Electronic Effect on the Molecular Motion of Aromatic Amides: Combined Studies Using VT-NMR and Quantum Calculations



Abstract
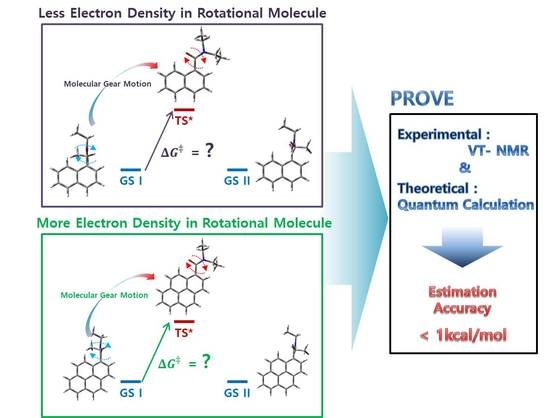
1. Introduction
2. Results
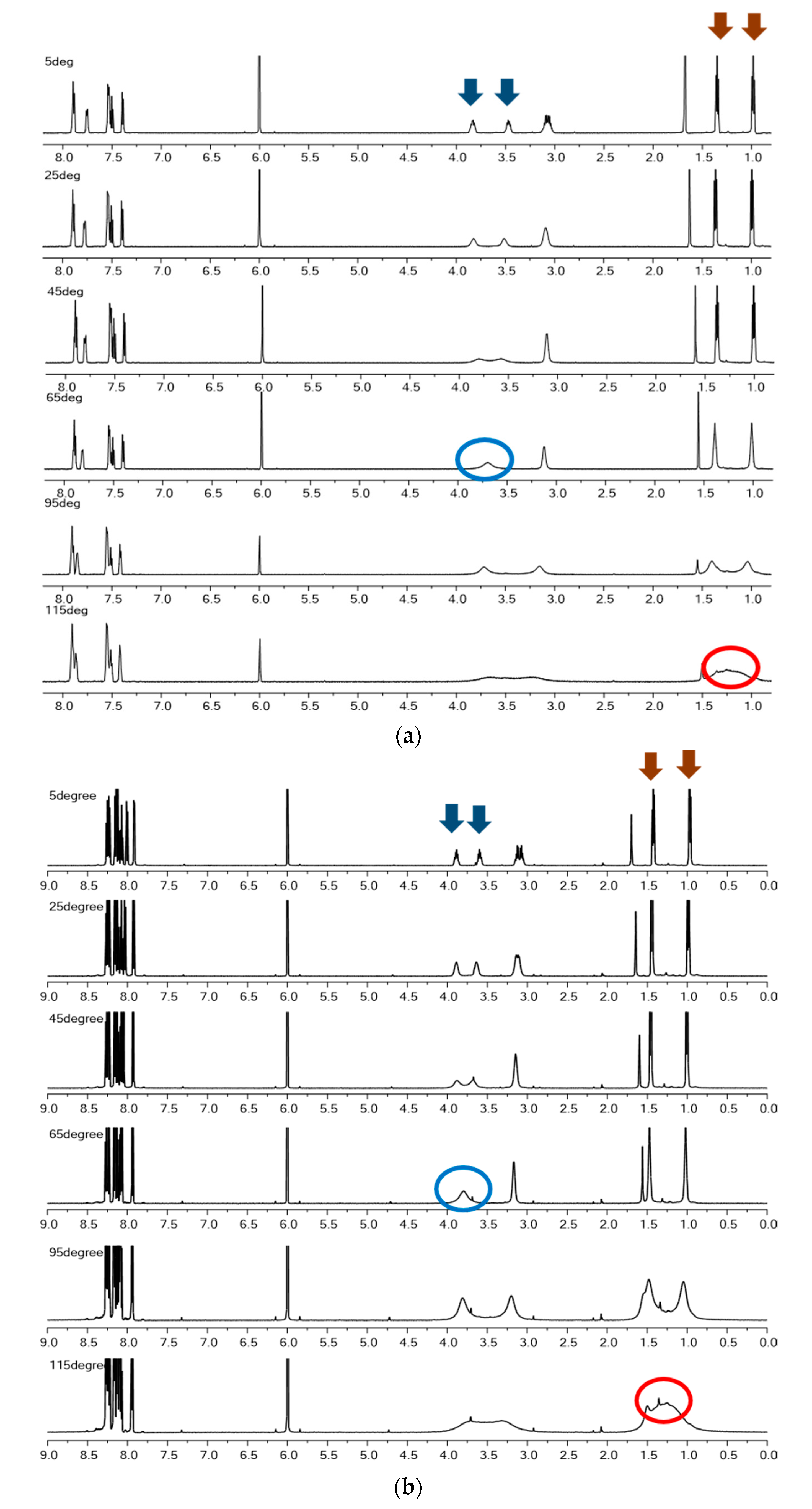
2.1. Interpretation of VT-NMR: Coalescence
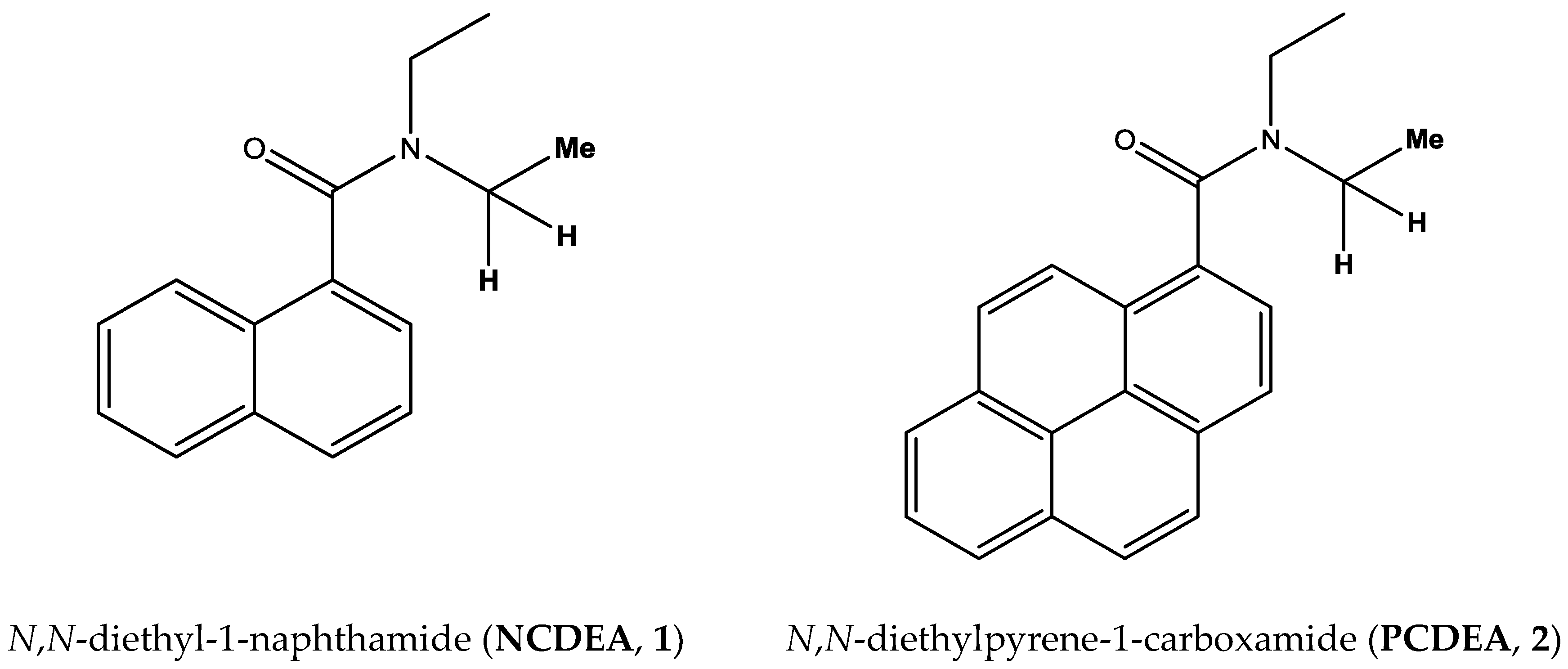
Characterization of N,N-diethylamide Derivatives (1), (2)
2.2. Theoretical Calculations
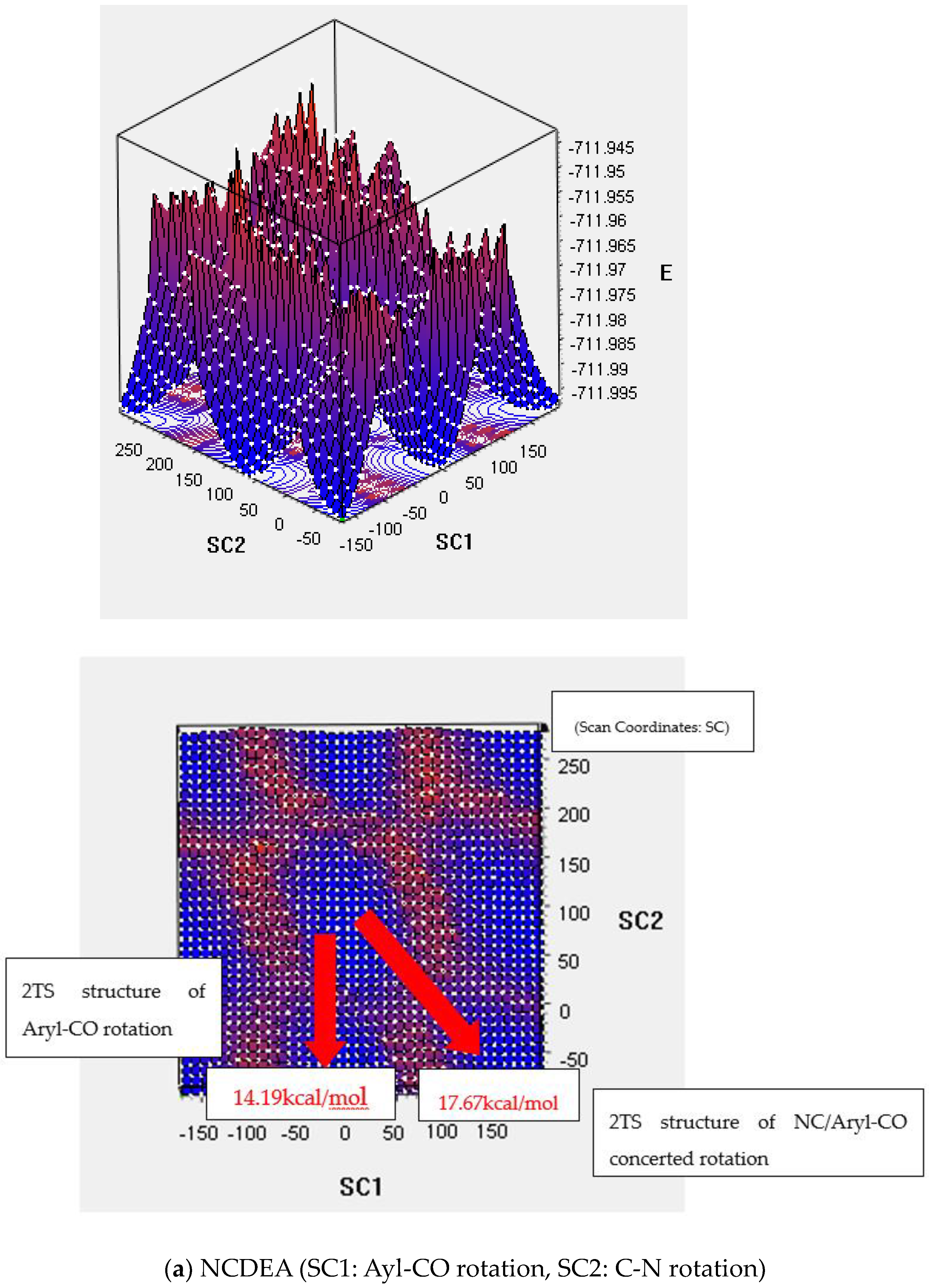
2.2.1. One-Dimensional Potential Energy Surface Scan for Aryl-CO and C-N Rotation
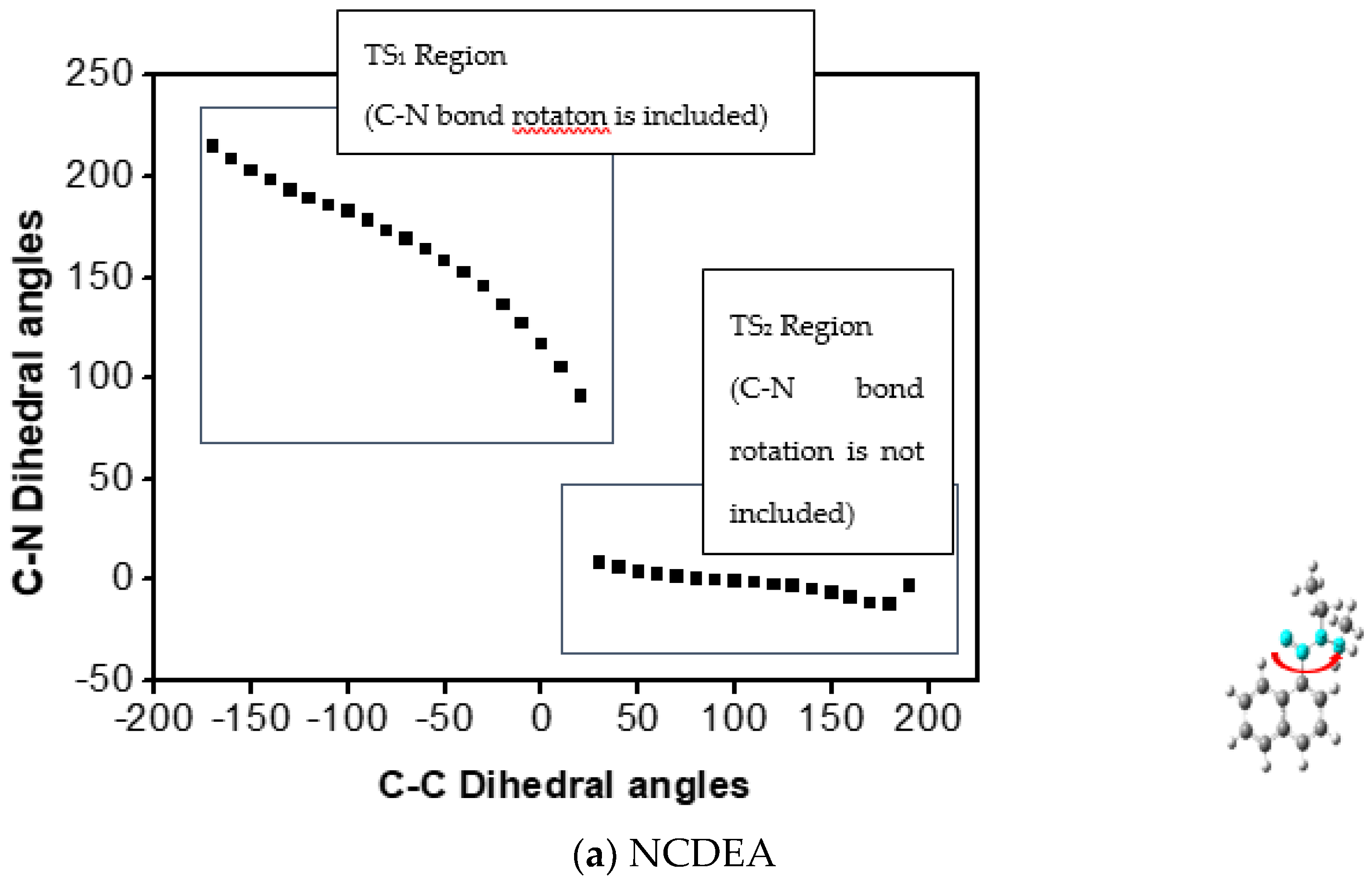
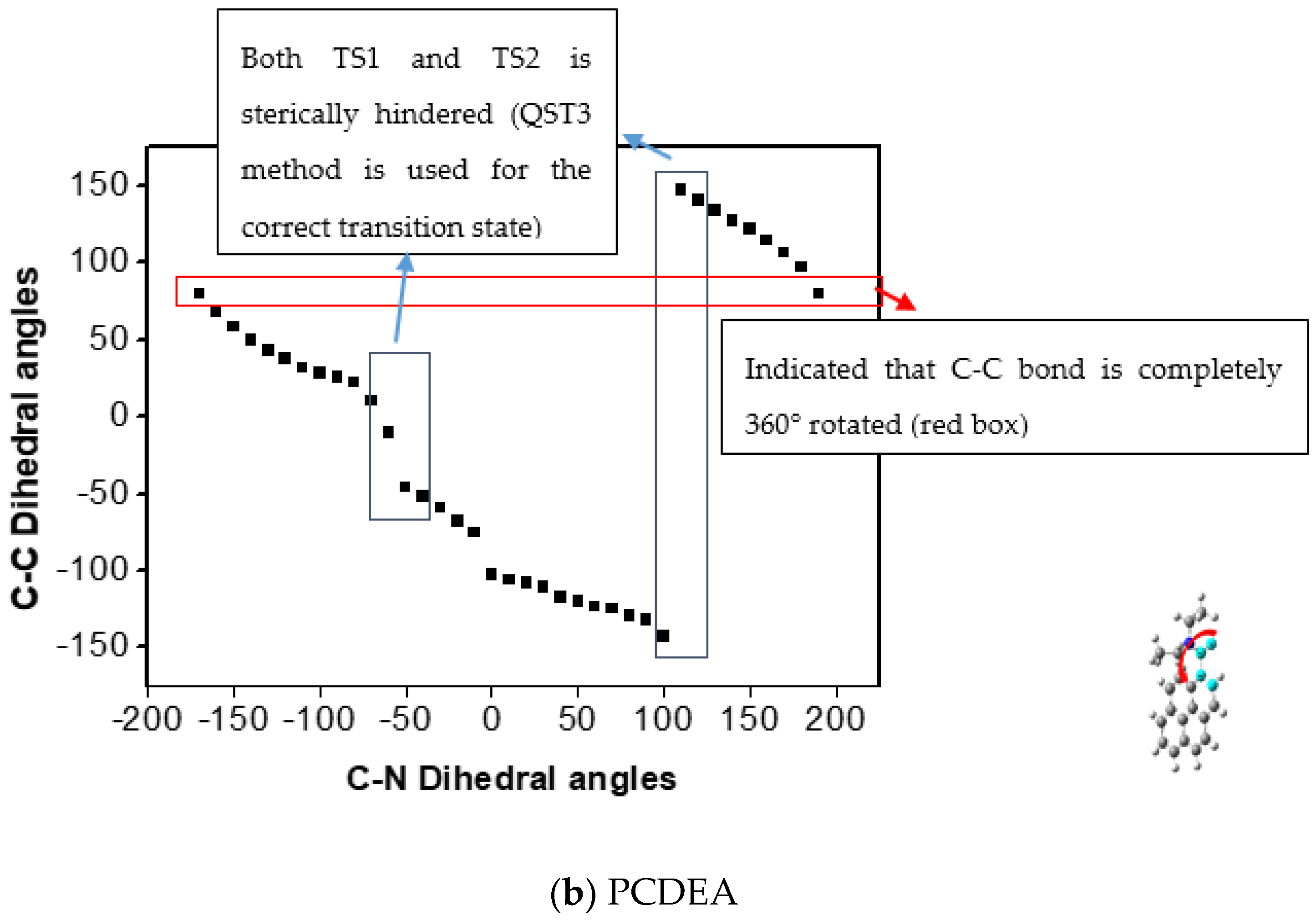
2.2.2. Calculated Result for QST3 and Changing Dihedral Angles of Di-ethyl Groups
Scheme for Interconversion in N,N-Diethylamide Derivatives (1), (2)
Transition States Optimized through the QST3 Method
Dihedral Angle Change in the Diethyl Conformation of Transition States of the Aryl-CO Bond in NCDEA and PCDEA
Transition State of the 2D PES
Quantum Theory of Atoms in Molecules (QTAIM) Study
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Experimental Methods
4.3. Computational Methodss
4.3.1. 1D PES Method
