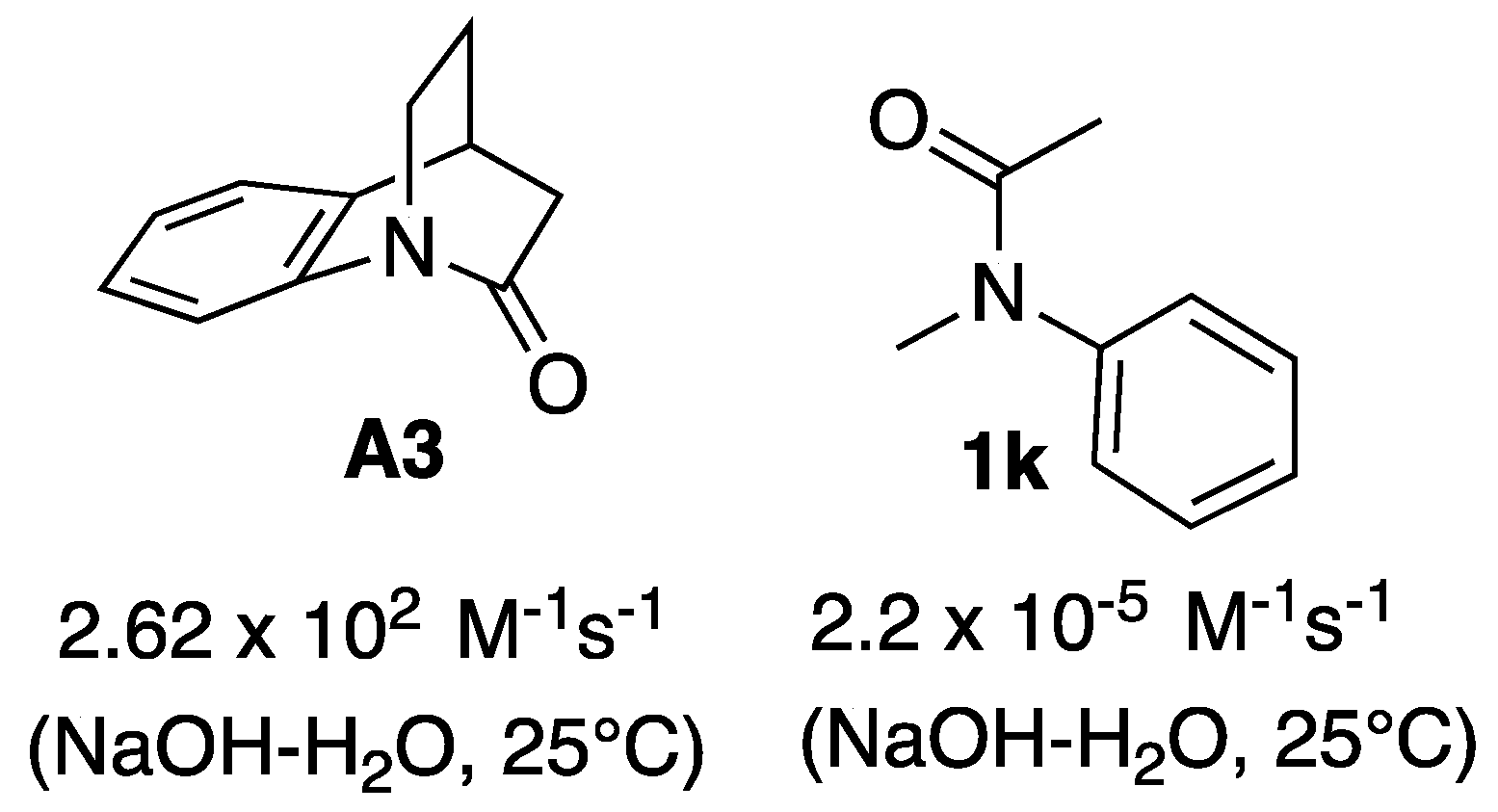
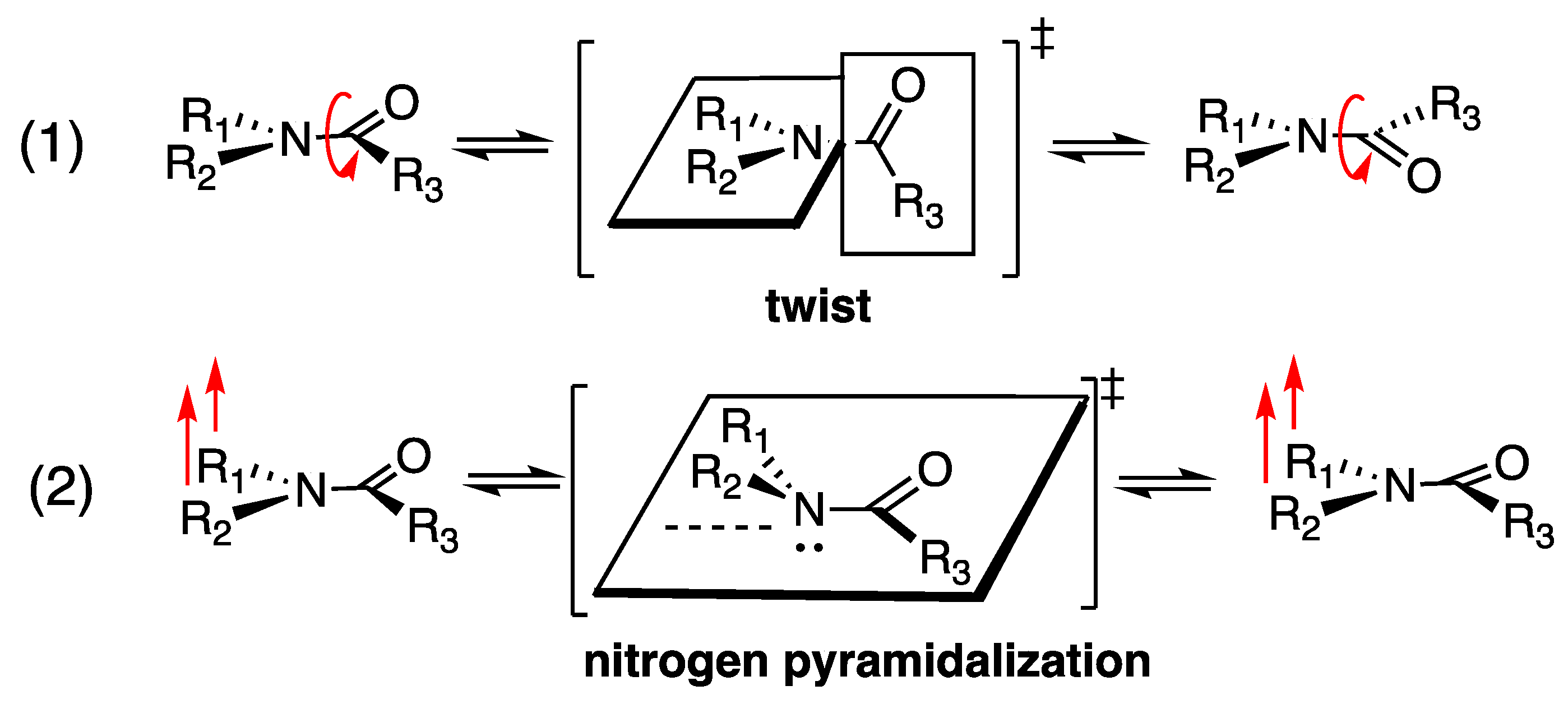
1. Introduction
2. Results and Discussion
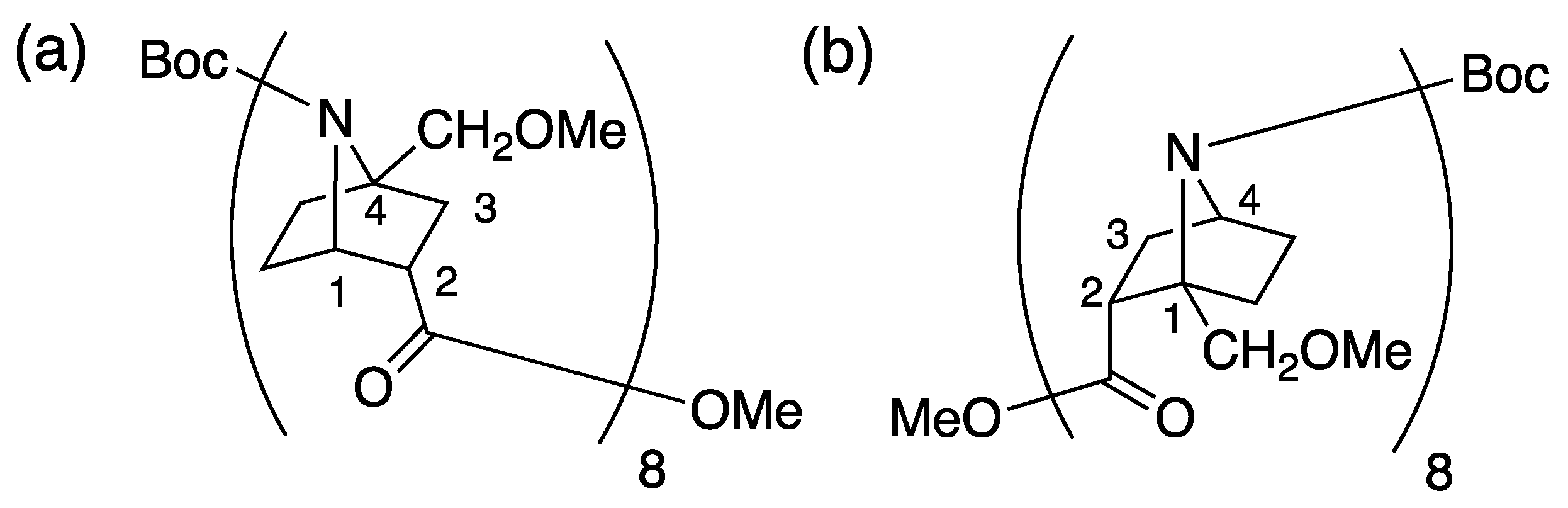
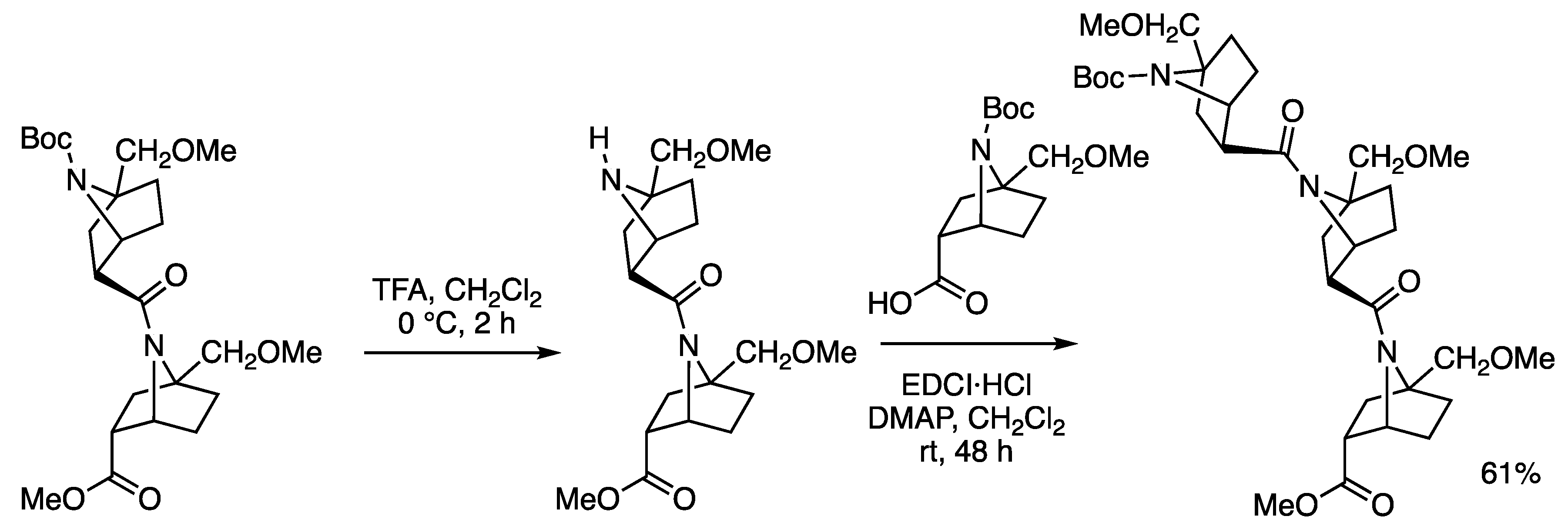
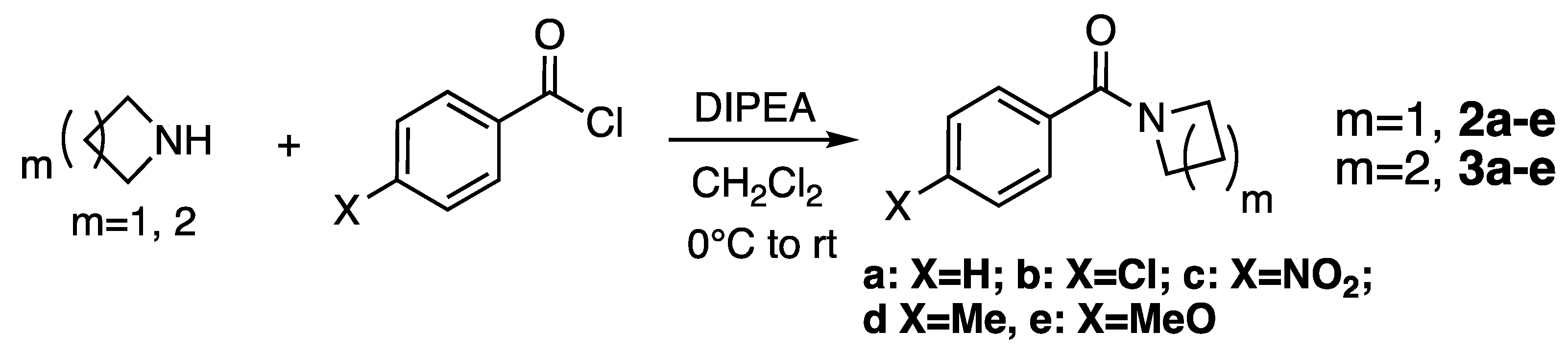
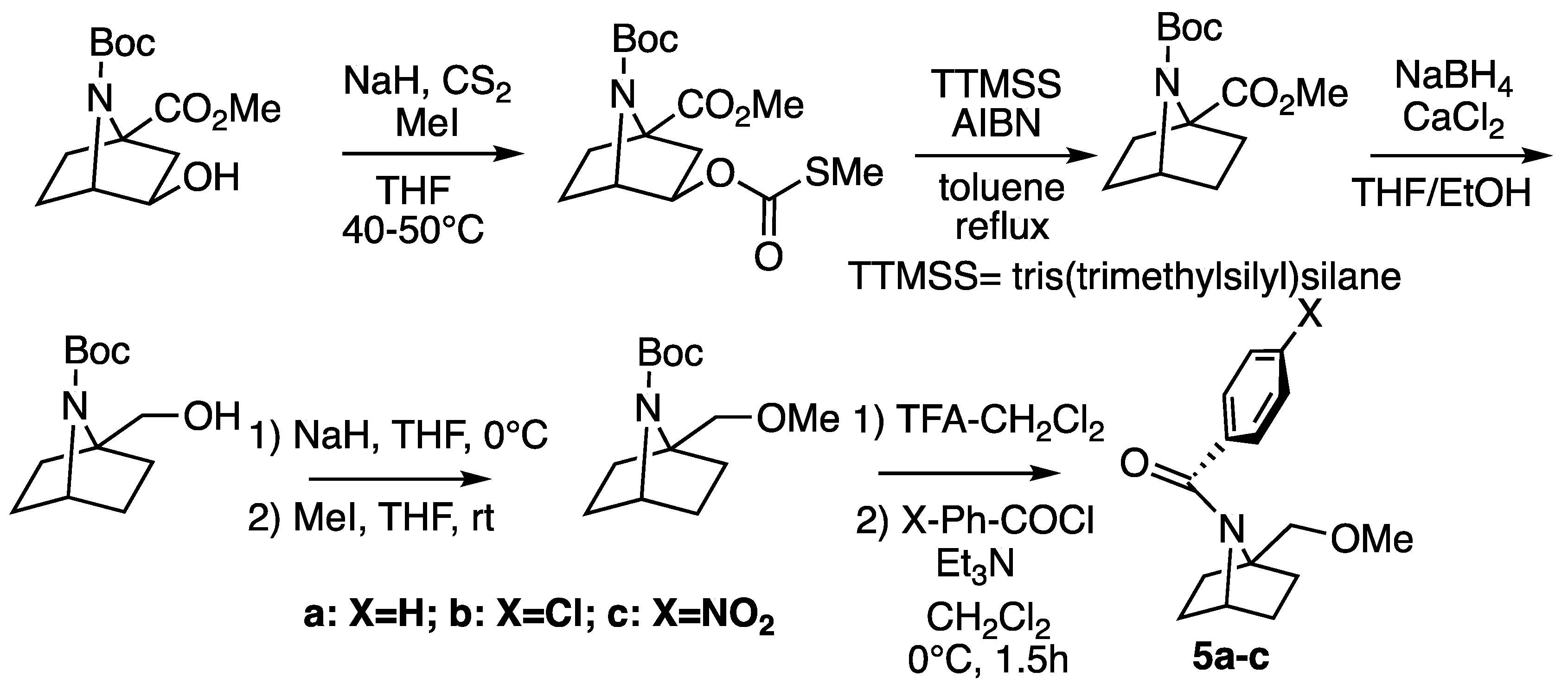
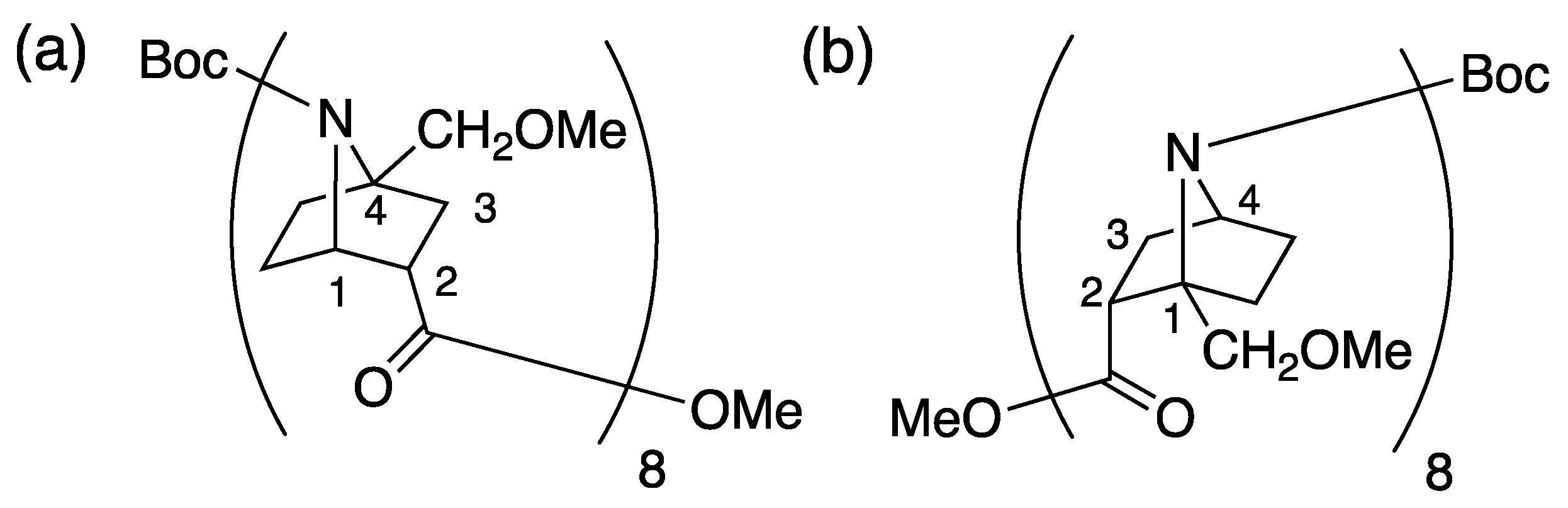
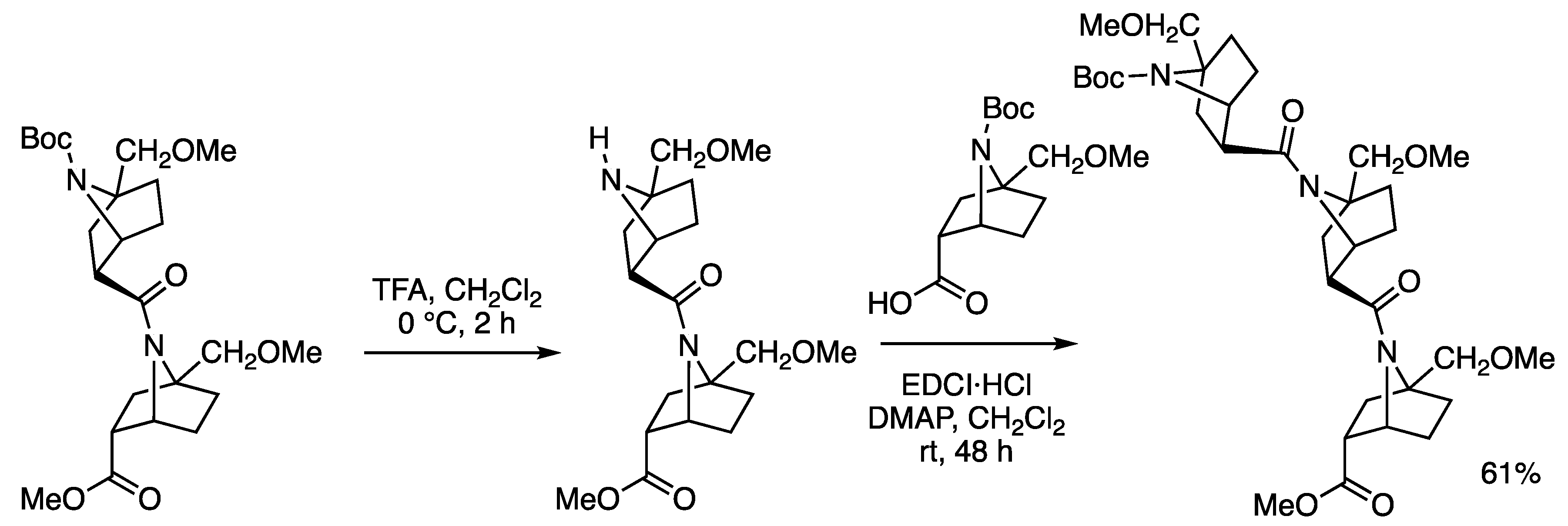
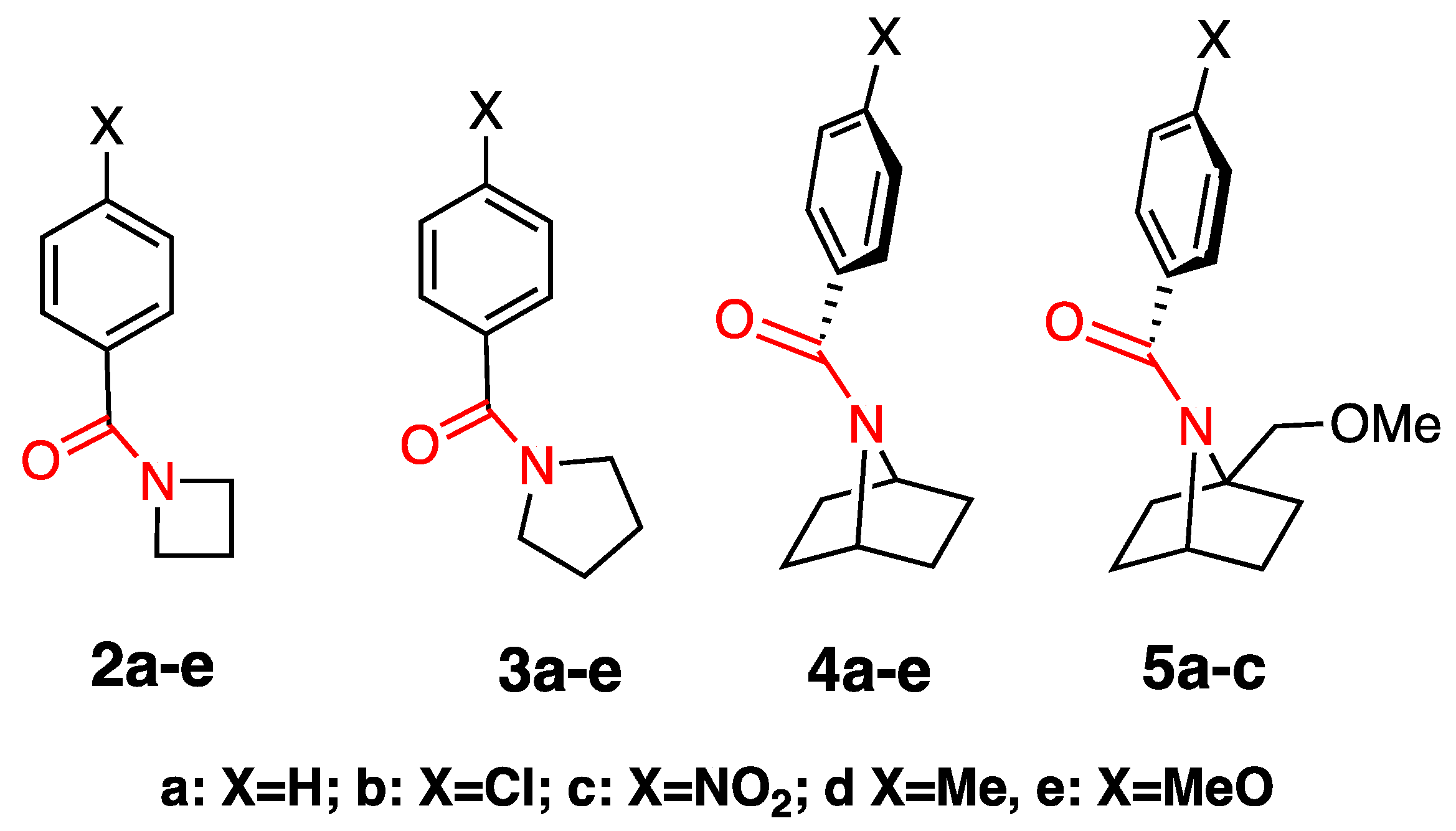
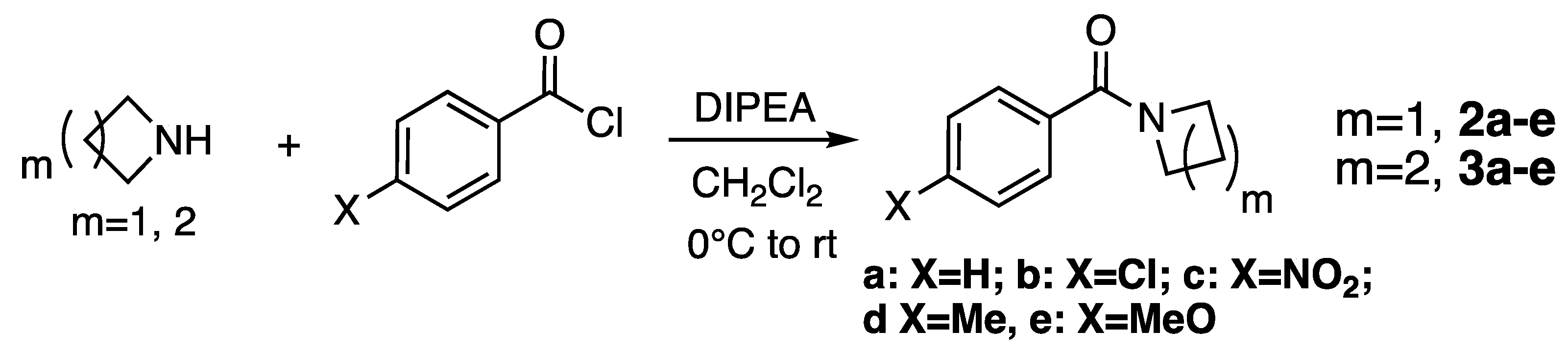
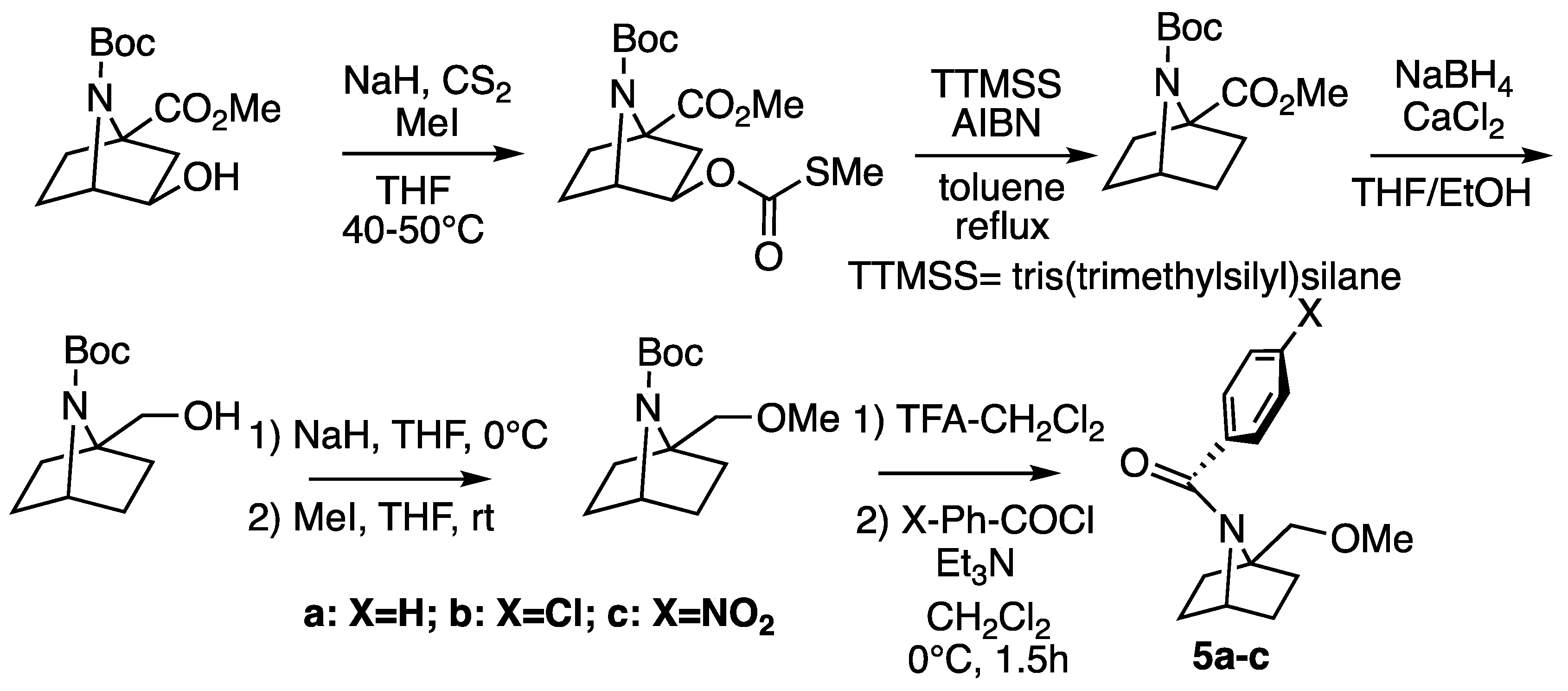
2.1. Synthesis
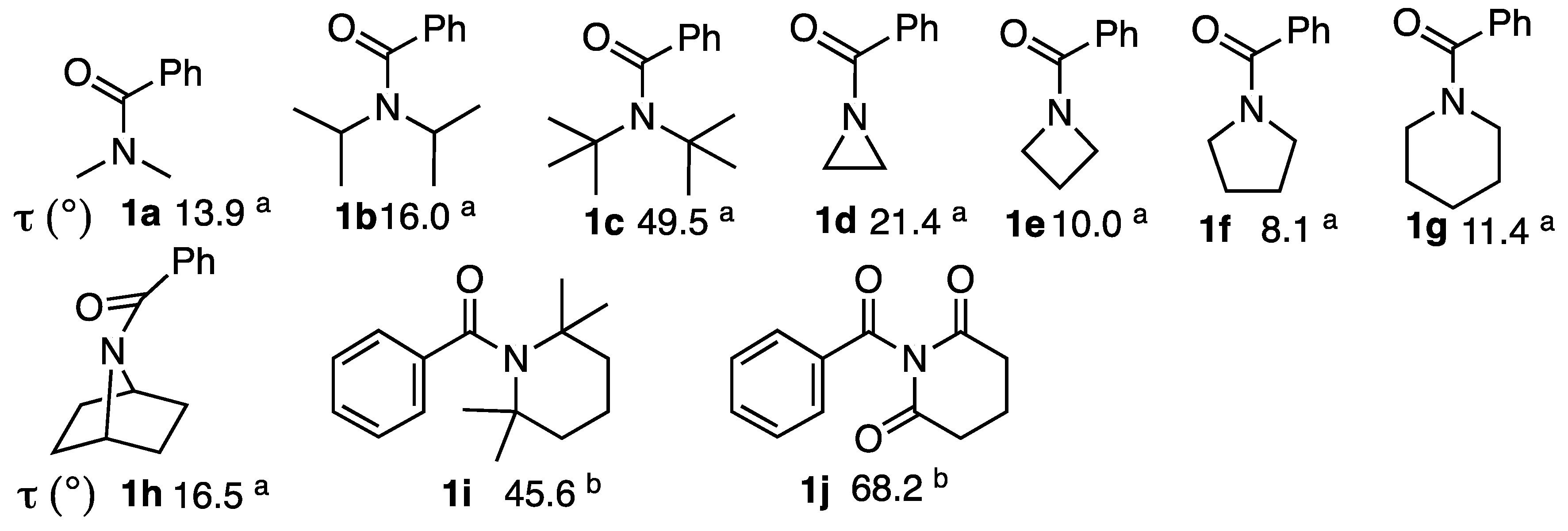
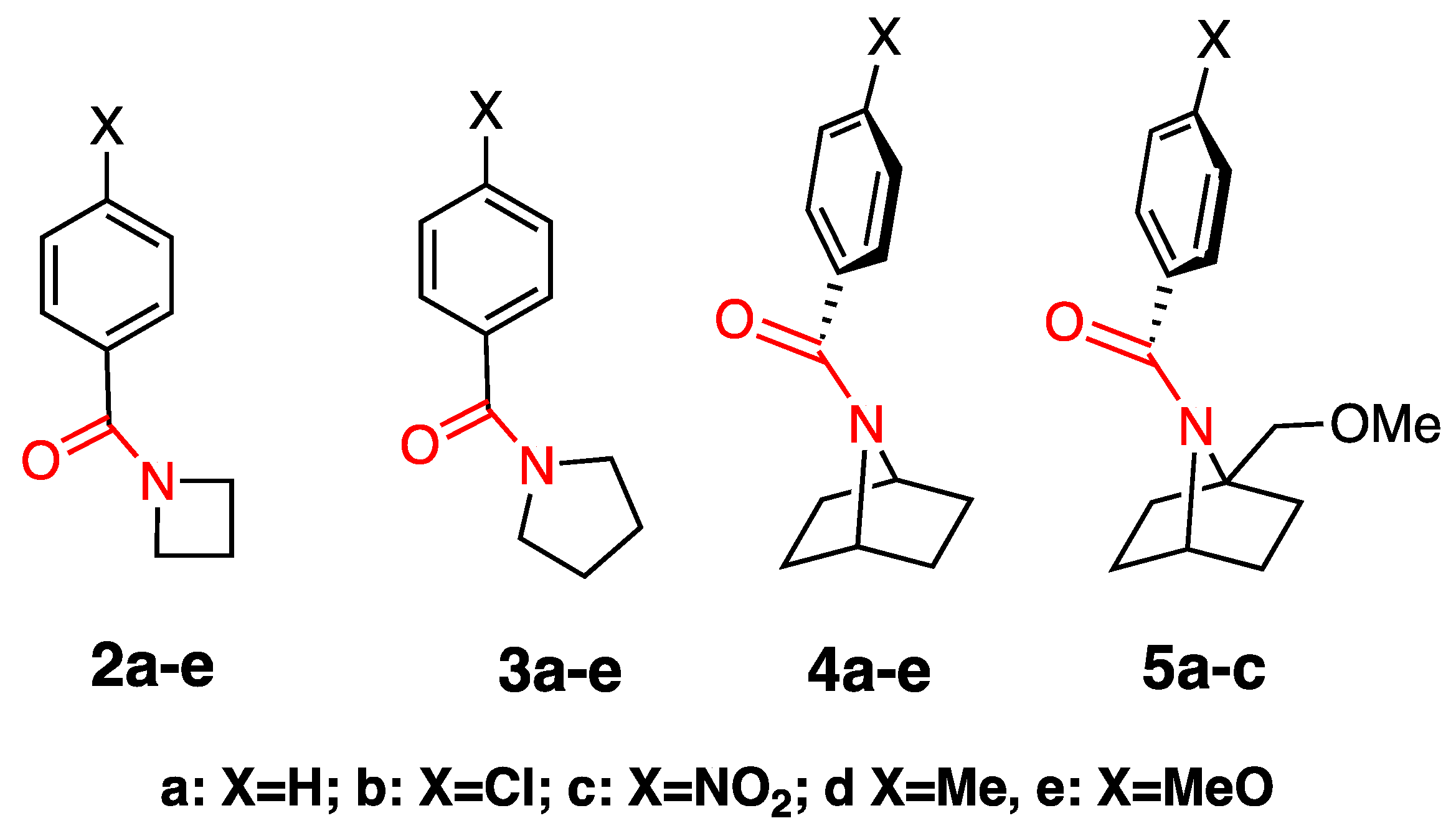
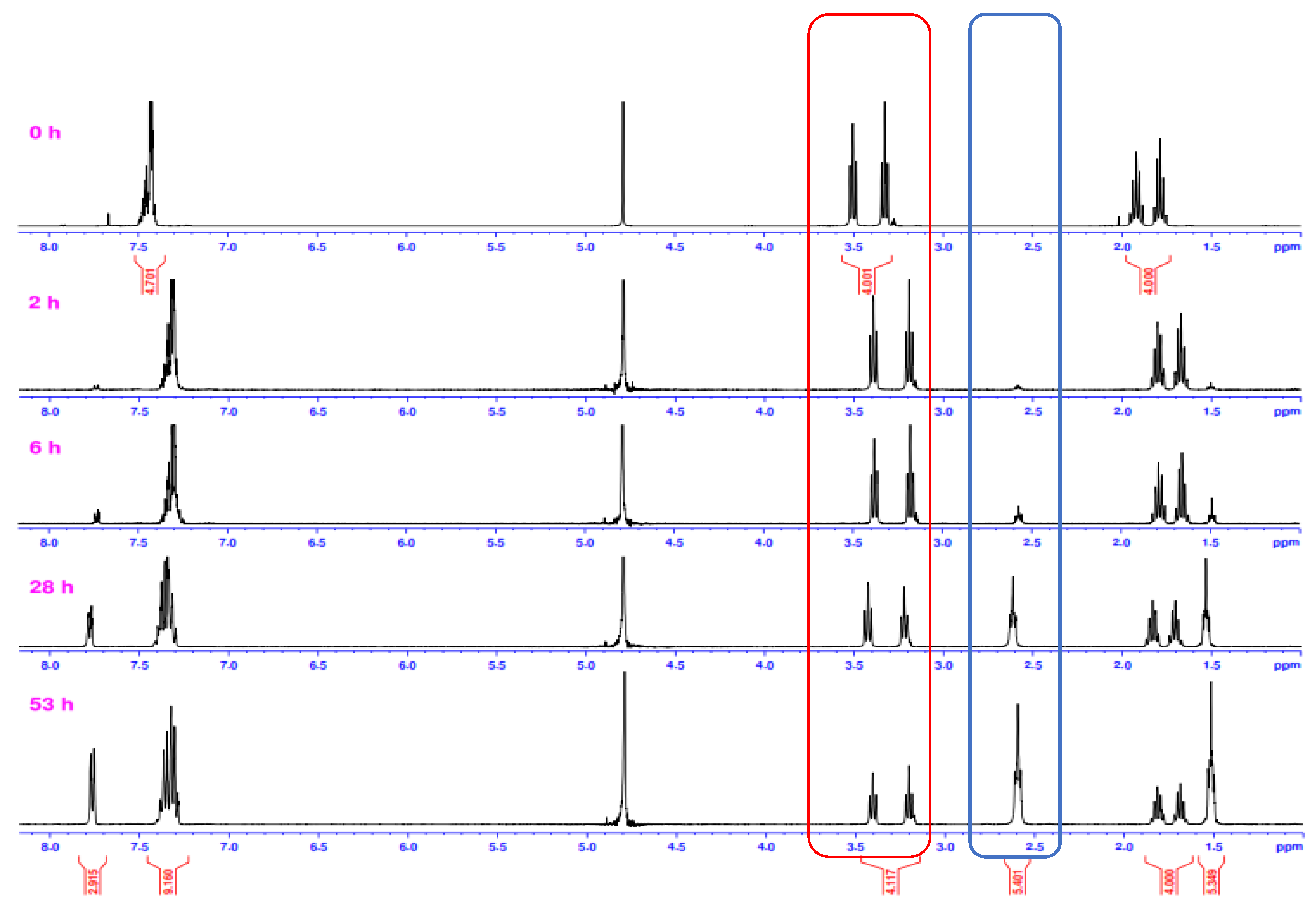
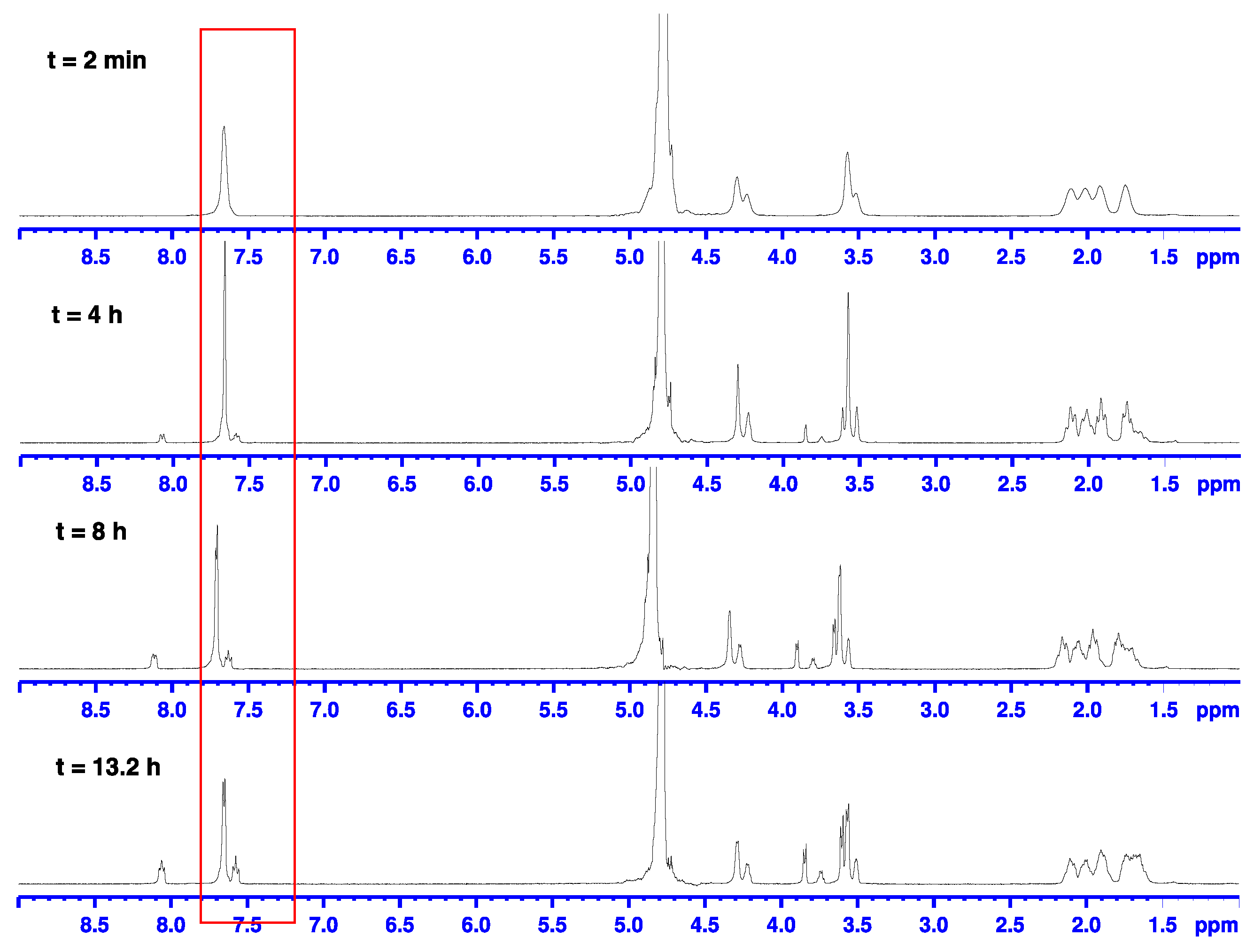
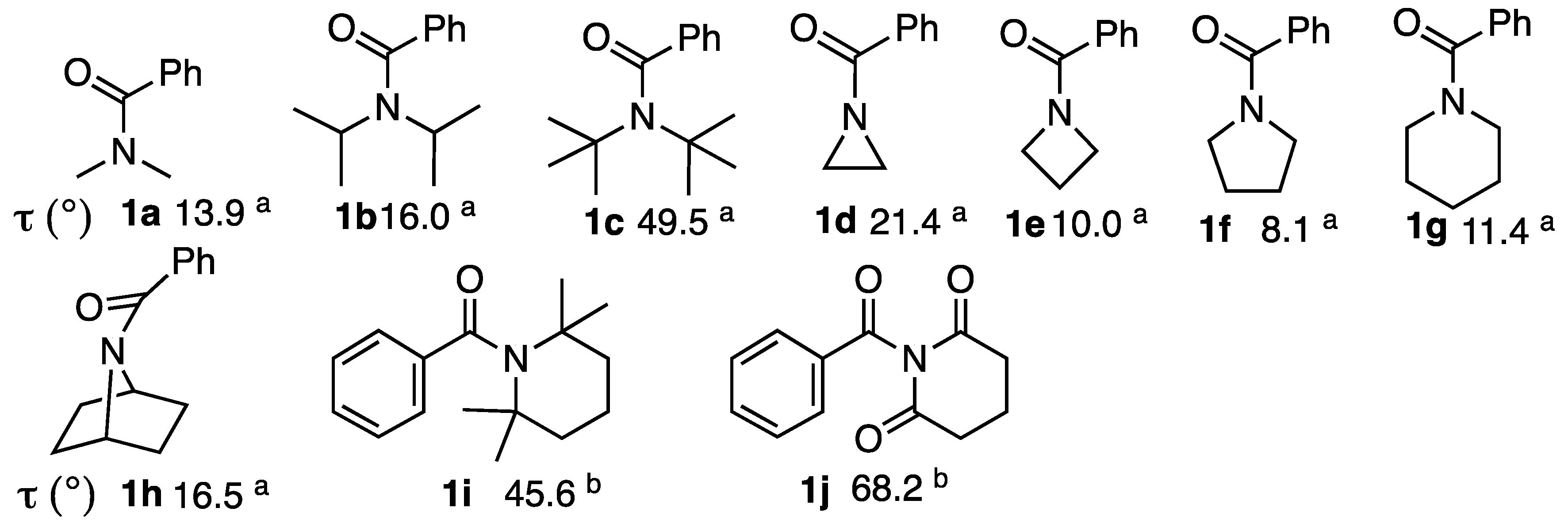
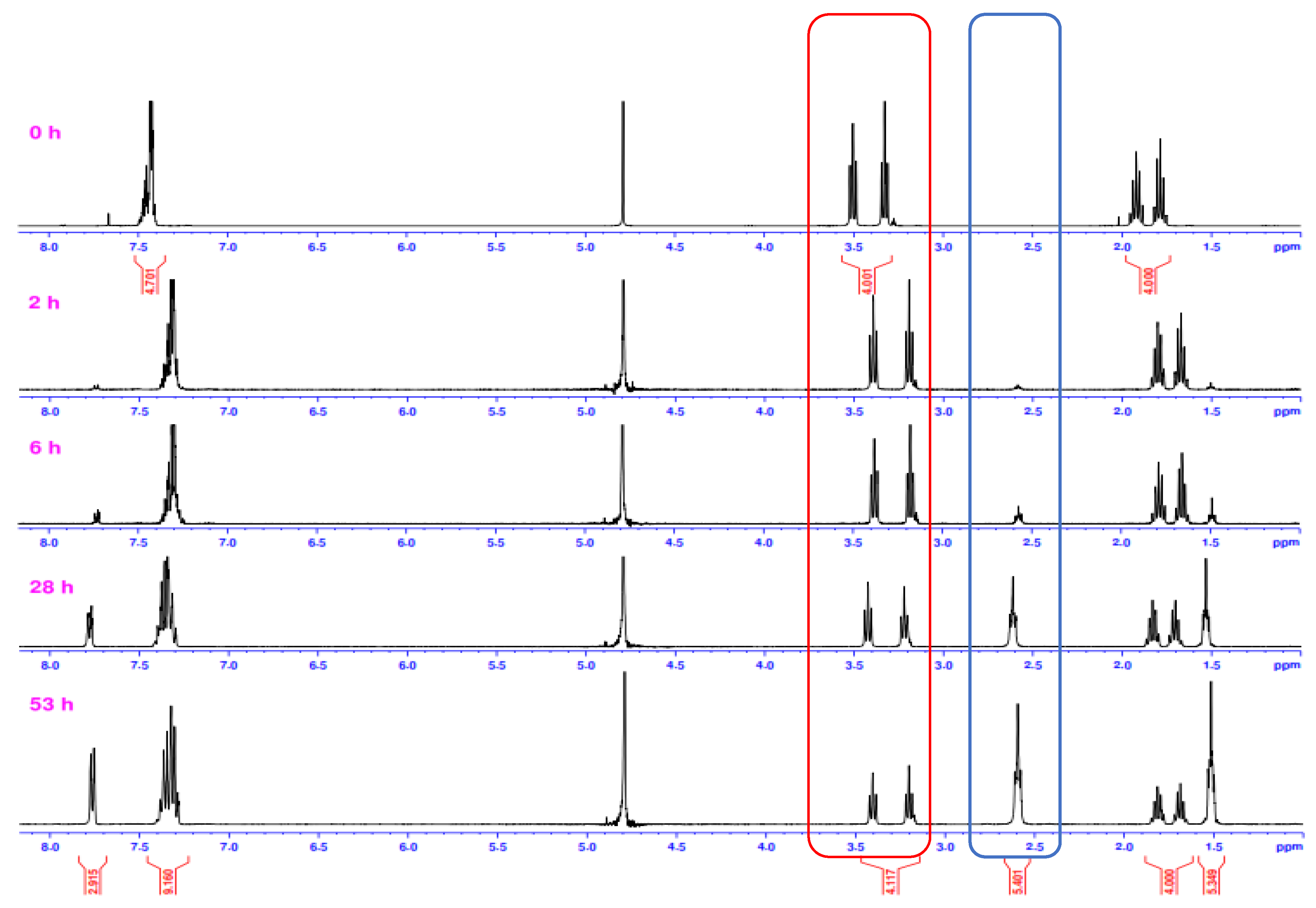
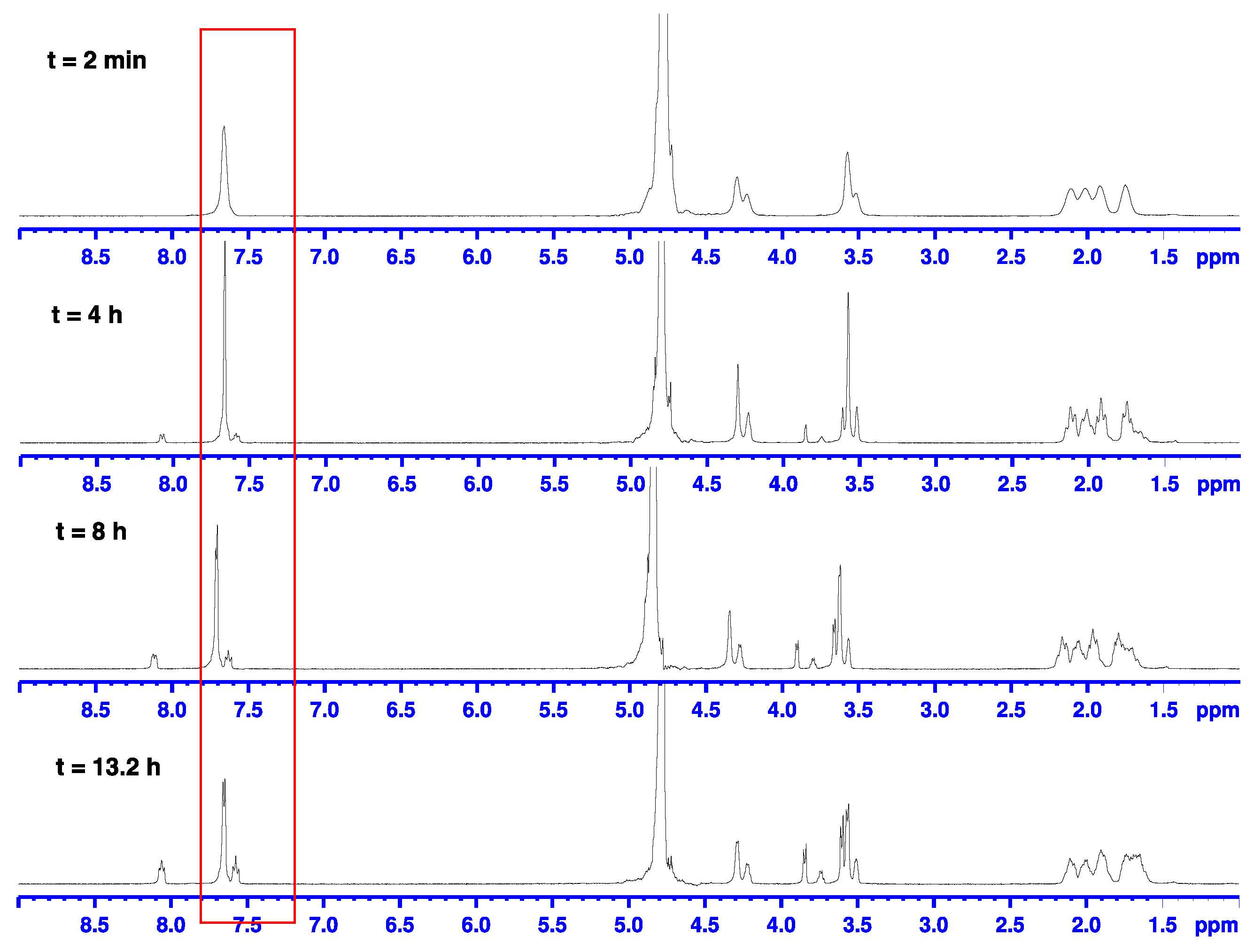
2.2. Alkaline Hydrolysis of Planar Amide 3a
2.2.1. Optimization of Reaction Conditions
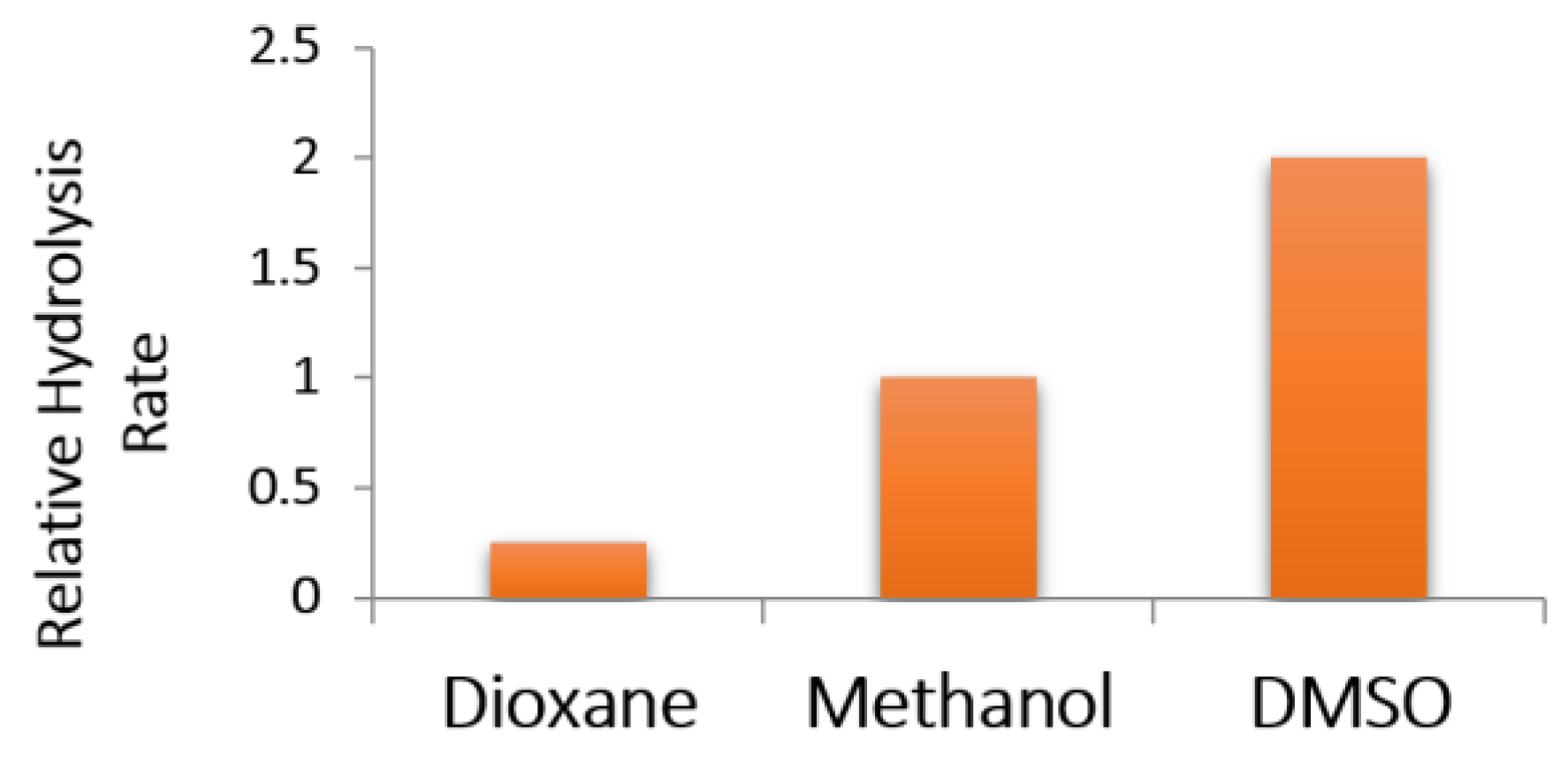
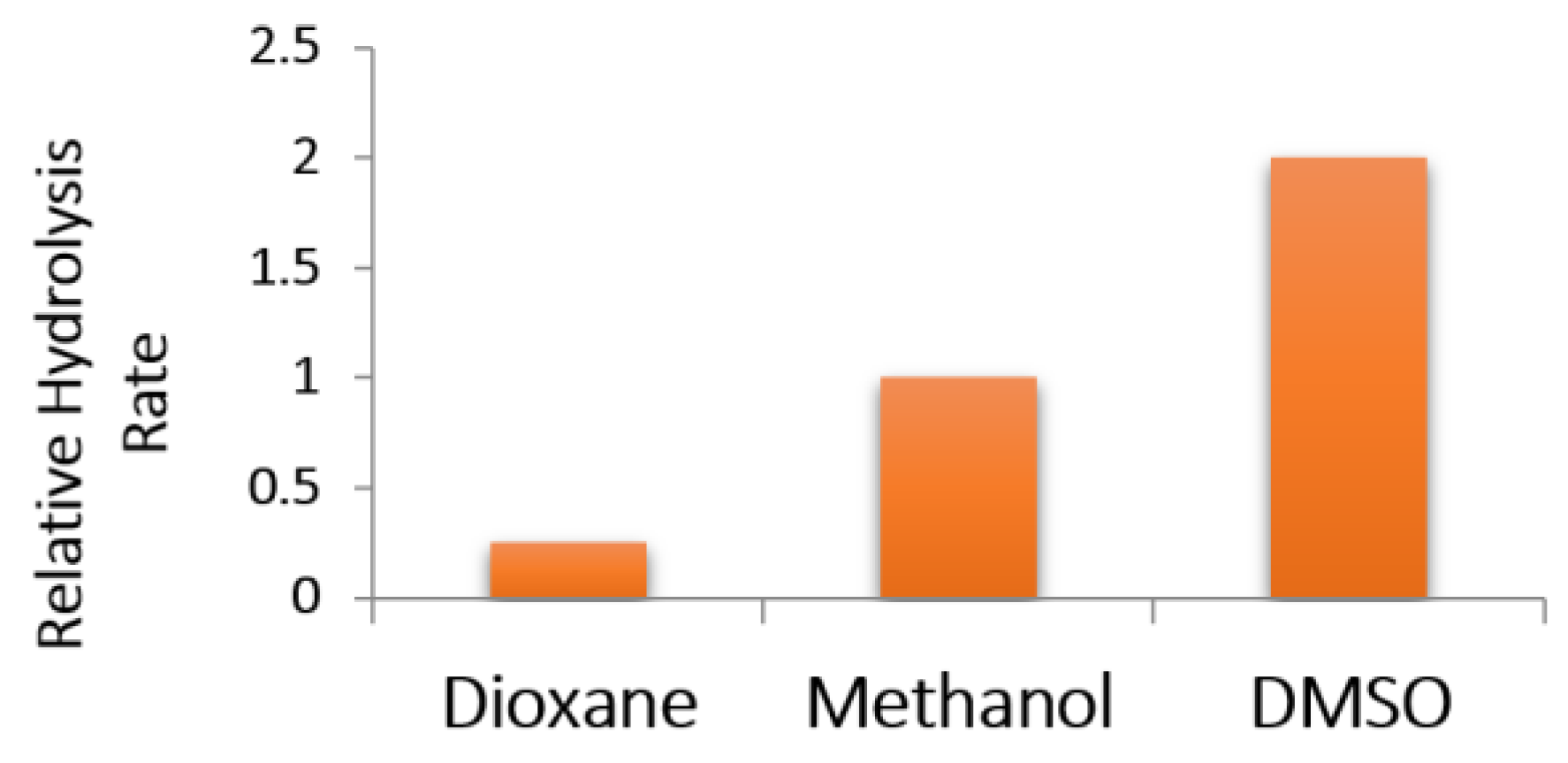
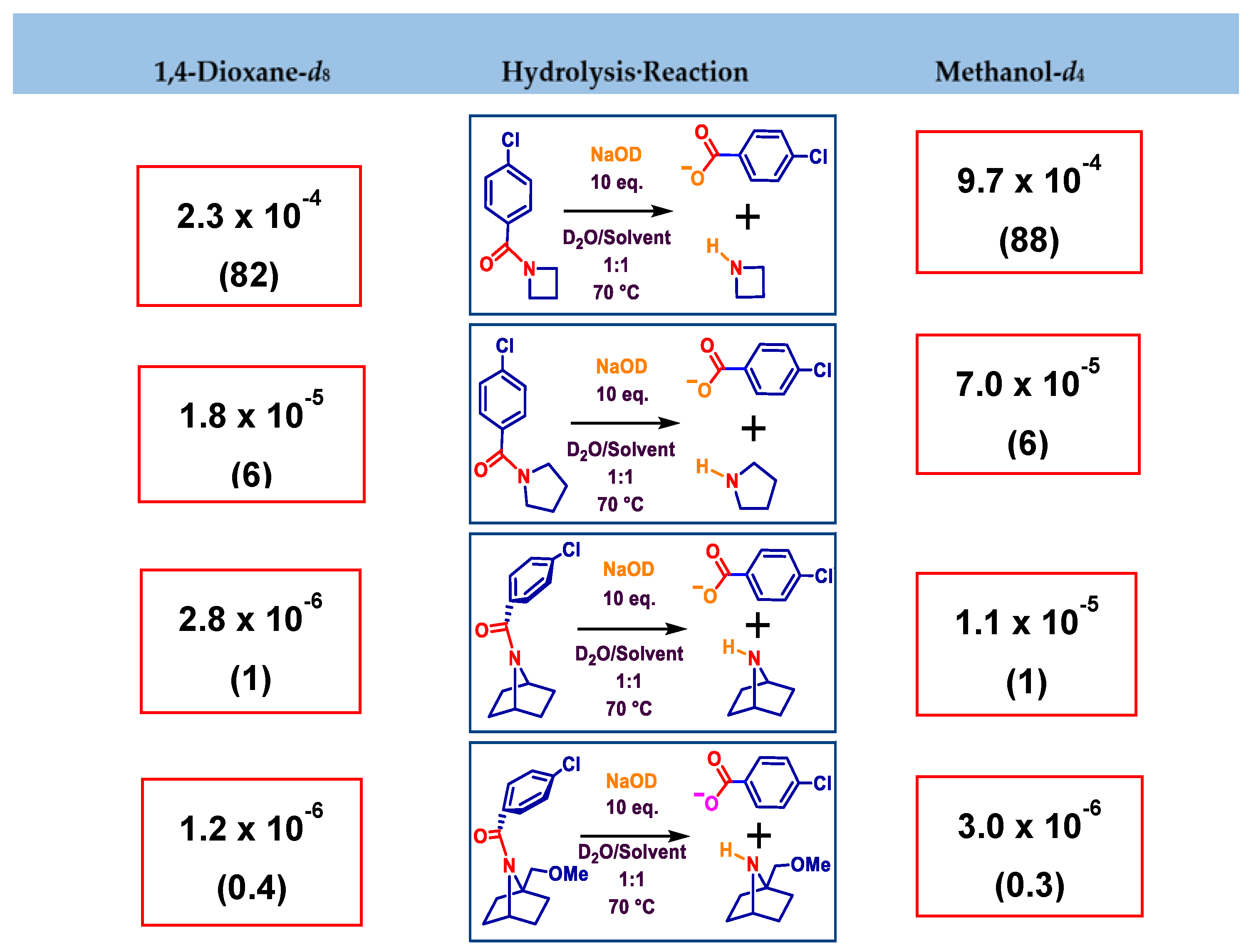
2.2.2. Alkaline Hydrolysis of Amides in Two Solvents
2.2.3. Alkaline Hydrolysis in Dioxane
2.2.4. Alkaline Hydrolysis in Methanol
2.2.5. Comparison of Kinetic Data
