
New Chromones from a Marine-Derived Fungus, Arthrinium sp., and Their Biological Activity

 ,
, 
Abstract
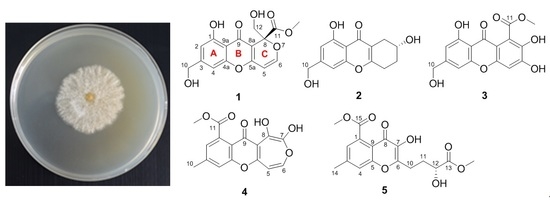
:
1. Introduction
2. Results and Discussion
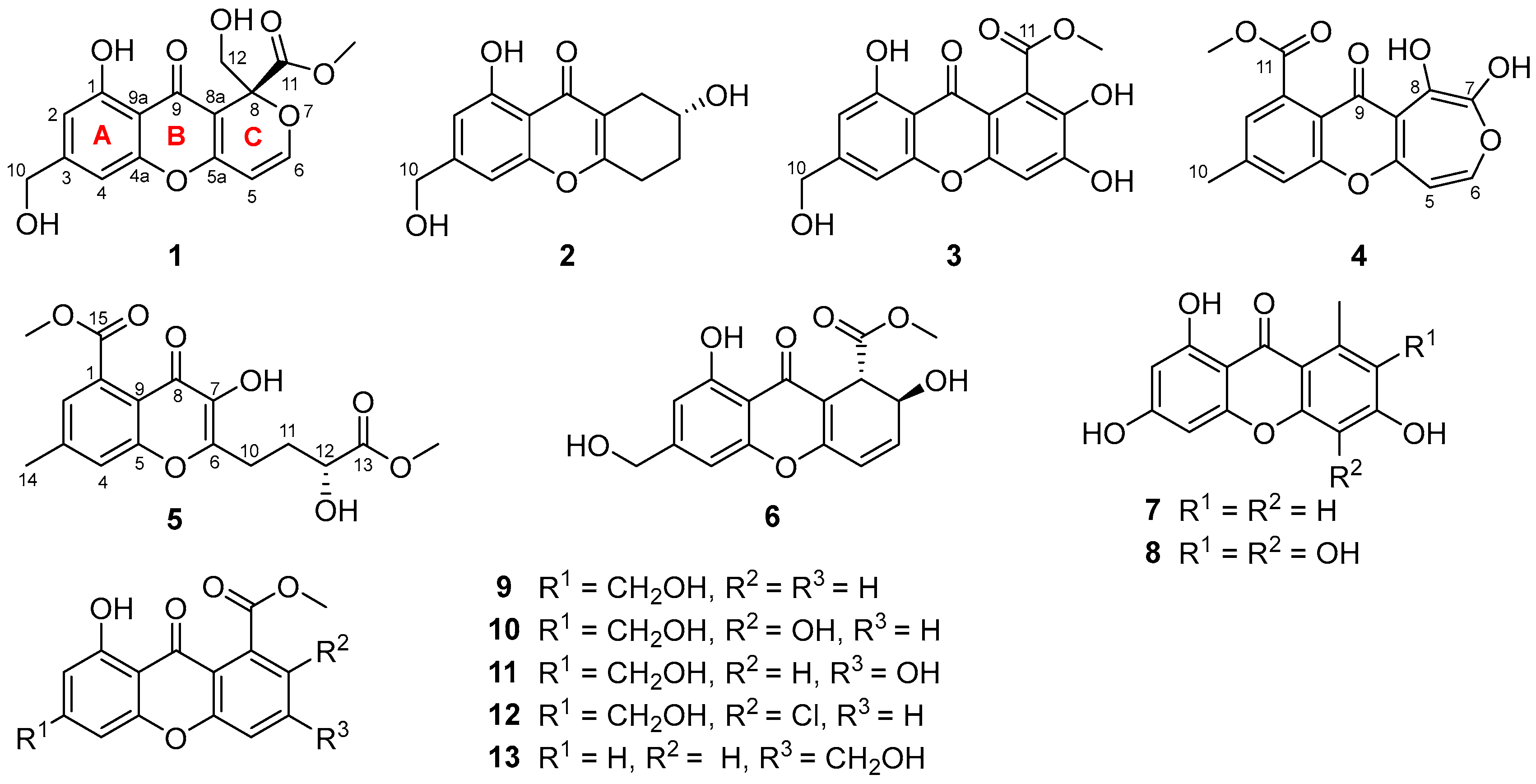
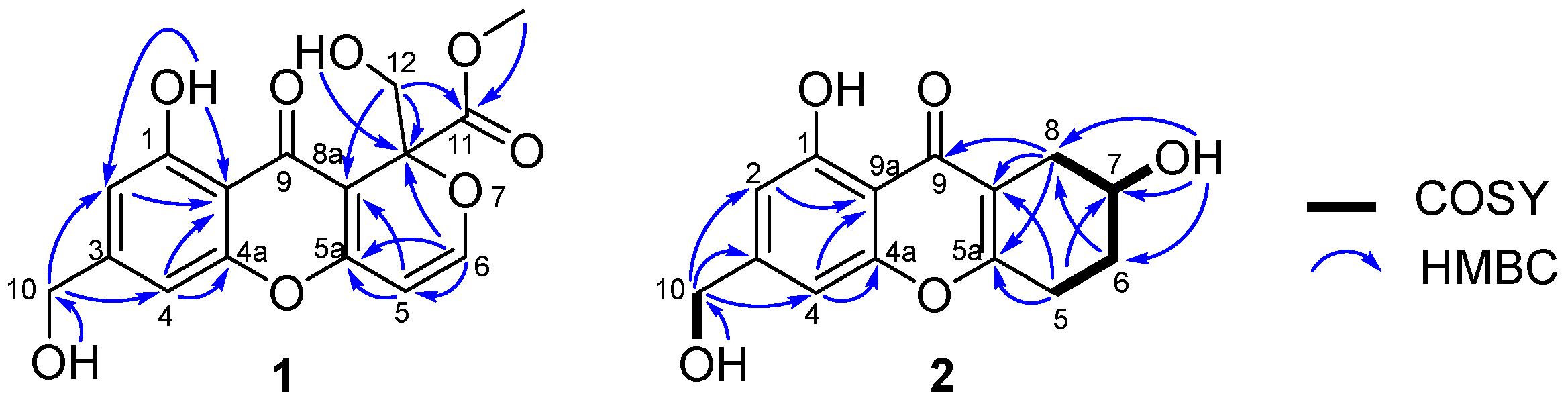
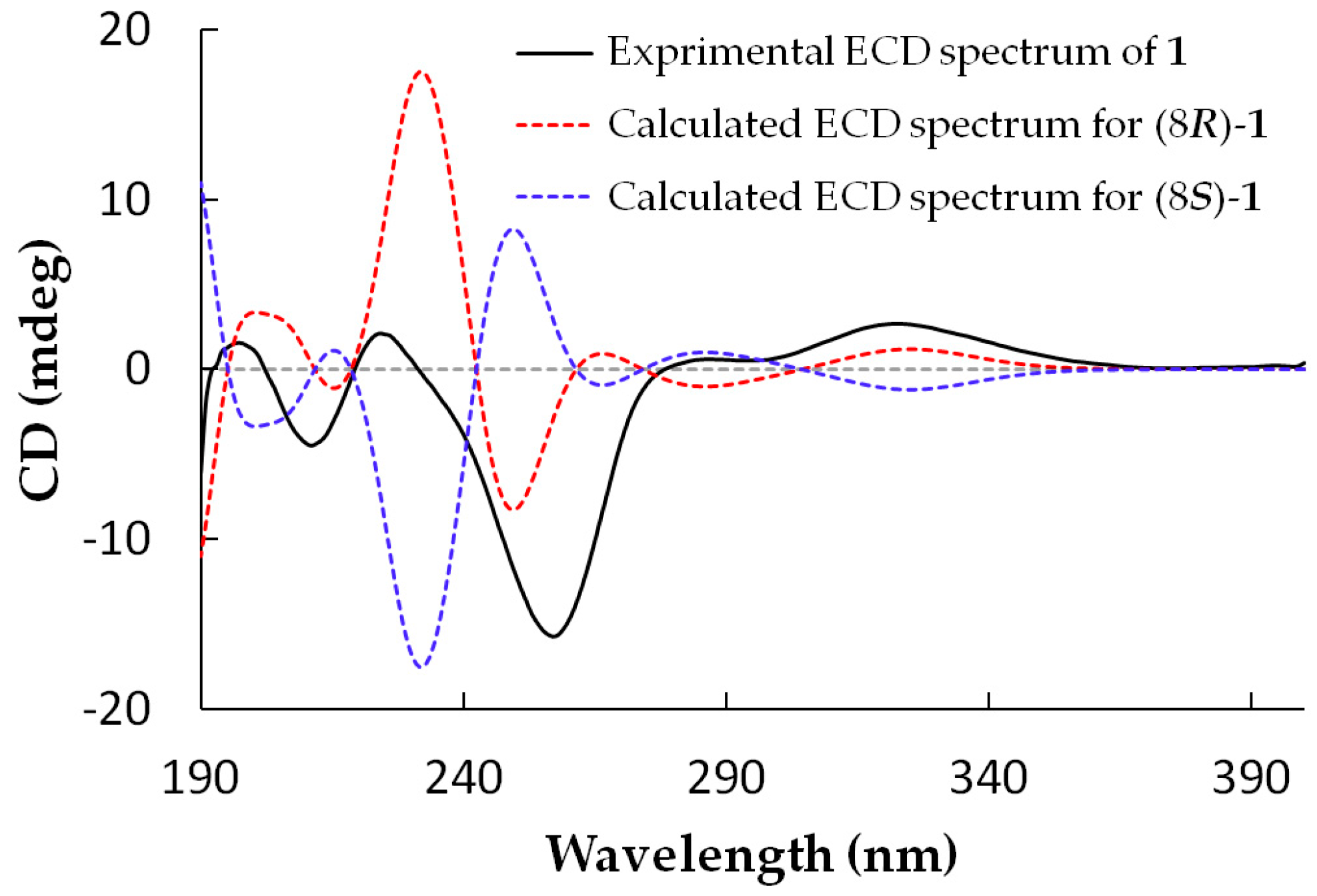
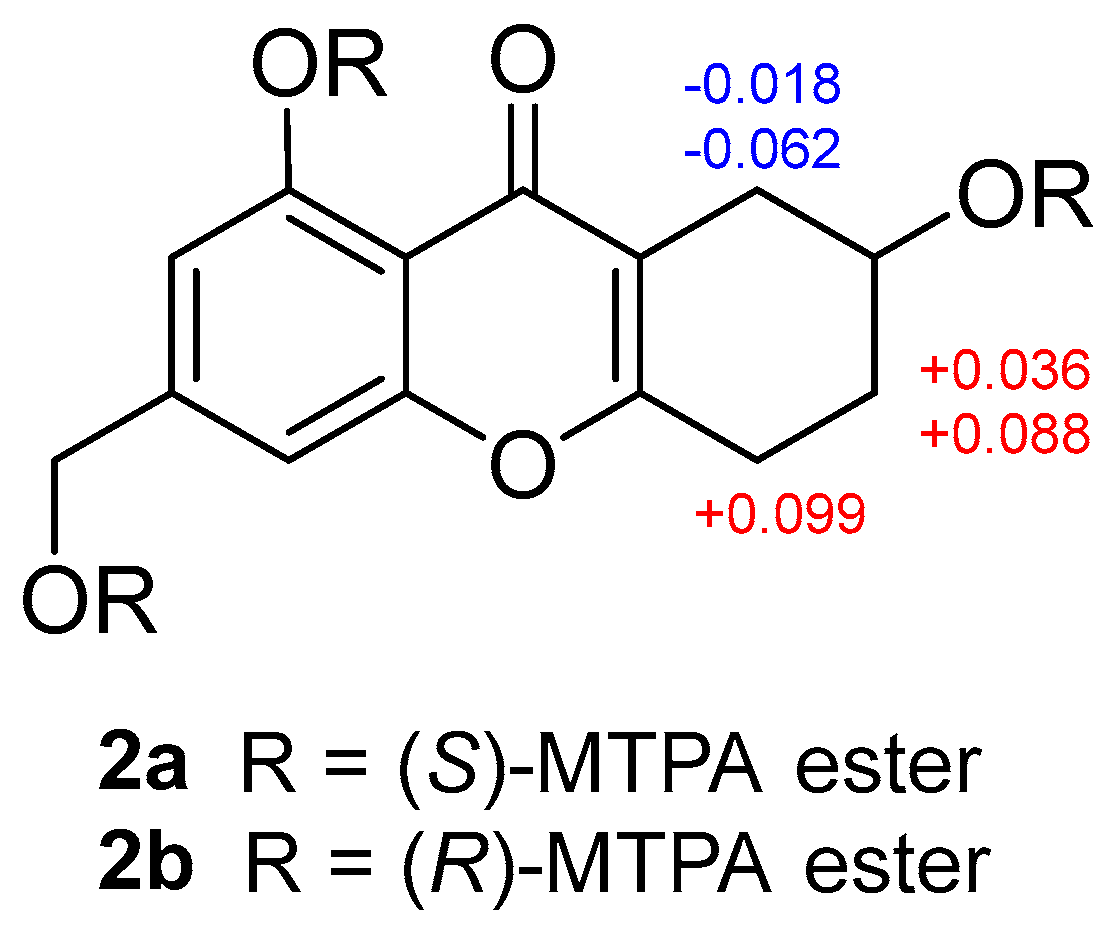
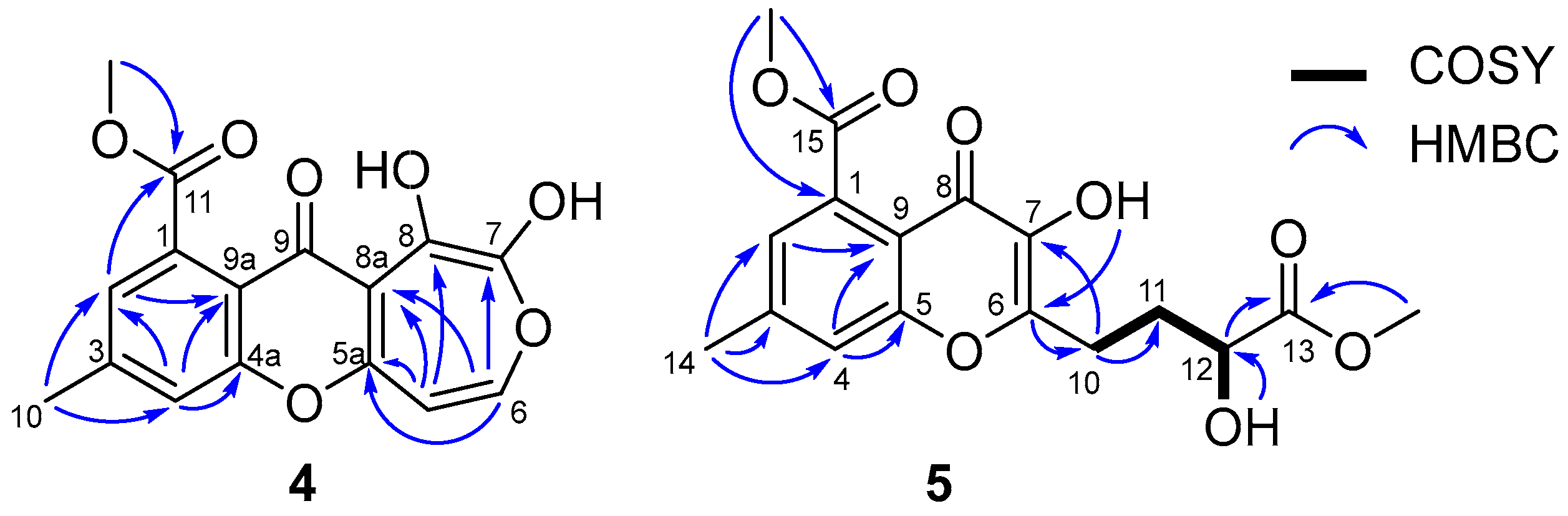
2.1. Structure Elucidation
2.2. Biological Activity
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.4.1. Arthone A (1)
3.4.2. Arthone B (2)
3.4.3. Arthone C (3)
3.4.4. Arthone D (4)
3.4.5. Arthone E (5)
3.5. Antioxidant Assay
3.6. Antimicrobial Assays
3.7. Anti-Inflammatory Assay
3.8. ECD Calculations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, M.; Tan, X.-M.; Liu, F.; Cai, L. Eight new Arthrinium species from China. Mycokeys 2018, 34, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Zhu, T.-H.; Zhu, H.-M.-Y.; Liang, M.; Qiao, T.-M.; Han, S.; Che, G.-N. Purification of protein AP-toxin from Arthrinium phaeospermum causing blight in Bambusa pervariabilis x Dendrocalamopisis grandis and its metabolic effects on four bamboo varieties. Phytopathology 2013, 103, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Li, B.J.; Liu, P.Q.; Jiang, Y.; Weng, Q.Y.; Chen, Q.H. First report of culm rot caused by Arthrinium phaeospennum on Phyllostachys viridis in China. Plant Dis. 2016, 100, 1013. [Google Scholar] [CrossRef]
