Further Studies on the Pyrolytic Domino Cyclization of Stabilized Phosphonium Ylides Bearing an Ortho-Aminophenyl Group



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
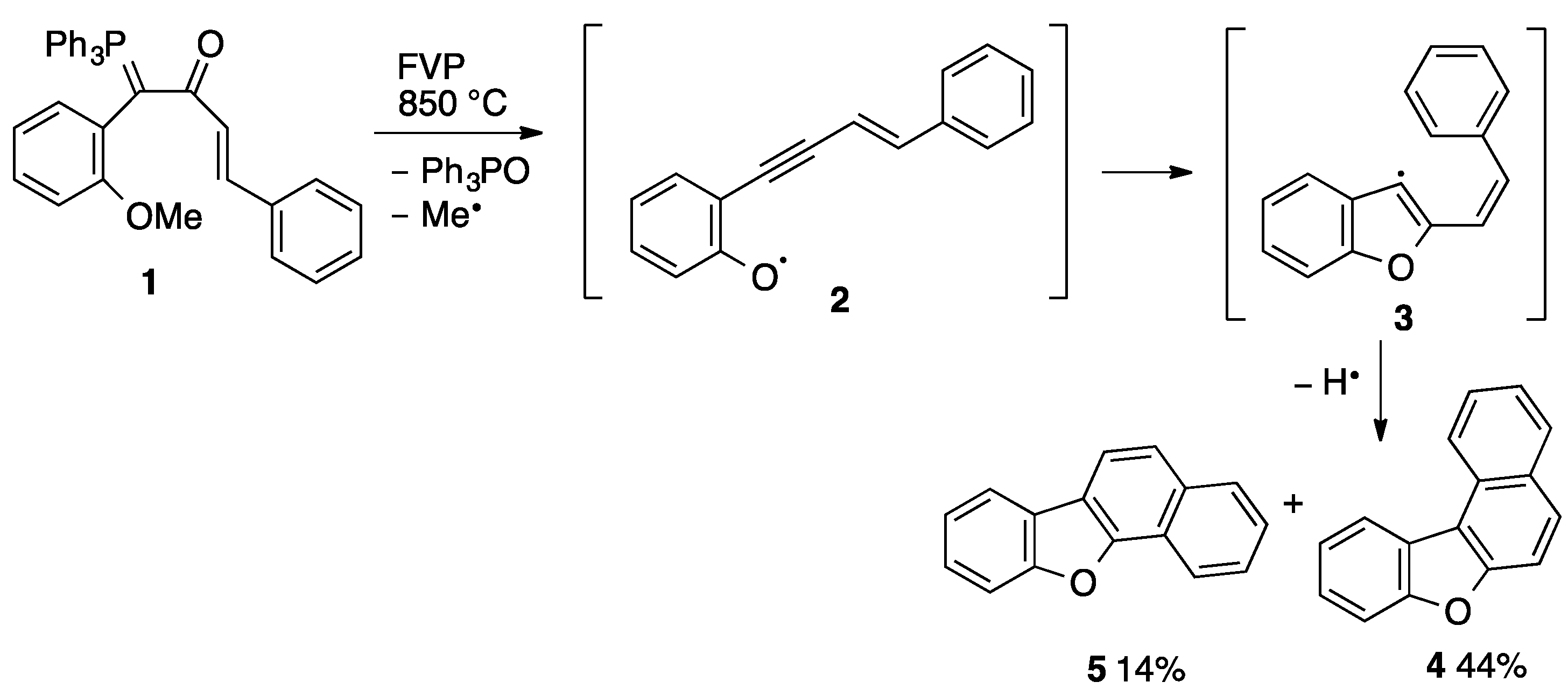
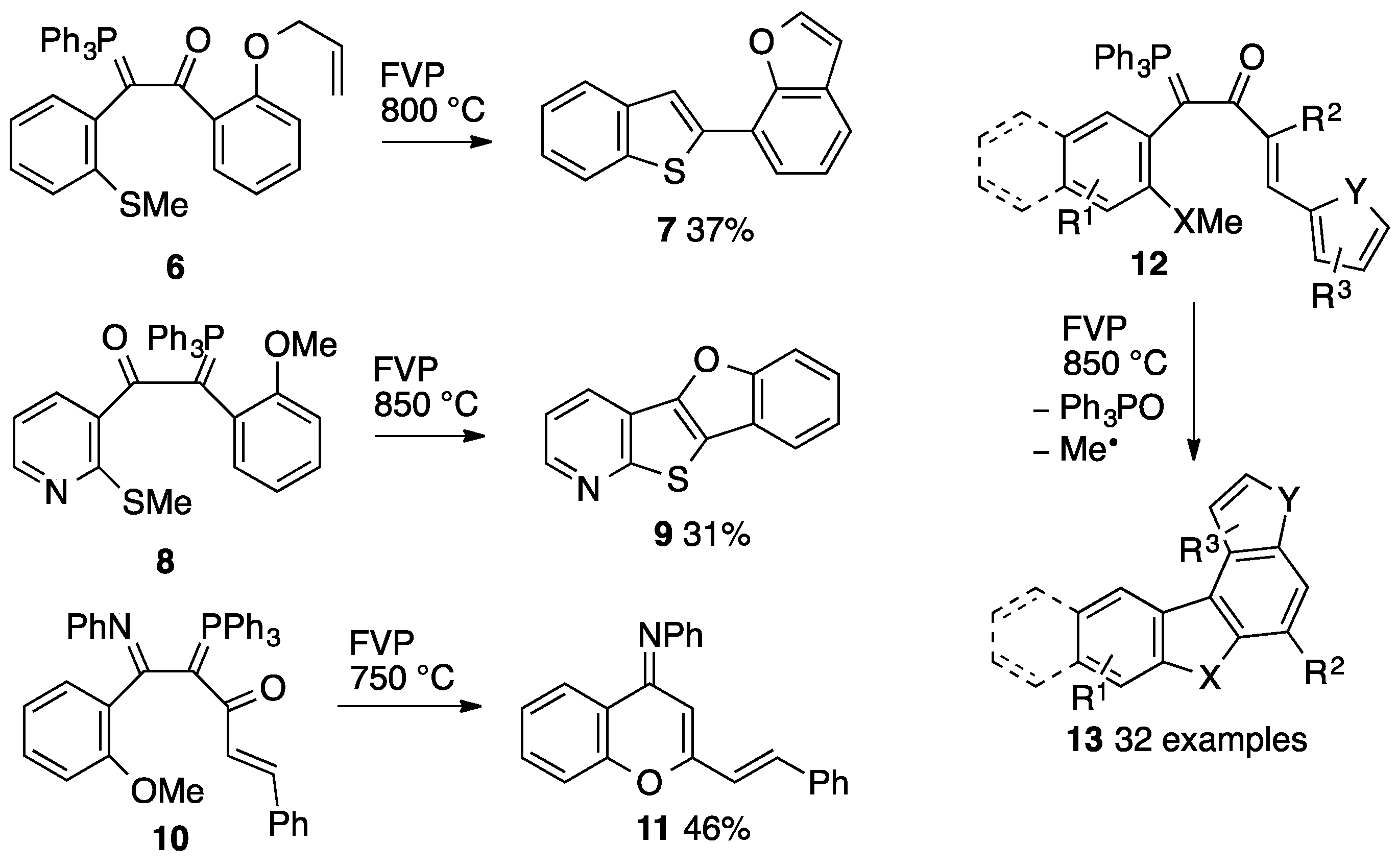
1. Introduction
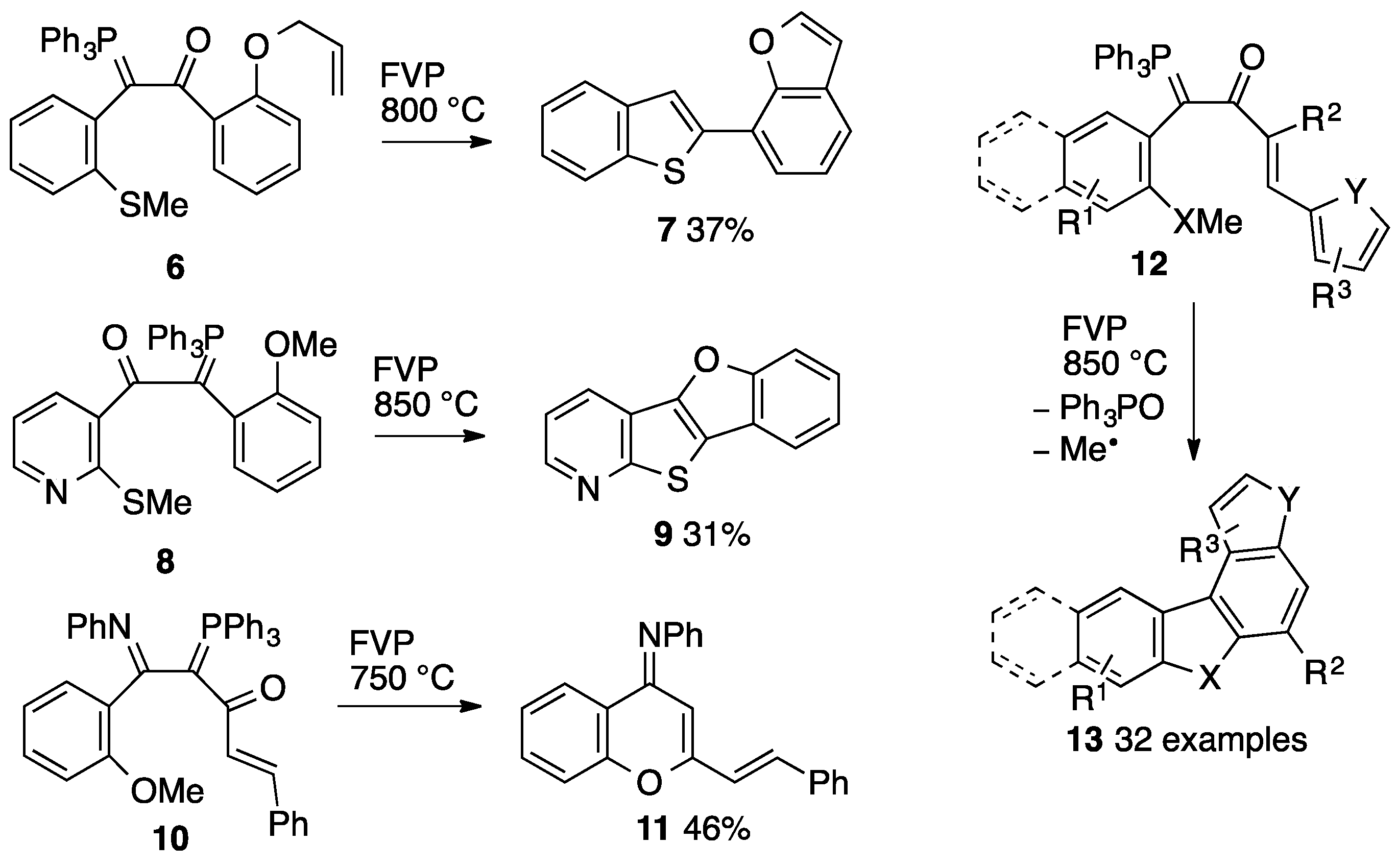
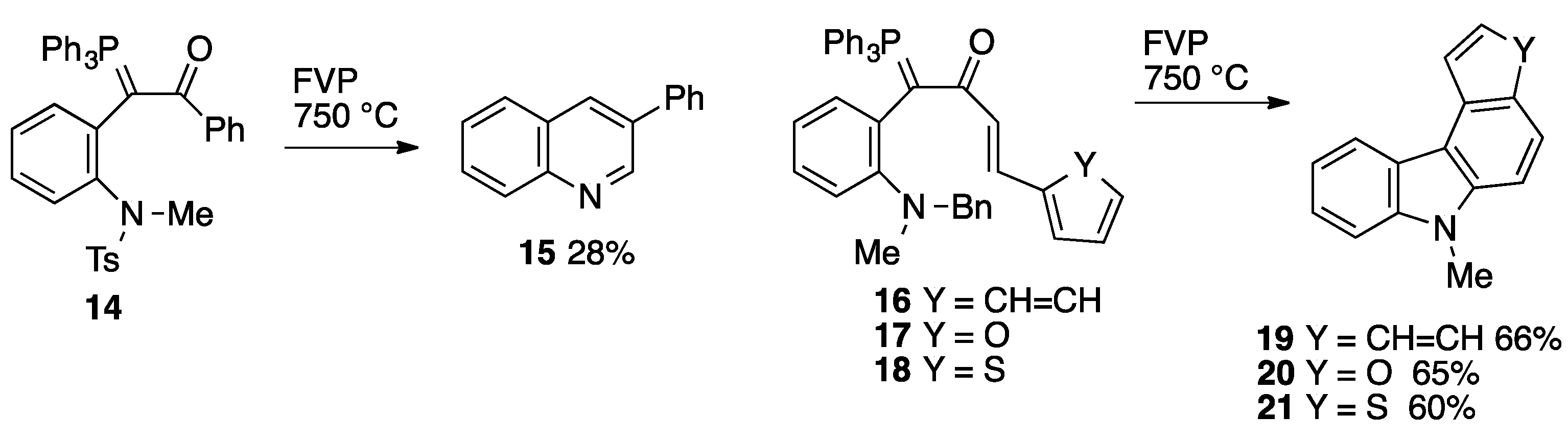
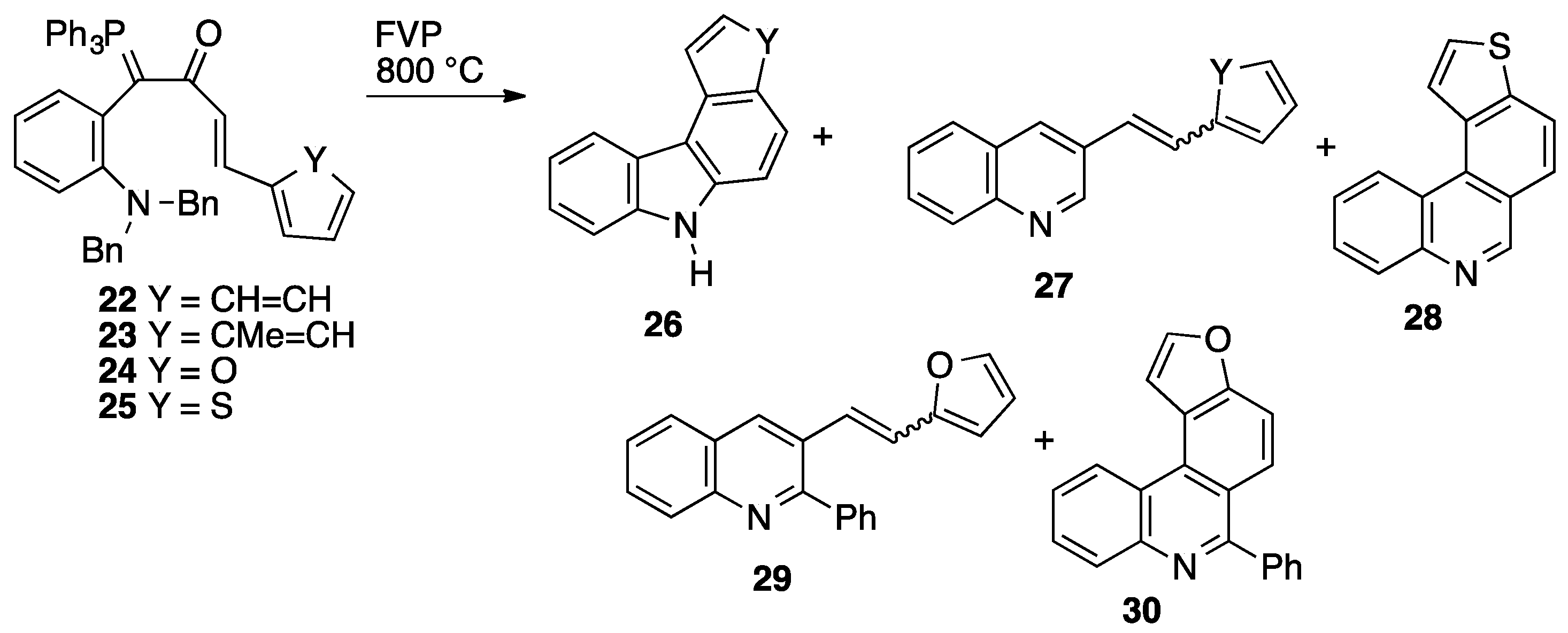
2. Results
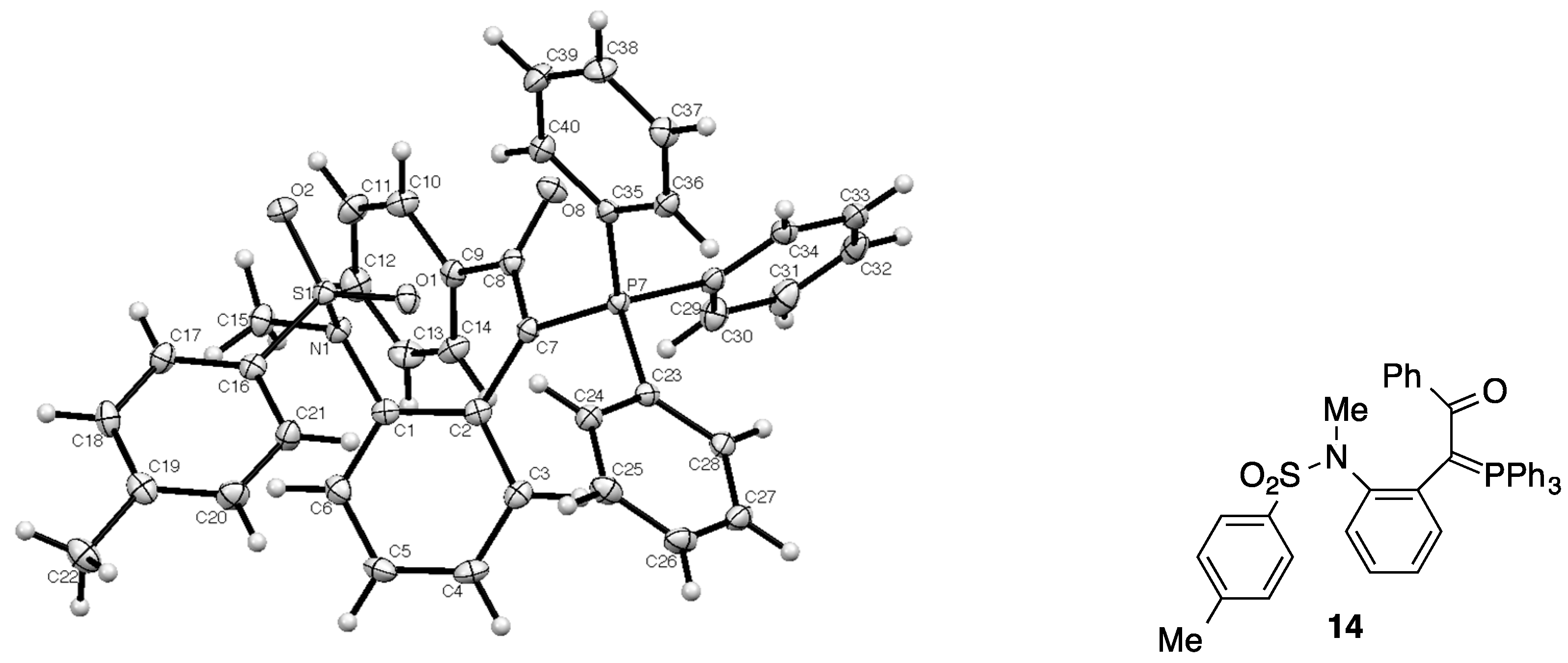
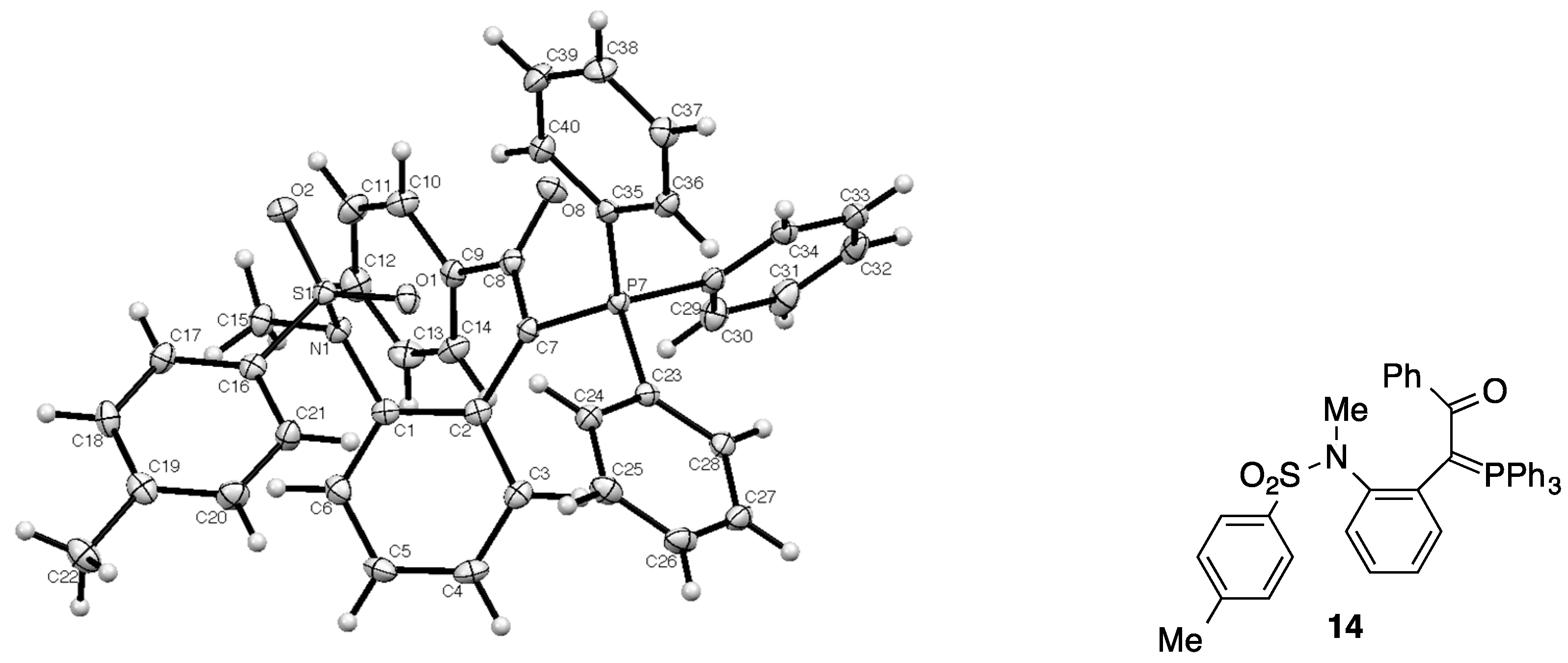
2.1. X-ray Structure Determination of Ylide 14
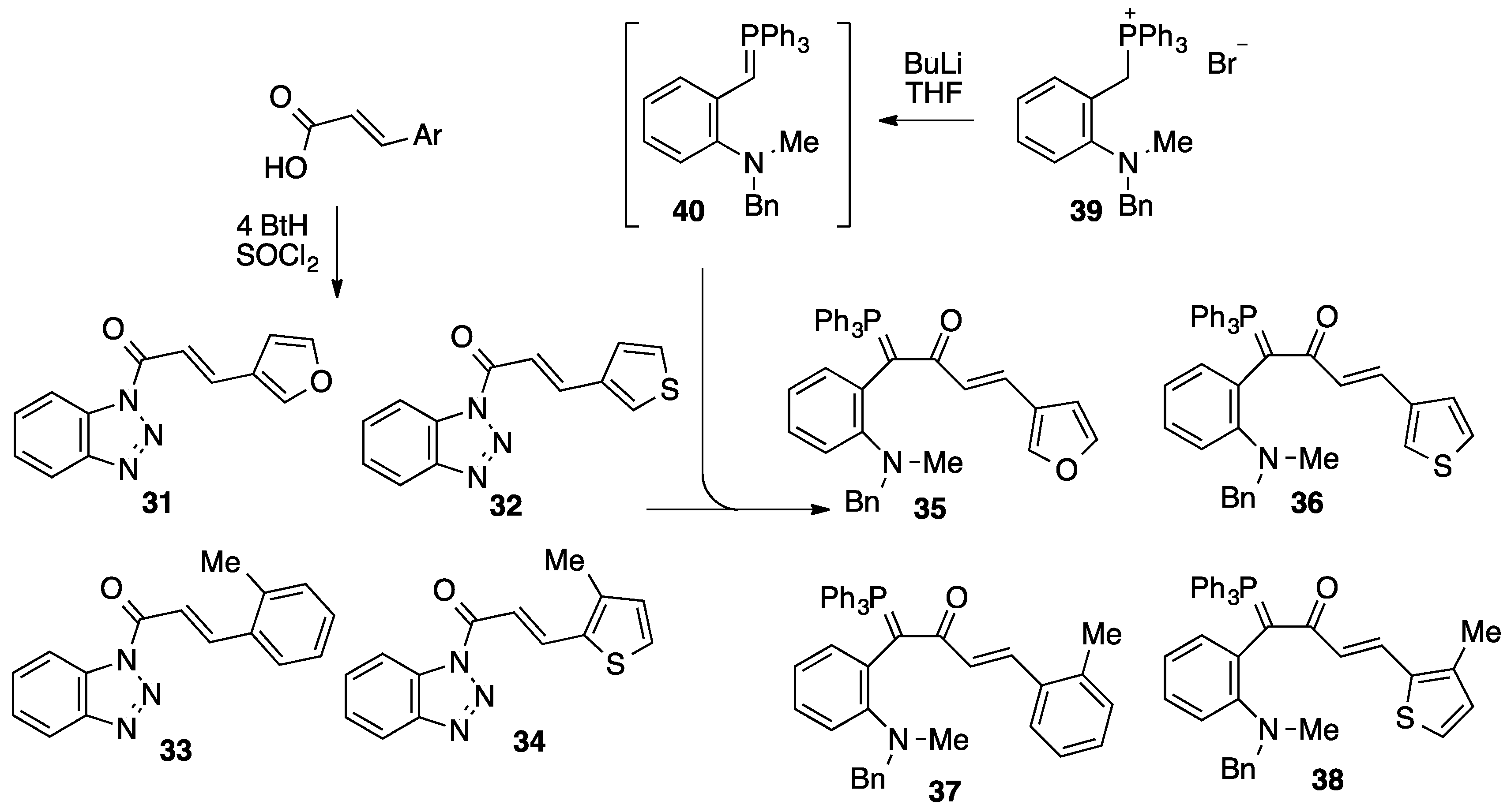
2.2. Preparation and Characterization of Ylides 35–38
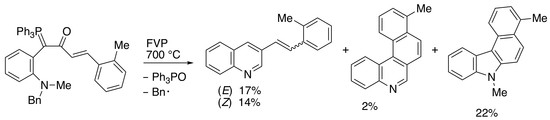
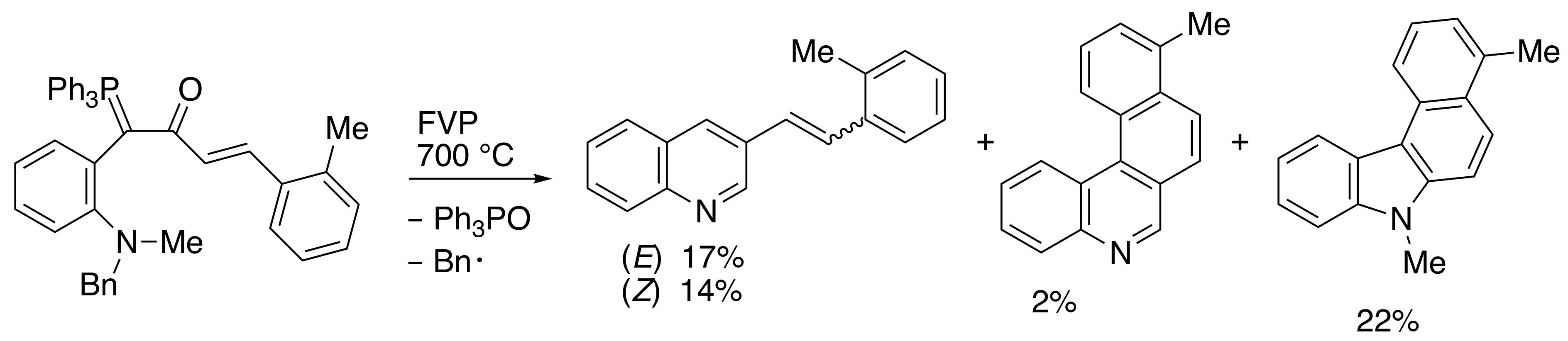
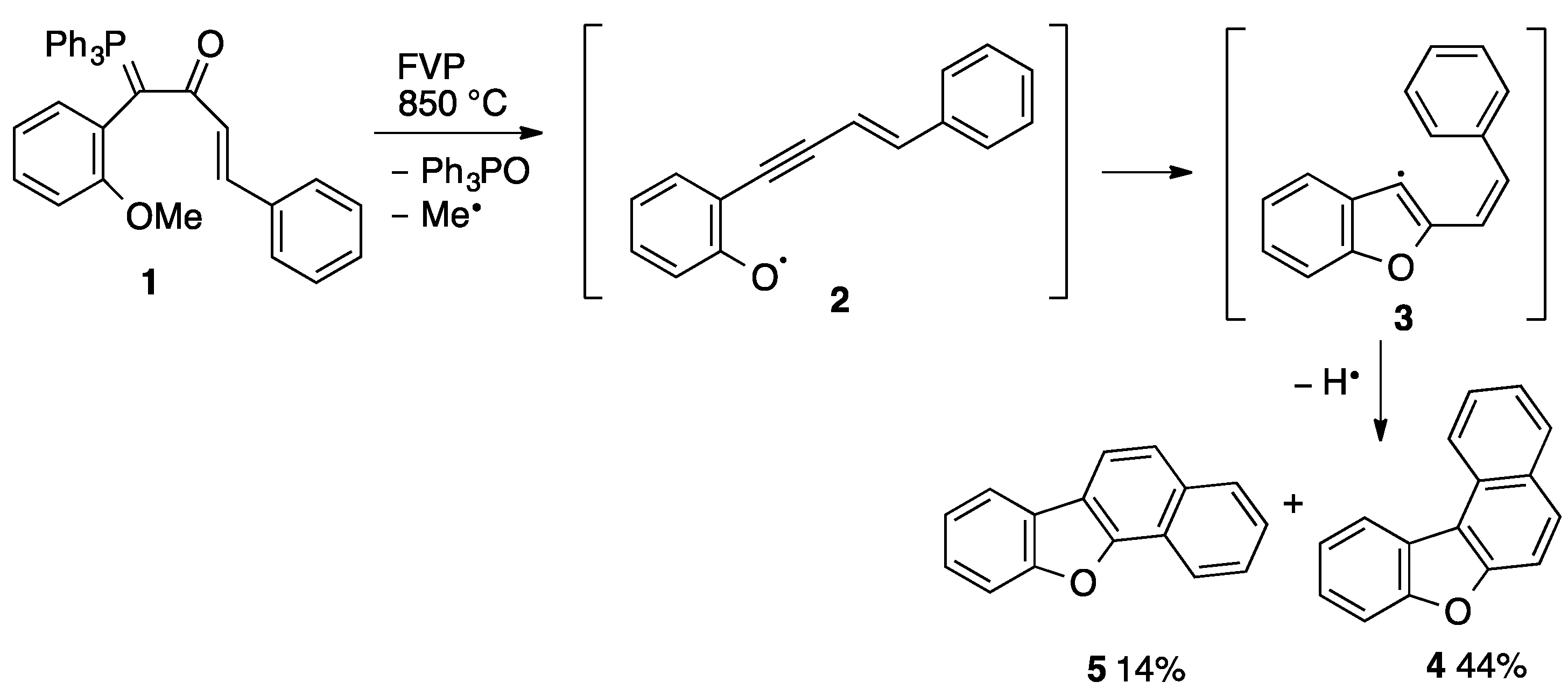
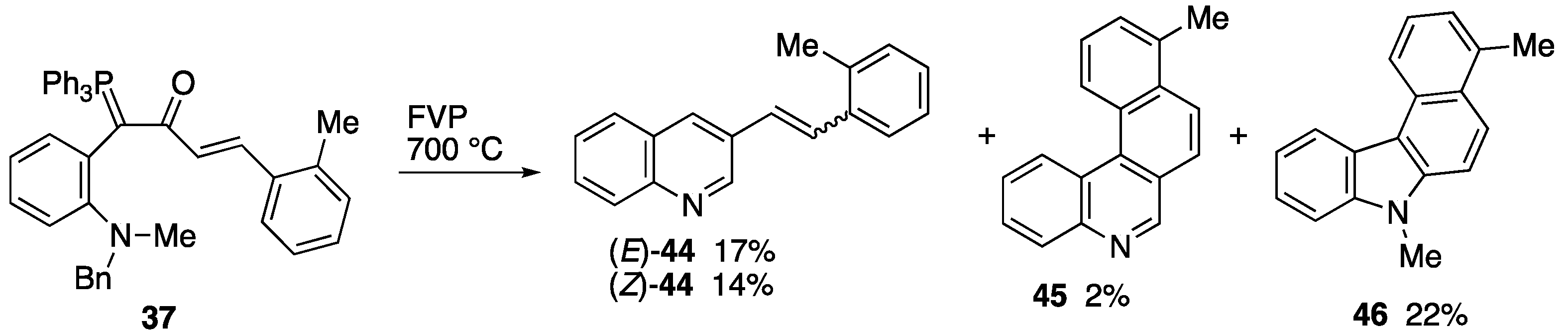
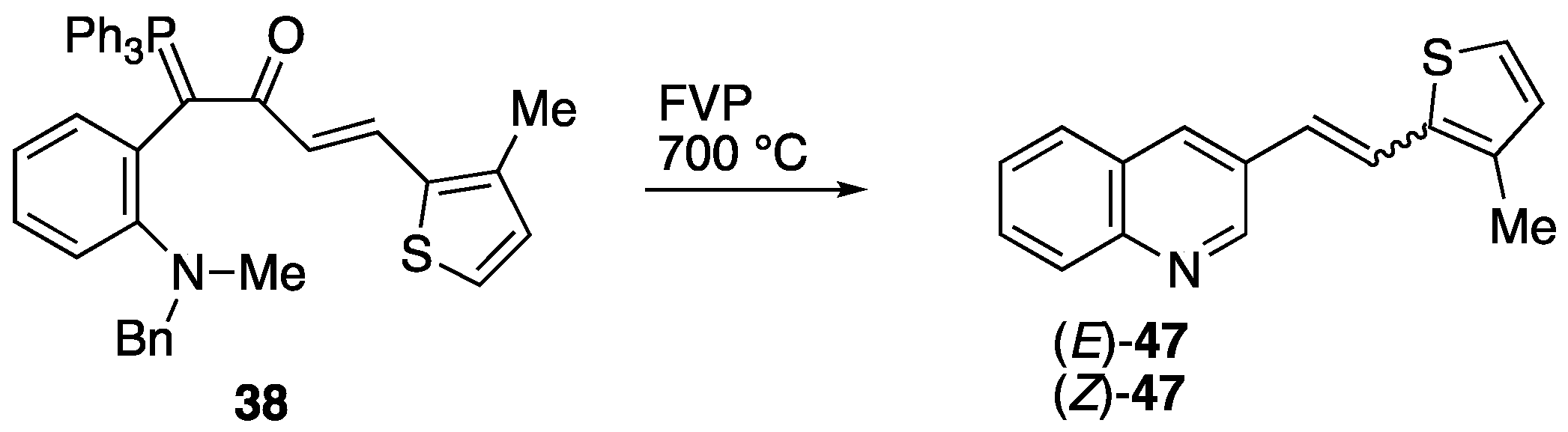
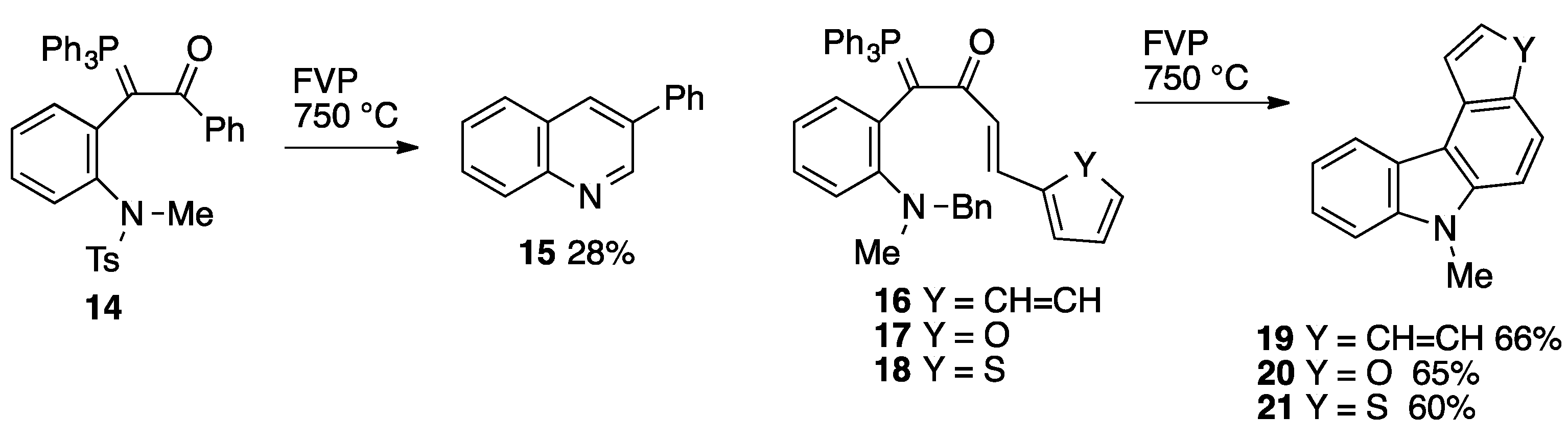
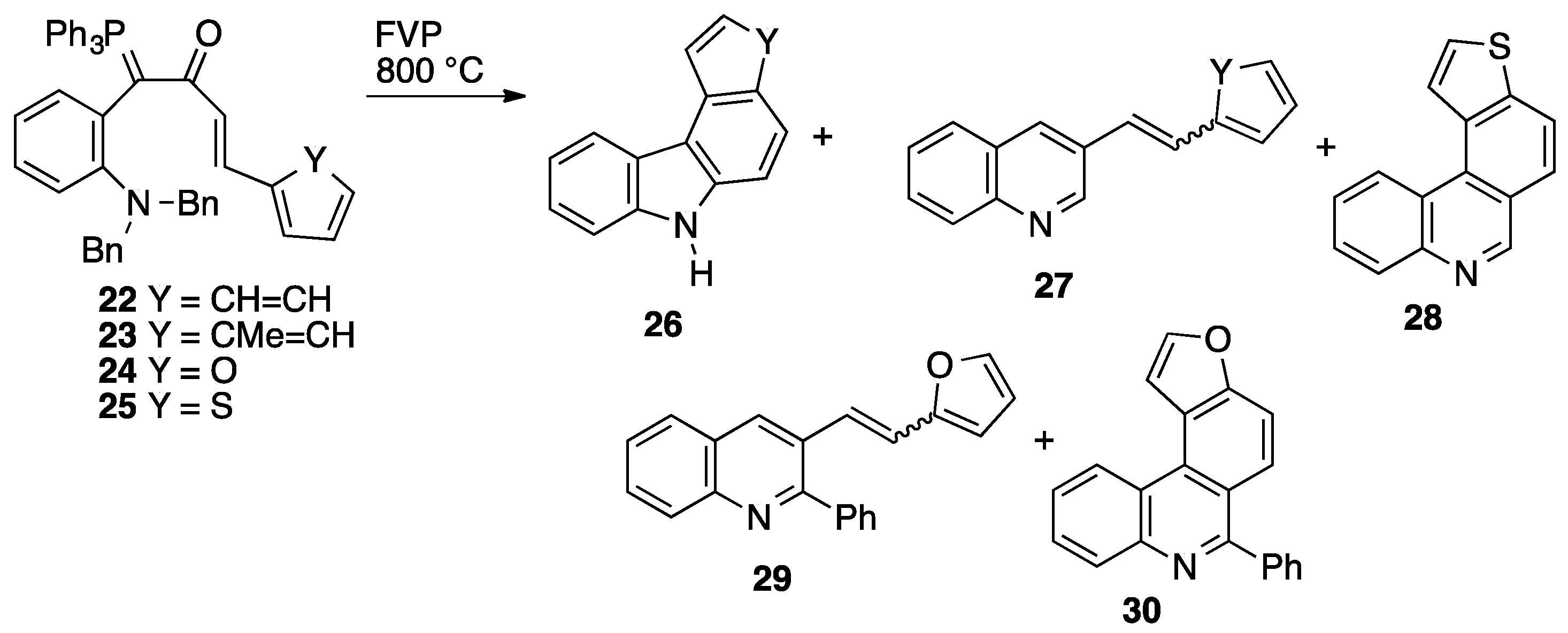
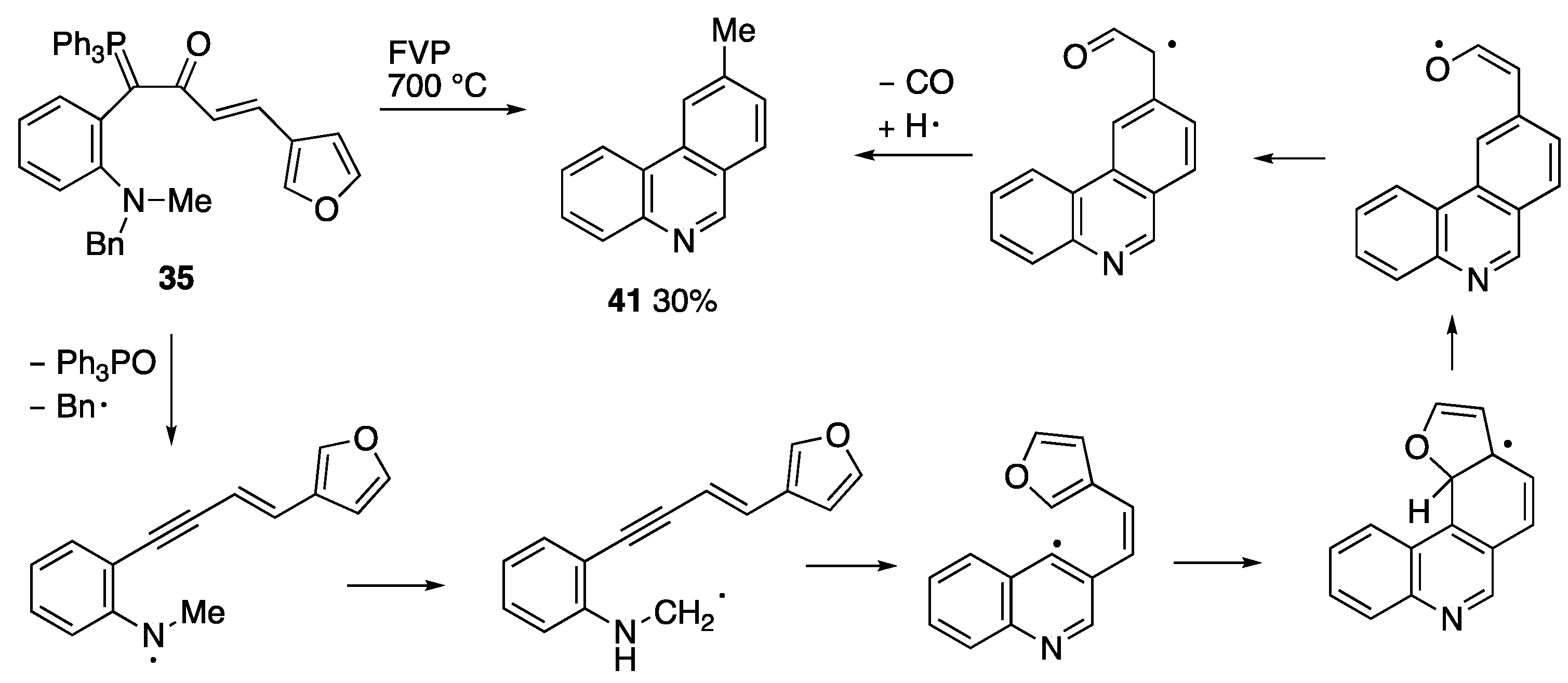