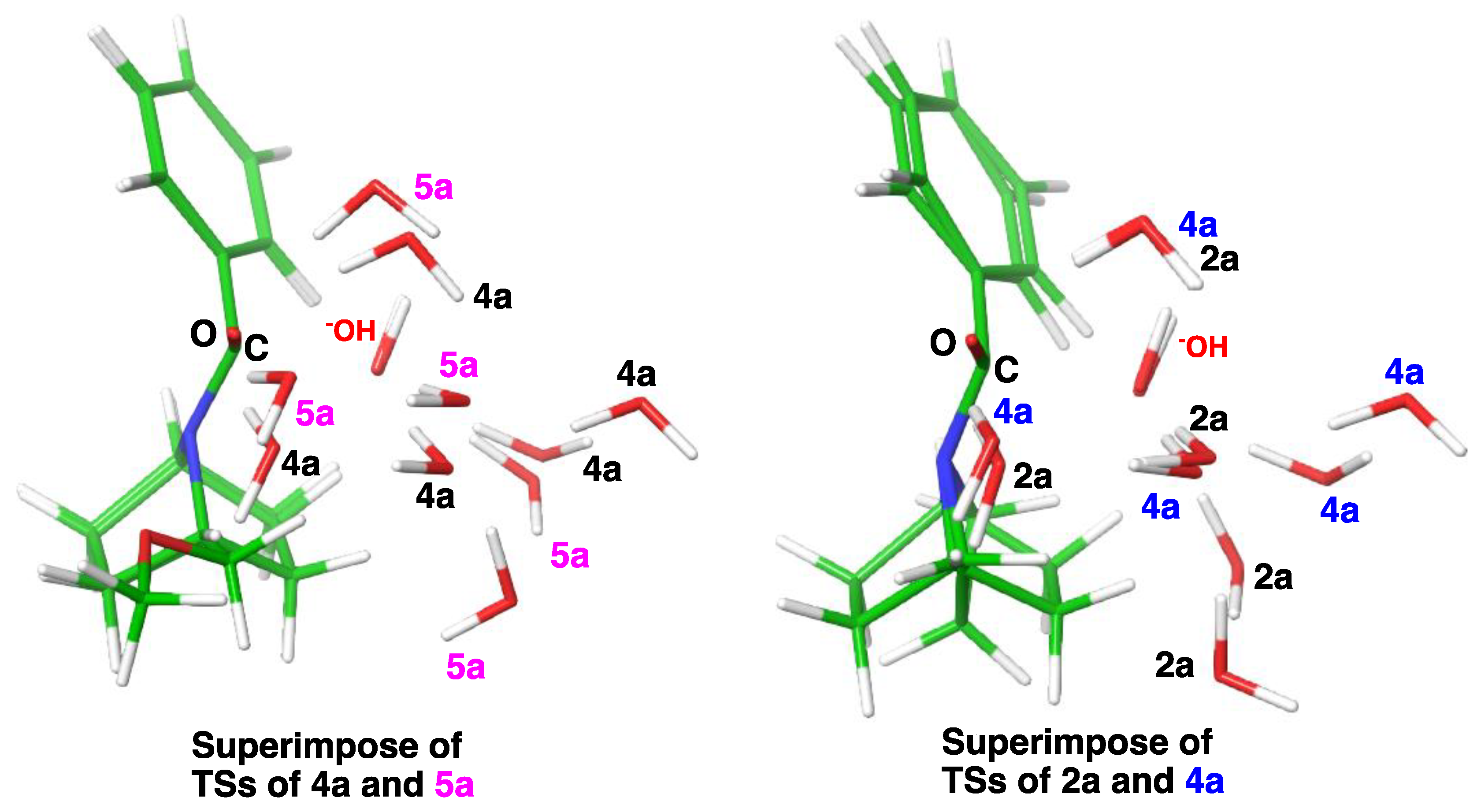
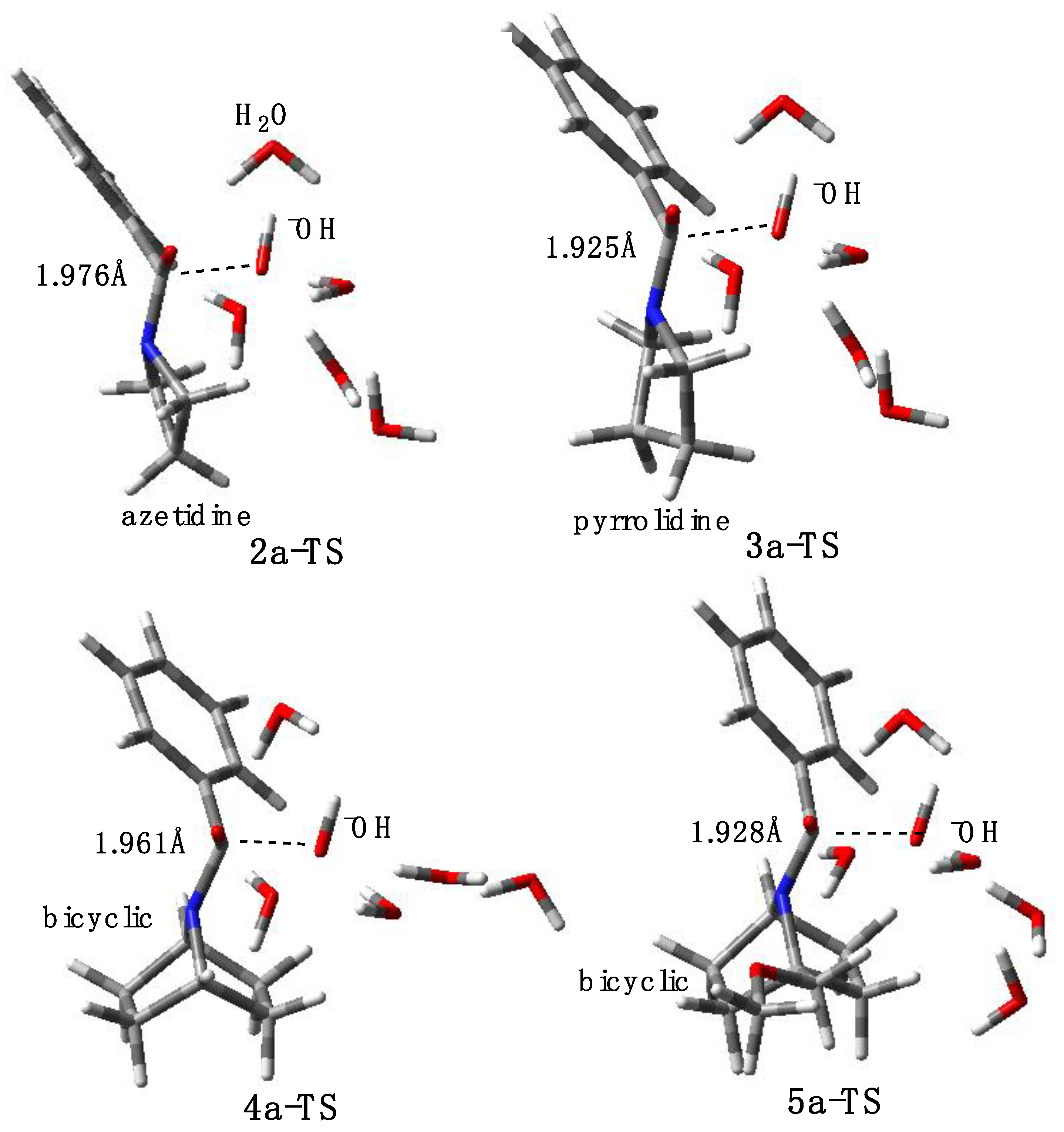
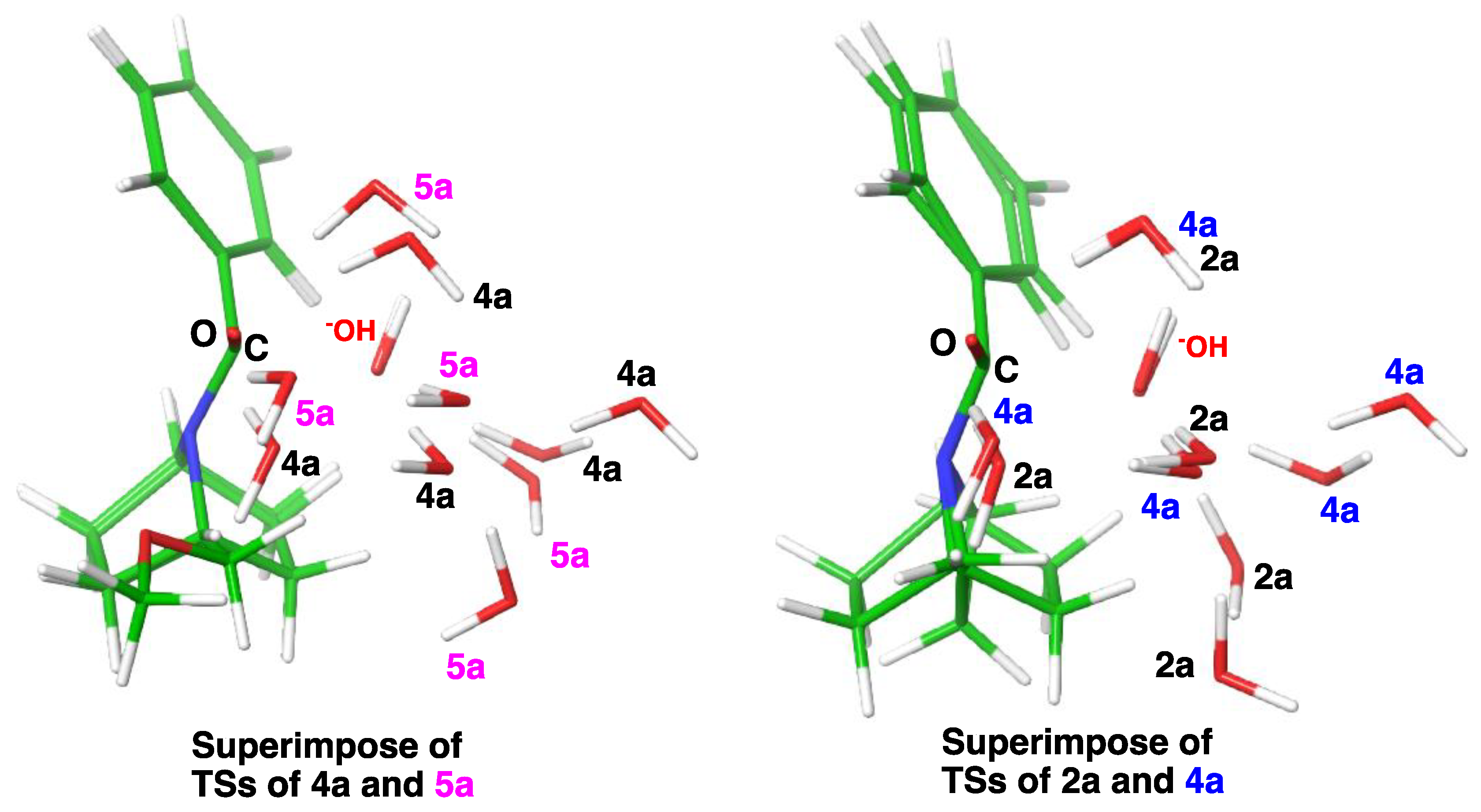
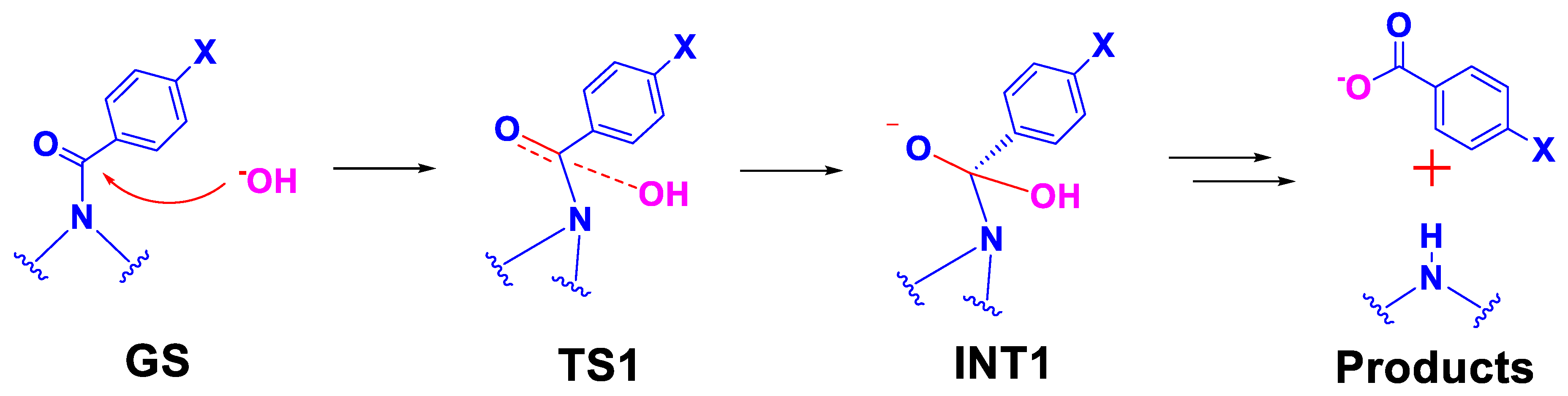
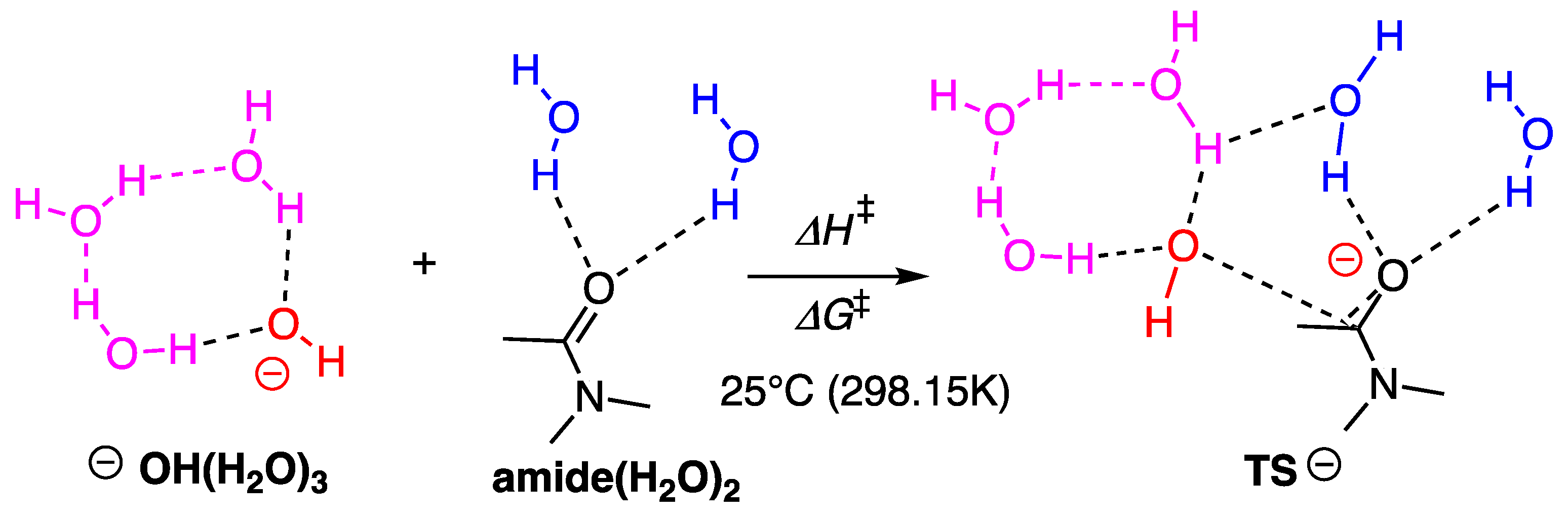
2.3. Computational Studies
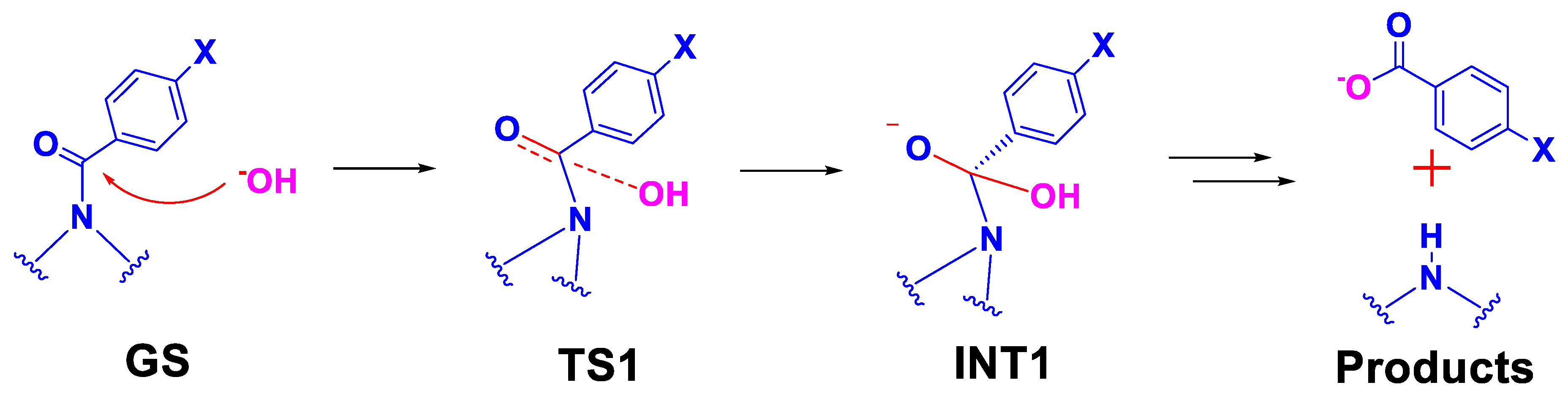
2.3.1. Reaction Model
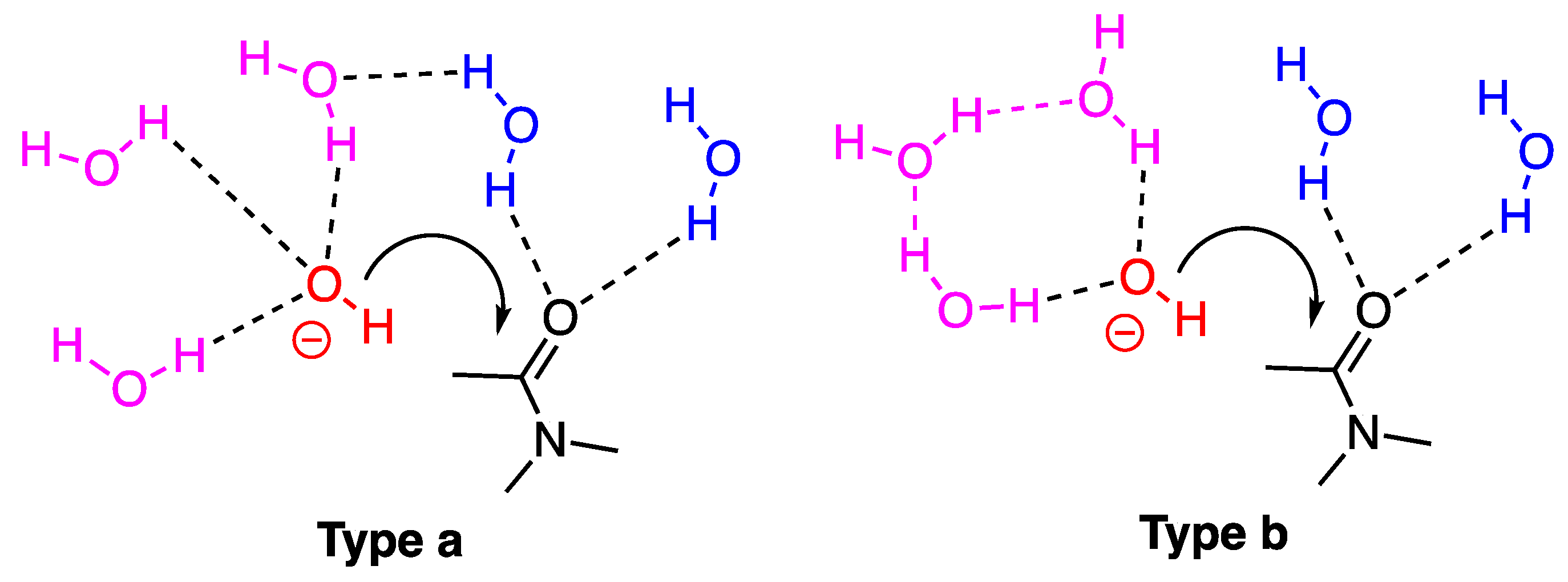
2.3.2. Model with Five Explicit Water Molecules
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis of Amides
Synthesis of N-Benzoylazetidines
Synthesis of N-Benzoylpyrrolidines
Synthesis of N-Benzoylazetidines
Synthesis of N-Benzoyl-1-(Methoxymethyl)-7-azabicyclo[2.2.1]heptanes
3.3. Kinetic Studies
3.4. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Greenberg, A.; Liebman, J.F. The Amide Linkage Structural Significance in Chemistry, Biochemistry and Materials Science; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Fischer, G. Chemical Aspects of Peptide Bond Isomerisation. Chem. Soc. Rev. 2000, 29, 119–127. [Google Scholar] [CrossRef]
- Thakkar, B.S.; Svendsen, J.-S.M.; Engh, R.A. Cis/Trans Isomerization in Secondary Amides: Reaction Paths, Nitrogen Inversion, and Relevance to Peptidic Systems. J. Phys. Chem. A. 2017, 121, 6830–6837. [Google Scholar] [CrossRef] [PubMed]