- Chen, K.; Wu, X.Q.; Huang, M.X.; Han, Y.Y. First report of brown culm streak of Phyllostachys praecox caused by Arthrinium arundinis in Nanjing, China. Plant Dis. 2014, 98, 1274. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z. A phylogenetic re-evaluation of Arthrinium. Ima Fungus 2013, 4, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Wang, Z.; Ju, Z.R.; Wan, J.T.; Liao, S.R.; Lin, X.P.; Zhang, T.Y.; Zhou, X.F.; Chen, H.; Tu, Z.C.; et al. Cytotoxic cytochalasins from marine-derived fungus Arthrinium arundinis. Planta Med. 2015, 81, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Wei, X.Y.; Qin, X.C.; Lin, X.P.; Zhou, X.F.; Liao, S.R.; Yang, B.; Liu, J.; Tu, Z.C.; Liu, Y.H. Arthpyrones A-C, pyridone alkaloids from a sponge-derived fungus Arthrinium arundinis ZSDS1-F3. Org. Lett. 2015, 17, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Wang, J.F.; He, W.J.; Lin, X.P.; Zhou, X.F.; Liu, Y.H. One strain-many compounds method for production of polyketide metabolites using the sponge-derived fungus Arthrinium arundinis ZSDS1-F3. Chem. Nat. Compd. 2017, 53, 373–374. [Google Scholar] [CrossRef]
- Wang, J.-F.; Xu, F.-Q.; Wang, Z.; Lu, X.; Wan, J.-T.; Yang, B.; Zhou, X.-F.; Zhang, T.-Y.; Tu, Z.