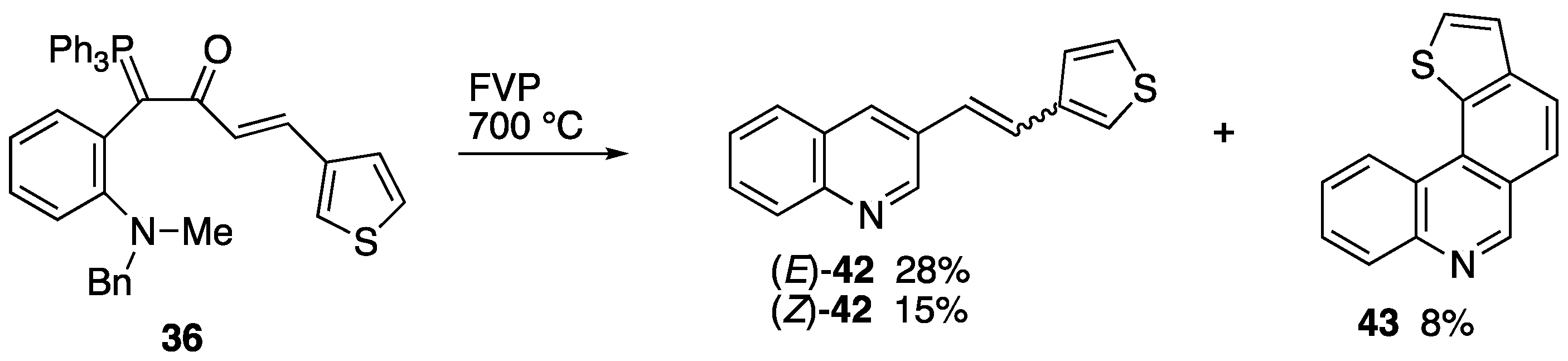
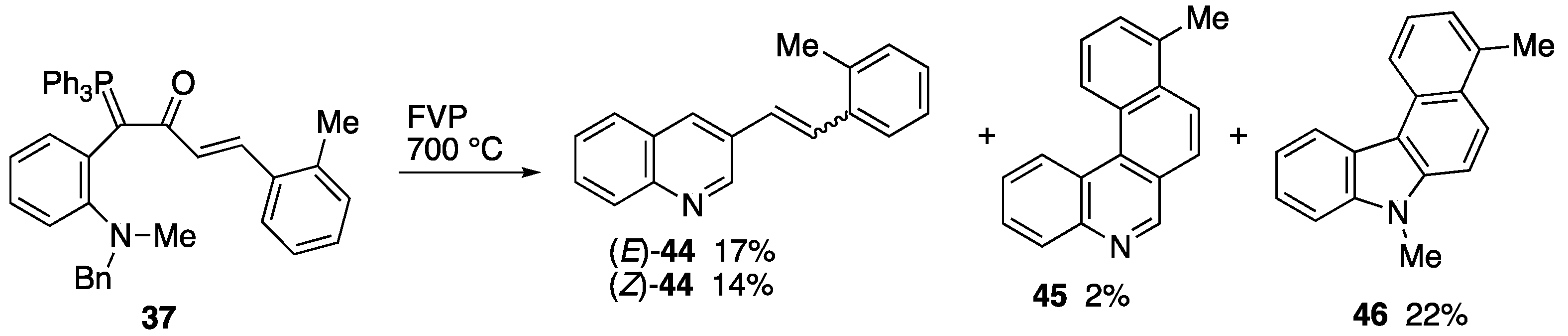
2.3. Flash Vacuum Pyrolysis to Give Heterocyclic Products
3. Experimental
3.1. General Experimental Details
3.2. X-ray Structure Determination for 14
3.3. Preparation of N-Acylbenzotriazoles
3.3.1. 1-(3-(3-Furyl)propenoyl)benzotriazole 31
3.3.2. 1-(3-(3-Thienyl)propenoyl)benzotriazole 32
3.3.3. 1-(3-(2-Methylphenyl)propenoyl)benzotriazole 33
3.3.4. 1-(3-(3-Methyl-2-thienyl)propenoyl)benzotriazole 34
3.4. Preparation of Ylides
3.4.1. [(2-(N-Methyl-N-benzylamino)phenyl)(3-(3-furyl)propenoyl)methylene]triphenylphosphorane 35
3.4.2. [(2-(N-Methyl-N-benzylamino)phenyl)(3-(3-thienyl)propenoyl)methylene]triphenylphosphorane 36
3.4.3. [(2-(N-Methyl-N-benzylamino)phenyl)(3-(2-methyl)phenylpropenoyl)methylene]triphenylphosphorane 37
3.4.4. [(2-(N-Methyl-N-benzylamino)phenyl)(3-(3-methyl-2-thienyl)propenoyl)methylene]triphenylphosphorane 38
3.5. Flash Vacuum Pyrolysis of Ylides