- Otani, Y.; Nagae, O.; Naruse, Y.; Inagaki, S.; Ohno, M.; Yamaguchi, K.; Yamamoto, G.; Uchiyama, M.; Ohwada, T. An Evaluation of Amide Group Planarity in 7-Azabicyclo[2.2.1]heptane Amides. Low Amide Bond Rotation Barrier in Solution. J. Am. Chem. Soc. 2003, 125, 15191–15199. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.K.; Manhas, M.S.; Bank, B.K.; Srirajan, V. B-Lactams: Cyclic amides of distinction, chapter 7. In The Amide Linkage Structural Significance in Chemistry, Biochemistry and Materials Science; Greenberg, A., Liebman, J.F., Eds.; Wiley: New York, NY, USA, 2000; pp. 157–214. [Google Scholar]
- Komarov, I.V.; Yanik, S.; Ishchenko, A.Y.; Davies, J.E.; Goodman, J.M.; Kirby, A.J. The Most Reactive Amide As a Transition-State Mimic For cis-trans Interconversion. J. Am. Chem. Soc. 2015, 137, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.P.; Bennet, A.J.; Brown, R.S.; Santarsiero, B.D. Distorted Amides as Models for Activated Peptide N-C(O) Units. 3. Synthesis, Hydrolytic Profile, and Molecular Structure of 2,3,4,5-Tetrahydro-2-oxo-1,5-propanobenzazepine. J. Am. Chem. Soc. 1991, 113, 5757–5765. [Google Scholar] [CrossRef]