-C.; Liu, Y. A new naphthalene glycoside from the sponge-derived fungus Arthrinium sp. ZSDS1-F3. Nat. Prod. Res. 2014, 28, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Cabello, M.A.; Platas, G.; Collado, J.; Diez, M.T.; Martin, I.; Vicente, F.; Meinz, M.; Onishi, J.C.; Douglas, C.; Thompson, J.; et al. Arundifungin, a novel antifungal compound produced by fungi: Biological activity and taxonomy of the producing organisms. Int. Microbiol. 2001, 4, 93–102. [Google Scholar] [PubMed]
- Wei, M.-Y.; Xu, R.-F.; Du, S.-Y.; Wang, C.-Y.; Xu, T.-Y.; Shao, C.-L. A new griseofulvin derivative from the marine-derived Arthrinium sp. fungus and its biological activity. Chem. Nat. Compd. 2016, 52, 1011–1014. [Google Scholar] [CrossRef]
- Tsukada, M.; Fukai, M.; Miki, K.; Shiraishi, T.; Suzuki, T.; Nishio, K.; Sugita, T.; Ishino, M.; Kinoshita, K.; Takahashi, K.; et al. Chemical constituents of a marine fungus, Arthrinium sacchari. J. Nat. Prod. 2011, 74, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.P.; Simao, M.R.; de Souza, J.M.; Magalhaes, L.G.; Rodrigues, V.; Ambrosio, S.R.; Said, S. Evaluation of dihydroisocoumarins produced by the endophytic fungus Arthrinium state of Apiospora montagnei against Schistosoma mansoni. Nat. Prod. Res. 2013, 27, 2240–2243. [Google Scholar] [CrossRef] [PubMed]
- Monggoot, S.; Popluechai, S.; Gentekaki, E.; Pripdeevech, P. Fungal endophytes: An alternative source for production of volatile compounds from agarwood oil of Aquilaria subintegra. Microb. Ecol. 2017, 74, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhai, H.J.; Zhu, K.K.; Yu, J.