3.5.1. FVP of Ylide 35
3.5.2. FVP of Ylide 36
3.5.3. FVP of Ylide 37
3.5.4. FVP of Ylide 38
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wentrup, C. Flash vacuum pyrolysis: Techniques and reactions. Angew. Chem. Int. Ed. Engl. 2017, 56, 14808–14835. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Boubalouta, Y. Recent advances in the synthesis of heterocyclic compounds using flash vacuum pyrolysis. Adv. Heterocycl. Chem. 2015, 115, 93–150. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.; Brasche, G.; Gericke, K.M. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006; ISBN 978-3-527-29060-4. [Google Scholar]
- Tietze, L.F. (Ed.) Domino Reactions: Concepts for Efficient Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014; ISBN 978-3-527-33432-2. [Google Scholar]
- Aitken, R.A.; Bradbury, C.K.; Burns, G.; Morrison, J.J. Tandem radical cyclisation in the flash vacuum pyrolysis of 2-methoxyphenyl and 2-methylthiophenyl substituted phosphorus ylides. Synlett 1995, 53–54. [Google Scholar] [CrossRef]
- Aitken, R.A.; Burns, G.; Morrison, J.J. Flash vacuum pyrolysis of stabilised phosphorus ylides. Part 14. Tandem cyclisation of intermediate aryloxy and arylthio radicals leading to tri- and tetra-cyclic aromatic heterocycles. J. Chem. Soc. Perkin Trans. 1 1998, 3937–3941. [Google Scholar] [CrossRef]
- Aitken, R.A.; Garnett, A.N. An eight-stage gas-phase cascade reaction leading to 7-(2-benzothienyl)benzofuran and 2,7′-bi(benzofuran). Synlett 2001, 228–229. [Google Scholar] [CrossRef]
- Aitken, R.A.; Garnett, A.N. Cascade synthesis of new tetracyclic heteroaromatic thieno[2,3-b]pyridine-containing ring systems. New J. Chem. 2009, 33, 2402–2404. [Google Scholar] [CrossRef]
- Aitken, R.A.; Chang, D. New gas-phase domino processes leading to benzopyranones and benzofurans. Heterocycles 2016, 93, 164–1284. [Google Scholar] [CrossRef]