- Aubé, J. A New Twist on Amide Solvolysis. Angew. Chem. Int. Ed. 2012, 51, 3063–3065. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S. Structure and Reactivity of a Highly Twisted Amide. Angew. Chem. Int. Edit. Engl. 1993, 32, 1083–1085. [Google Scholar] [CrossRef]
- Alkorta, I.; Cativela, C.; Elguero, J.; Gil, A.M.; Jiménez, A.I. A Theoretical Study of the Influence of Nitrogen Angular Constraints on the Properties of Amides: Rotation/Inversion Barriers and Hydrogen Bond Accepting Abilities of N-Formylaziridine and—Azirine. New J. Chem. 2005, 29, 1450–1453. [Google Scholar] [CrossRef]
- Yamamoto, G.; Nakajo, F.; Tsubai, N.; Murakami, H.; Mazaki, Y. Structures and Stereodynamics of N-9-Triptycylacetamide and Its N-Alkyl Derivatives. Bull. Chem. Soc. Jpn. 1999, 72, 2315–2326. [Google Scholar] [CrossRef]
- Glover, S.A.; White, J.M.; Rosser, A.A.; Digianantonio, K.M. Structures of N,N-Dialkoxyamides: Pyramidal Anomeric Amides with Low Amidicity. J. Org. Chem. 2011, 76, 9757–9763. [Google Scholar] [CrossRef] [PubMed]