-H.; Zhang, Y.Y.; Wang, Y.Y.; Jiang, C.-S.; Zhang, X.Y.; Zhang, Y.; Zhang, H. Bioactive pyridone alkaloids from a deep-sea-derived fungus Arthrinium sp. UJNMF0008. Mar. Drugs 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Park, I.Y.; Park, Y.J.; Lee, J.H.; Hong, Y.S.; Lee, J.J. A novel dihydroxanthenone, AGI-B4 with inhibition of VEGF-induced endothelial cell growth. J. Antibiot. 2002, 55, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Mutanyatta, J.; Matapa, B.G.; Shushu, D.D.; Abegaz, B.M. Homoisoflavonoids and xanthones from the tubers of wild and in vitro regenerated Ledebouria graminifolia and cytotoxic activities of some of the homoisoflavonoids. Phytochemistry 2003, 62, 797–804. [Google Scholar] [CrossRef]
- Abdel-Lateff, A.; Klemke, C.; Konig, G.M.; Wright, A.D. Two new xanthone derivatives from the algicolous marine fungus Wardomyces anomalus. J. Nat. Prod. 2003, 66, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, T.; Sato, Y.; Hatsuda, Y. Structure of sydowinin A, sydowinin B, and sydowinol, metabolites from Aspergillus sydowi. Agric. Biol. Chem. 1975, 39, 2341–2345. [Google Scholar] [CrossRef]