4.3.2. QST3 Method and Changing Dihedral Angles of Diethyl Groups Method
4.3.3. 2D PES Method
4.3.4. QTAIM Study
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chakrabarti, P.; Dunitz, J.D. Structural characteristics of the carboxylic amide group. Helv. Chim. Acta 1982, 65, 1555–1562. [Google Scholar] [CrossRef]
- Hirota, E.; Sugisaki, R.; Nielsen, C.J.; Sørensen, G.O. Molecular structure and internal motion of formamide from microwave spectrum. J. Mol. Spectrosc. 1974, 49, 251–267. [Google Scholar] [CrossRef]
- Kottas, G.S.; Clarke, L.I.; Horinek, D.; Michl, J. Artificial molecular rotors. Chem. Rev. 2005, 105, 1281–1376. [Google Scholar] [CrossRef] [PubMed]
- Bragg, R.A.; Clayden, J.; Morris, G.A.; Pink, J.H. Stereodynamics of bond rotation in tertiary aromatic amides. Chem. Eur. J. 2002, 8, 1279–1289. [Google Scholar] [CrossRef]
- Clayden, J.; Pink, J.H. Concerted rotation in a tertiary aromatic amide: Towards a simple molecular gear. Angew. Chem. Int. Ed. 1998, 37, 1937–1939. [Google Scholar] [CrossRef]
- Bragg, R.A.; Clayden, J. Using symmetry to monitor geared bond rotation in aromatic amides by dynamic NMR. Org. Lett. 2000, 2, 3351–3354. [Google Scholar] [CrossRef] [PubMed]