- Ohwada, T.; Achiwa, T.; Okamoto, I.; Shudo, K. On the Planarity of Amide Nitrogen. Intrinsic Pyramidal Nitrogen of N-Acyl-7Azabicyclo[2.2.1]heptanes. Tetrahedron Lett. 1998, 39, 865–868. [Google Scholar] [CrossRef]
- Mujika, J.I.; Matxain, J.M.; Eriksson, L.A.; Lopez, X. Resonance Structures of the Amide Bond: The Advantages of Planarity. Chem. Eur. J. 2006, 12, 7215–7224. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E.; Piontek, A.; Dziuk, B.; Szostak, R.; Szostak, M. Barriers to Rotation in ortho-Substituted Tertiary Aromatic Amides: Effect of Chloro-Substitution on Resonance and Distortion. J. Org. Chem. 2018, 83, 3159–3163. [Google Scholar] [CrossRef] [PubMed]
- Clayden, J.; Foricher, Y.J.Y.; Lam, H.K. Intermolecular Dearomatising Addition of Organolithium Compounds to N-Benzoylamides of 2,2,6,6-Tetramethylpiperidine. Eur. J. Chem. 2002, 3558–3565. [Google Scholar] [CrossRef]
- Hutchby, M.; Houlden, C.E.; Haddow, M.F.; Tyler, S.N.G.; Llloyd-Jones, G.C.; Booker-Milburn, K.I. Switching Pathways: Room-Temperature Neutral Solvolysis and Substitution of Amides. Angew. Chem. Int. Ed. 2012, 51, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Meng, G.; Szostak, M. Synthesis of Biaryls through Nickel-Catalyzed Suzuki–Miyaura