- Little, A.; Porco, J.A., Jr. Total syntheses of graphisin A and sydowinin B. Org. Lett. 2012, 14, 2862–2865. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Zheng, Z.H.; Liu, S.C.; Zhang, H.; Li, E.W.; Guo, L.D.; Che, Y.S. Oxepinochromenones, furochromenone, and their putative precursors from the endolichenic fungus Coniochaeta sp. J. Nat. Prod. 2010, 73, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.F.; Wang, J.; Zhang, X.Y.; Nong, X.H.; Xu, X.Y.; Qi, S.H. Cytotoxic polyketides from the deep-sea-derived fungus Engyodontium album DFFSCS021. Mar. Drugs 2014, 12, 5902–5915. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Umeokoli, B.O.; Eze, P.; Heering, C.; Janiak, C.; Mueller, W.E.G.; Orfali, R.S.; Hartmann, R.; Dai, H.F.; Lin, W.H.; et al. Secondary metabolites of the lichen-associated fungus Apiospora montagnei. Tetrahedron Lett. 2017, 58, 1702–1705. [Google Scholar] [CrossRef]
- Leon, F.; Gao, J.T.; Dale, O.R.; Wu, Y.S.; Habib, E.; Husni, A.S.; Hill, R.A.; Cutler, S.J. Secondary metabolites from Eupenicillium parvum and their in vitro binding affinity for human opioid and cannabinoid receptors. Planta Med. 2013, 79, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Di, B.L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-L.; Bao, J.; Liu, K.-S.; Zhang, X.-Y.; He, F.; Wang, Y.-F.; Nong, X.-H.; Qi, S.-H. Cytotoxic dihydrothiophene-condensed chromones from the marine-derived fungus Penicillium oxalicum. Planta Med. 2013, 79, 1474–1479. [Google Scholar] [CrossRef] [PubMed]