- Aitken, R.A.; Garnett, A.N. Versatile pyrolytic synthesis of fused polycyclic heteroaromatic compounds. Synthesis 2017, 49, 4955–4977. [Google Scholar] [CrossRef]
- Aitken, R.A.; Murray, L. New gas-phase cascade reactions of stabilised phosphorus ylides leading to ring-fused indoles and to quinolines. J. Org. Chem. 2008, 73, 9781–9783. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Murray, L. Gas-phase domino cyclisation of phosphonium ylides leading to the total synthesis of Eustifoline D. Tetrahedron Lett. 2017, 58, 4328–4332. [Google Scholar] [CrossRef]
- Aitken, R.A.; Boubalouta, Y.; Chang, D.; Cleghorn, L.P.; Gray, I.P.; Karodia, N.; Reid, E.J.; Slawin, A.M.Z. The value of 2JP-CO as a diagnostic parameter for the structure and thermal reactivity of carbonyl-stabilised phosphonium ylides. Tetrahedron 2017, 73, 6275–6285. [Google Scholar] [CrossRef]
- Aitken, R.A.; Karodia, N.; Lightfoot, P. The solid-state conformation of oxo stabilised ylides: X-ray structures of four new polyoxo phosphorus ylides. J. Chem. Soc. Perkin Trans. 2 2000, 333–340. [Google Scholar] [CrossRef]