Coupling of Amides by Carbon–Nitrogen Bond Cleavage. Angew. Chem. Int. Ed. 2016, 55, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Szostak, M. Twisted Amides; From Obscurity to Broadly Useful Transition-Metal-Catalyzed Reactions by N-C Amide Bond Activation. Chem. Eur. J. 2017, 23, 7157–7173. [Google Scholar] [CrossRef] [PubMed]
- Otani, Y.; Futaki, S.; Kiwada, T.; Sugiura, Y.; Muranaka, A.; Kobayashi, N.; Uchiyama, M.; Yamaguchi, K.; Ohwada, T. Oligomers of β-Amino Acid Bearing Non-Planar Amides Form Ordered Structures. Tetrahedron 2006, 62, 11635–11644. [Google Scholar] [CrossRef]
- Hori, T.; Otani, Y.; Kawahata, M.; Yamaguchi, K.; Ohwada, T. Non-planar Structures of Thiomides Derived from 7-Azabicyclo[2.2.1]heptane. Electronically Tunable Planarity of Thioamides. J. Org. Chem. 2008, 73, 9102–9108. [Google Scholar] [CrossRef] [PubMed]
- Otani, Y.; Watanabe, S.; Ohwada, T.; Kitao, A. Molecular Dynamics Study of Nitrogen-Pyramidalized Bicyclic β-Proline Oligomers: Length-Dependent Convergence to Organized Structure. J. Phys. Chem. B 2017, 121, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Alemán, C.; Jiménez, A.I.; Cativela, C.; Pérez, J.J.; Casanovas, J. Unusually