- Lösgen, S.; Magull, J.; Schulz, B.; Draeger, S.; Zeeck, A. Isofusidienols: Novel chromone-3-oxepines produced by the endophytic fungus Chalara sp. Eur. J. Org. Chem. 2010, 2008, 698–703. [Google Scholar] [CrossRef]
- Sun, R.-R.; Miao, F.-P.; Zhang, J.; Wang, G.; Yin, X.-L.; Ji, N.-Y. Three new xanthone derivatives from an algicolous isolate of Aspergillus wentii. Magn. Reson. Chem. 2013, 51, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-K.; Li, H.-J.; Song, Z.-F.; Li, Y.; Huai, Q.-Y. Synthesis and biological evaluation of novel curcuminoid derivatives. Molecules 2014, 19, 16349–16372. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Riceevans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 2013, 26, 1231–1237. [Google Scholar] [CrossRef]
- MacroModel, 9.7.211; Schrödinger: New York, NY, USA, 2009.
- Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010.
Sample Availability: Samples of the compounds 1–13 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | Positon | 5 |
|---|---|---|---|---|---|---|
| 2 | 6.74, brs | 6.70, s | 6.70, brs | 7.27, d (0.9) | 2 | 7.23, brs |
| 4 | 6.96, brs | 6.91, s | 6.94 a, brs | 7.56, brs | 4 | 7.54, brs |
| 5 | 5.72, d (5.9) | 2.79, dt (18.4, 6.4) 2.71, dt (18.4, 6.4) | 6.94 a, s | 6.98, d (8.9) | 10 | 2.84, t (7.9) |
| 6 | 7.33, d (5.9) | 1.87, m 1.80, m | 7.34, d (8.9) | 11 | 2.08, m 1.93, m | |
| 7 | 3.99, m | 12 | 4.14, m | |||
| 8 | 2.61, dd (16.4, 4.2) 2.32, dd (16.4, 5.8) | 14 | 2.44, brs | |||