- Vargas, R.; Garza, J.; Dixon, D.; Hay, B.P. C(sp2)−C(aryl) bond rotation barrier in N-methylbenzamide. J. Phys. Chem. 2001, 105, 774–778. [Google Scholar] [CrossRef]
- Hammaker, R.M.; Gugler, B.A. An NMR study of hindered internal rotation in N,N-dialkyl amides. J. Mol. Spectrosc. 1965, 17, 356–364. [Google Scholar] [CrossRef]
- Olsen, R.A.; Liu, L.; Ghaderi, N.; Johns, A.; Hatcher, M.E.; Mueller, L.J. The amide rotational barriers in picolinamide and nicotinamide: NMR and ab initio studies. J. Am. Chem. Soc. 2003, 125, 10125–10132. [Google Scholar] [CrossRef] [PubMed]
- Skorupska, E.A.; Nazarski, R.B.; Ciechańska, M.; Jóźwiak, A.; Kłys, A. Dynamic 1 h NMR spectroscopic study of hindered internal rotation in selected N,N-dialkyl isonicotinamides: An experimental and DFT analysis. Tetrahedron 2013, 69, 8147–8154. [Google Scholar] [CrossRef]
- Ahmed, A.; Bragg, R.A.; Clayden, J.; Lai, L.W.; McCarthy, C.; Pink, J.H.; Westlund, N.; Yasin, S.A. Barriers to rotation about the chiral axis of tertiary aromatic amides. Tetrahedron 1998, 54, 13277–13294. [Google Scholar] [CrossRef]