High Pyramidal Geometry of the Bicyclic Amide Nitrogen in a Complex 7-Azabicyclo[2.2.1]heptane Derivative: Theoretical Analysis Using a Bottom-up Strategy. J. Phys. Chem. B 2005, 109, 11836–11841. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, M.; Otani, Y.; Kawahata, M.; Yamaguchi, K.; Ohwada, T. Water-Stable Helical Structure of Tertiary Amides of Bicyclic β-Amino Acid Bearing 7-Azabicyclo[2.2.1]heptane. Full Control of Amide Cis-Trans Equilibrium by Bridgehead Substitution. J. Am. Chem. Soc. 2010, 132, 14780–14789. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Otani, Y.; Liu, X.; Masatoshi, K.; Yamaguchi, K.; Ohwada, T. Robust trans-Amide Helical Structure of Oligomers of Bicyclic Mimics of β-Proline: Impact of Positional Switching of Bridgehead Substituent on Amide cis-trans Equilibrium. J. Org. Chem. 2014, 79, 5287–5300. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Wang, S.; Nara, M.; Takeuchi, K.; Shimada, I.; Otani, Y.; Ohwada, T. Application of C-terminal 7-azabicyclo[2.2.1]heptane to stabilize β-strand-like extended conformation of a neighboring α-amino acid. J. Org. Chem. Accepted for publication.