- Aitken, R.A.; Boeters, C.; Morrison, J.J. Novel Thermal Cyclisation of o-Methylstyrylalkynes to give 2-Alkenylnaphthalenes. Tetrahedron Lett. 1995, 36, 1303–1306. [Google Scholar] [CrossRef]
- Aitken, R.A.; Boeters, C.; Morrison, J.J. Flash vacuum pyrolysis of stabilised phosphorus ylides. Part 10. Generation of 2-methylstyrylalkynes and their thermal cyclization to 2-alkenylnaphthalenes. J. Chem. Soc. Perkin Trans. 1 1997, 2625–2631. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Vincek, A.; Suzuki, K. Microwave-assisted synthesis of peptidyl phosphorus ylides. Arkivoc 2005, v, 116–126. [Google Scholar] [CrossRef]
- Aitken, R.A.; Horsburgh, C.E.R. Flash Vacuum Pyrolysis of O-phenylene Sulfite: Formation and Purification of Cyclopentadienone Dimer. In Comprehensive Organic Chemistry Experiments for the Laboratory Classroom; Afonso, C.A.M., Candeias, N.R., Simao, D.P., Trinidade, A.F., Coelho, J.A.S., Tan, B., Franzén, R., Eds.; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 690–693. ISBN 978-1-84973-963-4. [Google Scholar]
- Shizuri, Y.; Ojika, M.; Yamada, K. Structure of an antitumor antibiotic, reductiomycin. Tetrahedron Lett. 1981, 22, 4291–4294. [Google Scholar] [CrossRef]
- Pardin, C.; Pelletier, J.N.; Lubell, W.D.; Keillor, J.W. Cinnamoyl inhibitors of tissue transglutaminase. J. Org. Chem. 2008, 73, 5766–5775. [Google Scholar] [CrossRef] [PubMed]
- Patra, P.K.; Suresh, J.R.; Ila, H.; Junjappa, H. A new regiospecific method for the synthesis of substituted phenanthridines and benzo[j]phenanthridines via aromatic annelation of 1-N-benzenesulfonyl-3-[bis(methylthio)methylene]-1,2,3,4-tetrahydroquinoline-4-one. Tetrahedron 1998, 54, 10167–10178. [Google Scholar] [CrossRef]
Sample Availability: Not Available. |


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aitken, R.A.; Murray, L.; Slawin, A.M.Z. Further Studies on the Pyrolytic Domino Cyclization of Stabilized Phosphonium Ylides Bearing an Ortho-Aminophenyl Group. Molecules 2018, 23, 2153. https://doi.org/10.3390/molecules23092153
Aitken RA, Murray L, Slawin AMZ. Further Studies on the Pyrolytic Domino Cyclization of Stabilized Phosphonium Ylides Bearing an Ortho-Aminophenyl Group. Molecules. 2018; 23(9):2153. https://doi.org/10.3390/molecules23092153
Chicago/Turabian StyleAitken, R. Alan, Lorna Murray, and Alexandra M. Z. Slawin. 2018. "Further Studies on the Pyrolytic Domino Cyclization of Stabilized Phosphonium Ylides Bearing an Ortho-Aminophenyl Group" Molecules 23, no. 9: 2153. https://doi.org/10.3390/molecules23092153
APA StyleAitken, R. A., Murray, L., & Slawin, A. M. Z. (2018). Further Studies on the Pyrolytic Domino Cyclization of Stabilized Phosphonium Ylides Bearing an Ortho-Aminophenyl Group. Molecules, 23(9), 2153. https://doi.org/10.3390/molecules23092153