- Kang, Y.K.; Park, H.S. Internal rotation about the C–N bond of amides. J. Mol. Struct. 2004, 676, 171–176. [Google Scholar] [CrossRef]
- Clayden, J.; Pink, J.H.; Yasin, S.A. Conformationally interlocked amides: Remote asymmetric induction by mechanical transfer of stereochemical information. Tetrahedron Lett. 1998, 39, 105–108. [Google Scholar] [CrossRef]
- Clayden, J.; Johnson, P.; Pink, J.H.; Helliwell, M. Atropisomeric amides as chiral ligands: Using (−)-sparteine-directed enantioselective silylation to control the conformation of a stereogenic axis. J. Org. Chem. 2000, 65, 7033–7040. [Google Scholar] [CrossRef] [PubMed]
- Reeves, L.W.; Shaddick, R.C.; Shaw, K.N. Nuclear magnetic resonance studies of multi-site chemical exchange. III. Hindered rotation in dimethylacetamide, dimethyl trifluoro-acetamide, and dimethyl benzamide. Can. J. Chem. 1971, 49, 3683–3691. [Google Scholar] [CrossRef]
- Campomanes, P.; Menéndez, M.I.; López, R.; Sordo, T.L. Stereodynamics of bond rotation in tertiary 1-naphthoic acid amides: A computational study. J. Comput. Chem. 2005, 26, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.V.; Thompson, W.B.; Maitra, K.; Maitra, S. Modulations in restricted amide rotation by steric induced conformational trapping. Chem. Phys. Lett. 2012, 523, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Bernhard Schlegel, H. Combining synchronous transit and quasi-newton methods to find transition states. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Ercolani, G. Determination of the rotational barrier in ethane by vibrational spectroscopy and statistical thermodynamics. J. Chem. Educ. 2005, 82, 1703. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Eor, S.; Hwang, J.; Choi, M.G.; Chang, S.-K. Fluorescent signaling of oxone by desulfurization of thioamide. Org. Lett. 2011, 13, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L.; Dwyer, T.J. Application of two-dimensional NMR to kinetics of chemical exchange. Chem. Rev. 1990, 90, 935–967. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.R.; Wüthrich, K. A two-dimensional nuclear overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent molecular orbital methods. XII. Further extensions of gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Wigner, E. The transition state method. Trans. Faraday Soc. 1938, 34, 29–41. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Signal | Chemical Shifts (δ)/ppm |
|---|---|---|
| NCDEA | H | 3.51/3.82 |
| NCDEA | Me | 0.98/1.34 |
| PCDEA | H | 3.64/3.89 |
| PCDEA | Me | 1.32/1.77 |
| Compound | Coalescing Signals | Δυ/Hz | Tc/°C | k/s−1 | |
|---|---|---|---|---|---|
| NCDEA | H-H | 175.43 | 60 | 602 | 15.40 |
| NCDEA | Me-Me | 273.93 | 115 | 389 | 18.00 |
| PCDEA | H-H | 219.62 | 57 | 478 | 15.62 |
| PCDEA | Me-Me | 220.28 | 115 | 478 | 17.65 |
| Variable Temperature 1H NMR | Theoretical Calculation (QST3//Di-ethyl Rotation *) | ||
|---|---|---|---|
| Compound | Bond | ||
| NCDEA | C-N/aryl-CO | 18.00 | 1TS: 20.61 ** 2TS: 17.13//16.09~20.04 2D TS: 17.67 |
| aryl-CO | 15.40 | 1TS: C-N bond rotation is included 2TS: 14.24//14.24~15.19 2D TS: 14.19 | |
| PCDEA | C-N/aryl-CO | 17.65 | 1TS: 19.94 ** 2TS: 17.04//15.97~19.92 2D TS: 17.57 |