- Marlier, J.F.; Dopke, N.C.; Johnstone, K.R.; Wirdzig, T.J. A Heavy-Atom Isotope Effect Study of the Hydrolysis of Formamide. J. Am. Chem. Soc. 1999, 121, 4356–4363. [Google Scholar] [CrossRef]
- Mujika, J.I.; Mercero, J.M.; Lopez, X. Water-Promoted Hydrolysis of a Highly Twisted Amides: Rate Acceleration Caused by the Twist of the Amide Bond. J. Am. Chem. Soc. 2005, 127, 4445–4453. [Google Scholar] [CrossRef] [PubMed]
- Gorb, L.; Asensio, A.; Tuñón, I.; Ruiz-López, M.F. The Mechanism of Formamide Hydrolysis in Water from Ab Initio Calculations and Simulations. Chem. Eur. J. 2005, 11, 6743–6753. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cao, Z. Mechanism of Acid-Catalyzed Hydrolysis of Formamide from Cluster-Continuum Model Calculations: Concerted versus Stepwise Pathway. J. Phys. Chem. A 2010, 114, 12918–12927. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Ueta, C. Computational Study of the Effects of Steric Hindrance on Amide Bond Cleavage. J. Phys. Chem. A 2014, 118, 8664–8675. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhan, C.-G. Theoretical Studies of Transition–State Structures and Free Energy Barriers for Base-Catalyzed Hydrolysis of amides. J. Phys. Chem. A 2006, 110, 12644–12652. [Google Scholar] [CrossRef] [PubMed]
- Ammann, C.; Meier, P.; Merbach, A.E. A simple multinuclear NMR thermometer. J. Mag. Reson. 1982, 46, 319–321. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
Sample Availability: Samples of the compounds 2, 3 and 4 are available from the authors. |
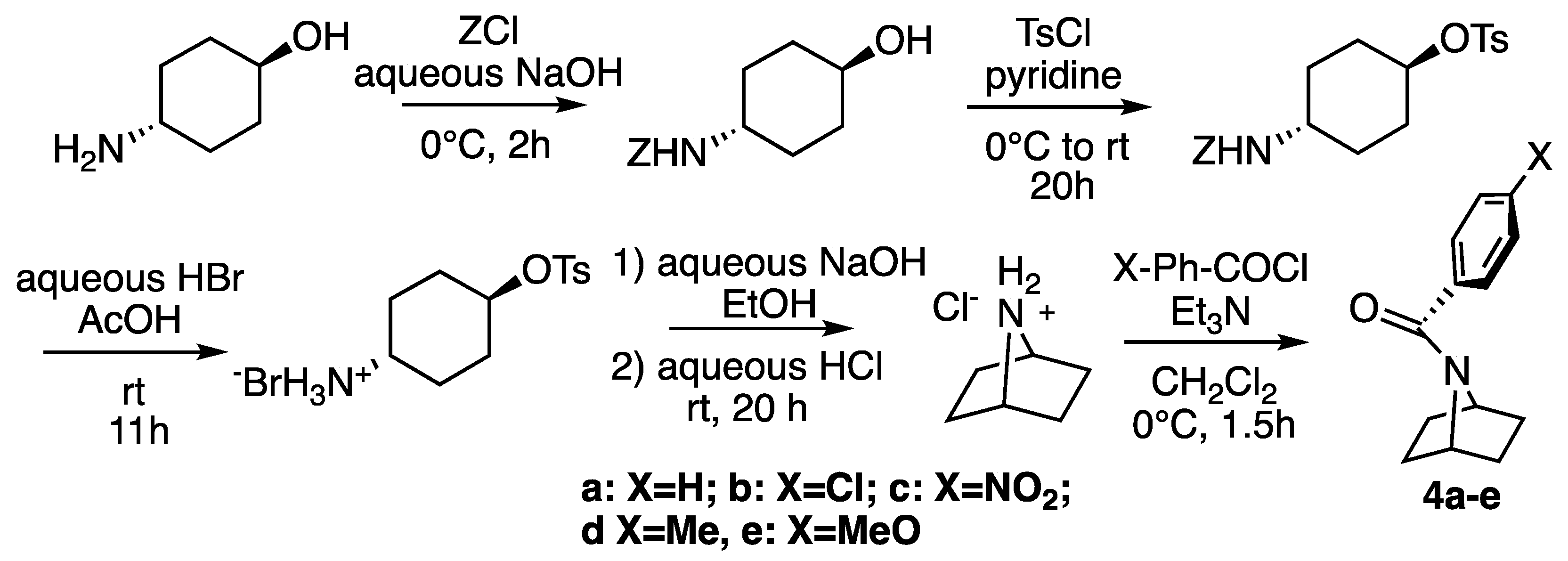




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
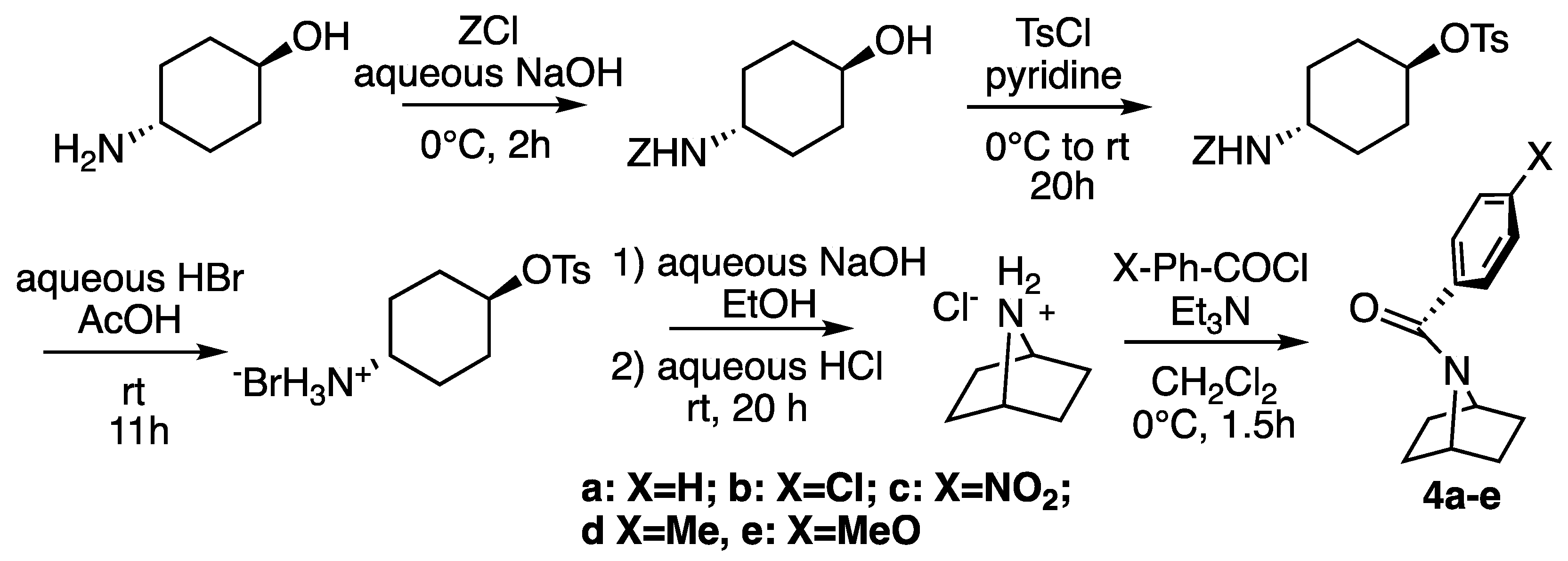{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1,4-Dioxane-d8/D2O (v/v%) | 3c | NaOD | Temperature | −kobs (s−1) |
|---|---|---|---|---|
| 25/75 | 0.05 mmol | 0.5 mmol | 70 °C | 3.0 × 10−4 |
| 50/50 | 0.05 mmol | 0.5 mmol | 70 °C | 9.6 × 10−5 |
| 75/25 | 0.05 mmol | 0.5 mmol | 70 °C | 2.3 × 10−6 |
| Starting Amide | NaOD 40% wt. | D2O | Co-solvent | Temperature |
|---|---|---|---|---|
| 0.05 mmol | 0.5 mmol | 250 µL | 250 µL | 70 °C |
| 1,4-Dioxane-D2O (1:1), NaOD, 70 °C a | Methanol-D2O (1:1), NaOD, 70 °C b | |||||||
|---|---|---|---|---|---|---|---|---|
| x= | 2x | 3x | 4x | 5x | 2x | 3x | 4x | 5x |
| a (H) | 2.1 × 10−5 (5.8) | 7.8 × 10−6 (2.2) | 3.6 × 10−6 (1) | 6.5 × 10−7 (0.2) | 1.1 × 10−4 | 2.9 × 10−5 | ND | ND |
| b (Cl) | 2.3 × 10−4 (82.1) | 1.8 × 10−5 (6.4) | 2.8 × 10−6 (1) | 1.2 × 10−6 (0.4) | 9.7 × 10−4 (88.2) | 7.0 × 10−5 (6.4) | 1.1 × 10−5(1) | 3.0 × 10−6 (0.3) |
| c (NO2) | 9.9 × 10−4 (33.0) | 1.0 × 10−4 (3.3) | 3.0 × 10−5 (1) | 3.7 × 10−5 (1.2) | 6.5 × 10−3 (92.9) | 4.2 × 10−4 (6.0) | 7.0 × 10−5(1) | 4.3 × 10−5 (0.6) |
| d (Me) | ND | 4.1 × 10−6 | NA | NE | ND | 1.7 × 10−5 (4.2) | 4.1 × 10−6(1) | NE |
| e (MeO) | ND | 4.8 × 10−6 | NE | NE | ND | 1.9 × 10−5 | NE | NE |
| Compound | ΔH‡ kcal/mol | −TΔS‡ kcal/mol | ΔS‡ cal/(mol·K) | ΔG‡25 °C kcal/mol |
|---|---|---|---|---|
| M06-2X/6-31+G(d) SMD=water b | ||||
| 2a | 2.51 | +17.38 | −58.29 | 19.89 |
| 3a | 5.03 | +17.03 | −57.12 | 22.06 |
| 4a | 6.80 | +16.57 | −55.58 | 23.37 |
| 5a | 4.66 | +17.95 | −60.20 | 22.61 |
| M06-2X/6-311++G(d,p) SMD=water c | ||||
| 2a | 4.26 | +16.16 | −54.20 | 20.42 |
| 3a | 7.13 | +16.03 | −53.76 | 23.16 |
| 4a | 8.80 | +15.76 | −52.86 | 24.56 |
| 5a | 7.50 | +15.44 | −51.79 | 22.94 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ocampo Gutiérrez de Velasco, D.A.; Su, A.; Zhai, L.; Kinoshita, S.; Otani, Y.; Ohwada, T. Unexpected Resistance to Base-Catalyzed Hydrolysis of Nitrogen Pyramidal Amides Based on the 7-Azabicyclic[2.2.1]heptane Scaffold. Molecules 2018, 23, 2363. https://doi.org/10.3390/molecules23092363
Ocampo Gutiérrez de Velasco DA, Su A, Zhai L, Kinoshita S, Otani Y, Ohwada T. Unexpected Resistance to Base-Catalyzed Hydrolysis of Nitrogen Pyramidal Amides Based on the 7-Azabicyclic[2.2.1]heptane Scaffold. Molecules. 2018; 23(9):2363. https://doi.org/10.3390/molecules23092363
Chicago/Turabian StyleOcampo Gutiérrez de Velasco, Diego Antonio, Aoze Su, Luhan Zhai, Satowa Kinoshita, Yuko Otani, and Tomohiko Ohwada. 2018. "Unexpected Resistance to Base-Catalyzed Hydrolysis of Nitrogen Pyramidal Amides Based on the 7-Azabicyclic[2.2.1]heptane Scaffold" Molecules 23, no. 9: 2363. https://doi.org/10.3390/molecules23092363
APA StyleOcampo Gutiérrez de Velasco, D. A., Su, A., Zhai, L., Kinoshita, S., Otani, Y., & Ohwada, T. (2018). Unexpected Resistance to Base-Catalyzed Hydrolysis of Nitrogen Pyramidal Amides Based on the 7-Azabicyclic[2.2.1]heptane Scaffold. Molecules, 23(9), 2363. https://doi.org/10.3390/molecules23092363