| 10 | 4.56, brd (5.6) | 4.54, d (5.8) | 4.56, s | 2.49, s | 13-OCH3 | 3.62, s |
| 12 | 4.12, dd (12.5, 5.8) 3.88, dd (12.5, 7.2) | 15-OCH3 | 3.83, s | |||
| OCH3 | 3.70, s | 3.83, s | 3.89, s | 7-OH | 9.00, brs | |
| 1-OH | 12.37, s | 12.67, s | 12.55, s | 12-OH | 5.63, d (4.3) | |
| 7-OH | 4.92, d (3.7) | 9.45, s | ||||
| 8-OH | 12.05, s | |||||
| 10-OH | 5.50, t (5.6) | 5.47, t (5.8) | ||||
| 12-OH | 5.29, dd (7.2, 5.8) |
| Position | 1 | 2 | 3 | 4 | Position | 5 |
|---|---|---|---|---|---|---|
| 1 | 159.4, C | 159.5, C | 160.5, C | 132.8, C | 1 | 132.7, C |
| 2 | 108.5, CH | 107.6, CH | 107.1, CH | 124.0, CH | 2 | 124.6, CH |
| 3 | 152.0, C | 151.8, C | 152.9, C | 147.7 b, C | 3 | 144.4, C |
| 4 | 104.1, CH | 103.8, CH | 103.8, CH | 119.1, CH | 4 | 119.6, CH |
| 4a | 154.7, C | 155.6, C | 155.3, C | 156.0, C | 5 | 155.0, C |
| 5 | 95.1, CH | 25.0, CH2 | 102.5, CH | 106.3, CH | 6 | 152.7, C |
| 5a | 160.6,C | 165.3, C | 155.2, C | 147.4 b, C | 7 | 138.8, C |
| 6 | 157.1, CH | 28.80 a, CH2 | 151.2, C | 124.6, CH | 8 | 171.1, C |
| 7 | 62.9, CH | 141.4, C | 147.9 b, C | 9 | 117.0, C | |
| 8 | 84.3, C | 28.78 a, CH2 | 117.6, C | 140.5, C | 10 | 24.8, CH2 |
| 8a | 103.8, C | 114.1, C | 108.6, C | 108.7, C | 11 | 31.2, CH2 |
| 9 | 178.2, C | 181.9, C | 179.1, C | 180.9, C | 12 | 69.7, CH |
| 9a | 108.8, C | 107.9, C | 106.3, C | 113.7, C | 13 | 174.6, C |
| 10 | 62.2, CH2 | 62.3, CH2 | 62.4, CH2 | 21.3, CH3 | 14 | 21.4, CH3 |