| aryl-CO | 15.62 | 1TS: C-N bond rotation is included 2TS: 13.52//13.38~15.74 2D TS: 15.15 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Kim, J.; Kim, J.; Won, D.; Chang, S.-K.; Cha, W.; Jeong, K.; Ahn, S.; Kwak, K. Electronic Effect on the Molecular Motion of Aromatic Amides: Combined Studies Using VT-NMR and Quantum Calculations. Molecules 2018, 23, 2294. https://doi.org/10.3390/molecules23092294
Kim S, Kim J, Kim J, Won D, Chang S-K, Cha W, Jeong K, Ahn S, Kwak K. Electronic Effect on the Molecular Motion of Aromatic Amides: Combined Studies Using VT-NMR and Quantum Calculations. Molecules. 2018; 23(9):2294. https://doi.org/10.3390/molecules23092294
Chicago/Turabian StyleKim, Sungsoo, Jungyu Kim, Jieun Kim, Daeun Won, Suk-Kyu Chang, Wansik Cha, Keunhong Jeong, Sangdoo Ahn, and Kyungwon Kwak. 2018. "Electronic Effect on the Molecular Motion of Aromatic Amides: Combined Studies Using VT-NMR and Quantum Calculations" Molecules 23, no. 9: 2294. https://doi.org/10.3390/molecules23092294
APA StyleKim, S., Kim, J., Kim, J., Won, D., Chang, S.-K., Cha, W., Jeong, K., Ahn, S., & Kwak, K. (2018). Electronic Effect on the Molecular Motion of Aromatic Amides: Combined Studies Using VT-NMR and Quantum Calculations. Molecules, 23(9), 2294. https://doi.org/10.3390/molecules23092294