| 11 | 167.9, C | 166.8, C | 168.8, C | 15 | 169.5, C | |
| 12 | 63.6, CH2 | 13-OCH3 | 52.0, CH3 | |||
| OCH3 | 52.8, CH3 | 52.2, CH3 | 52.7, CH3 | 15-OCH3 | 52.8, CH3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, J.; He, F.; Yu, J.-H.; Zhai, H.; Cheng, Z.-Q.; Jiang, C.-S.; Zhang, Y.; Zhang, Y.; Zhang, X.; Chen, G.; et al. New Chromones from a Marine-Derived Fungus, Arthrinium sp., and Their Biological Activity. Molecules 2018, 23, 1982. https://doi.org/10.3390/molecules23081982
Bao J, He F, Yu J-H, Zhai H, Cheng Z-Q, Jiang C-S, Zhang Y, Zhang Y, Zhang X, Chen G, et al. New Chromones from a Marine-Derived Fungus, Arthrinium sp., and Their Biological Activity. Molecules. 2018; 23(8):1982. https://doi.org/10.3390/molecules23081982
Chicago/Turabian StyleBao, Jie, Fei He, Jin-Hai Yu, Huijuan Zhai, Zhi-Qiang Cheng, Cheng-Shi Jiang, Yuying Zhang, Yun Zhang, Xiaoyong Zhang, Guangying Chen, and et al. 2018. "New Chromones from a Marine-Derived Fungus, Arthrinium sp., and Their Biological Activity" Molecules 23, no. 8: 1982. https://doi.org/10.3390/molecules23081982
APA StyleBao, J., He, F., Yu, J.-H., Zhai, H., Cheng, Z.-Q., Jiang, C.-S., Zhang, Y., Zhang, Y., Zhang, X., Chen, G., & Zhang, H. (2018). New Chromones from a Marine-Derived Fungus, Arthrinium sp., and Their Biological Activity. Molecules, 23(8), 1982. https://doi.org/10.3390/molecules23081982