Inhibitor of CBP Histone Acetyltransferase Downregulates p53 Activation and Facilitates Methylation at Lysine 27 on Histone H3


 ,
,
Abstract
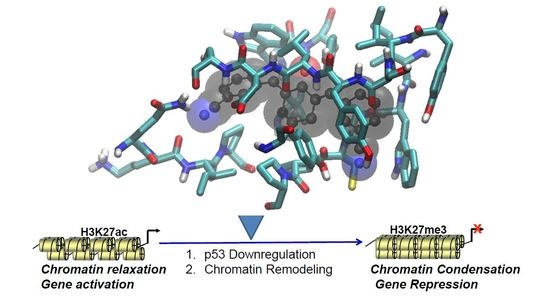
1. Introduction
2. Results
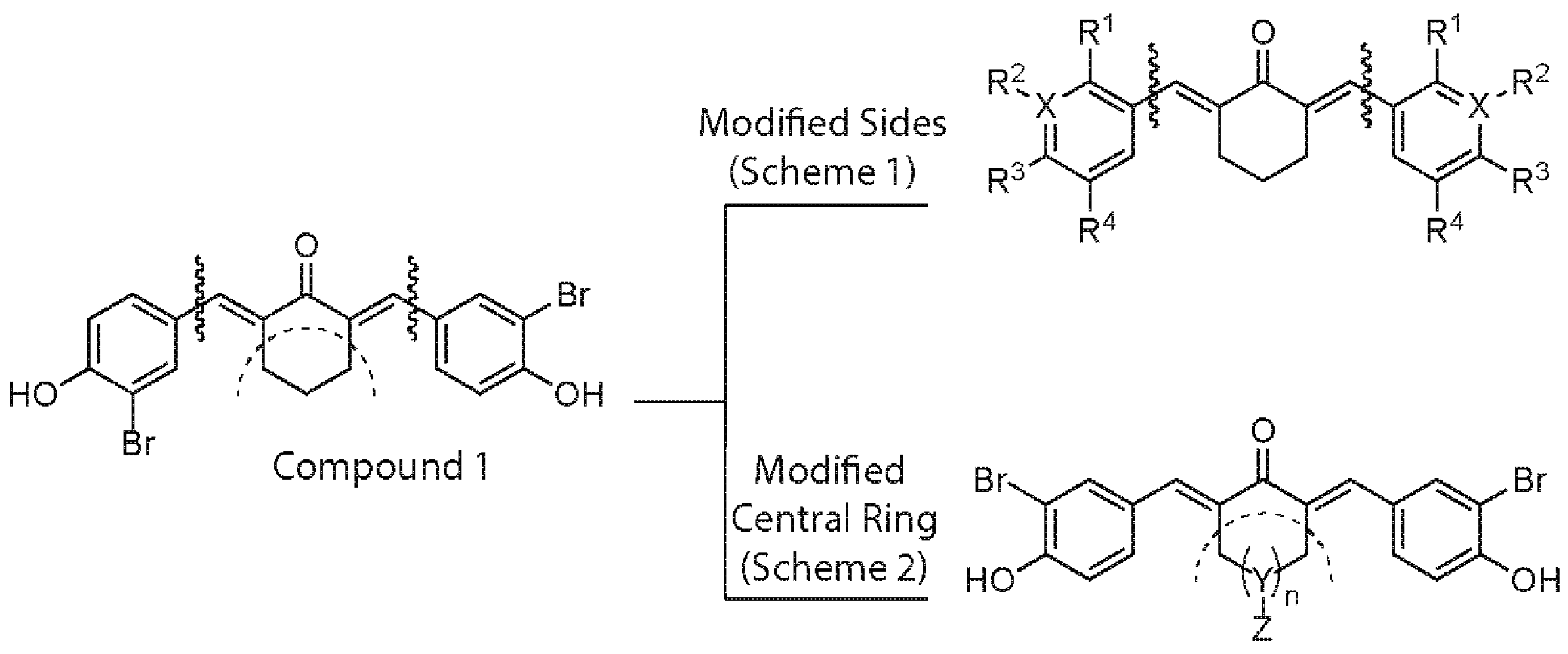
2.1. Structure–Activity Relationship Analysis of cy-C5-Curcuminoid Analogs
2.2. Biochemical Characterization of Potential Inhibitors of CBP HAT
2.2.1. IC50 Determination to Identify the Most Potent Inhibitor of CBP HAT Activity
2.2.2. Effects of NiCur on the CBP HAT Activity
2.2.3. Molecular Basis of Interaction between CBP HAT Domain and NiCur
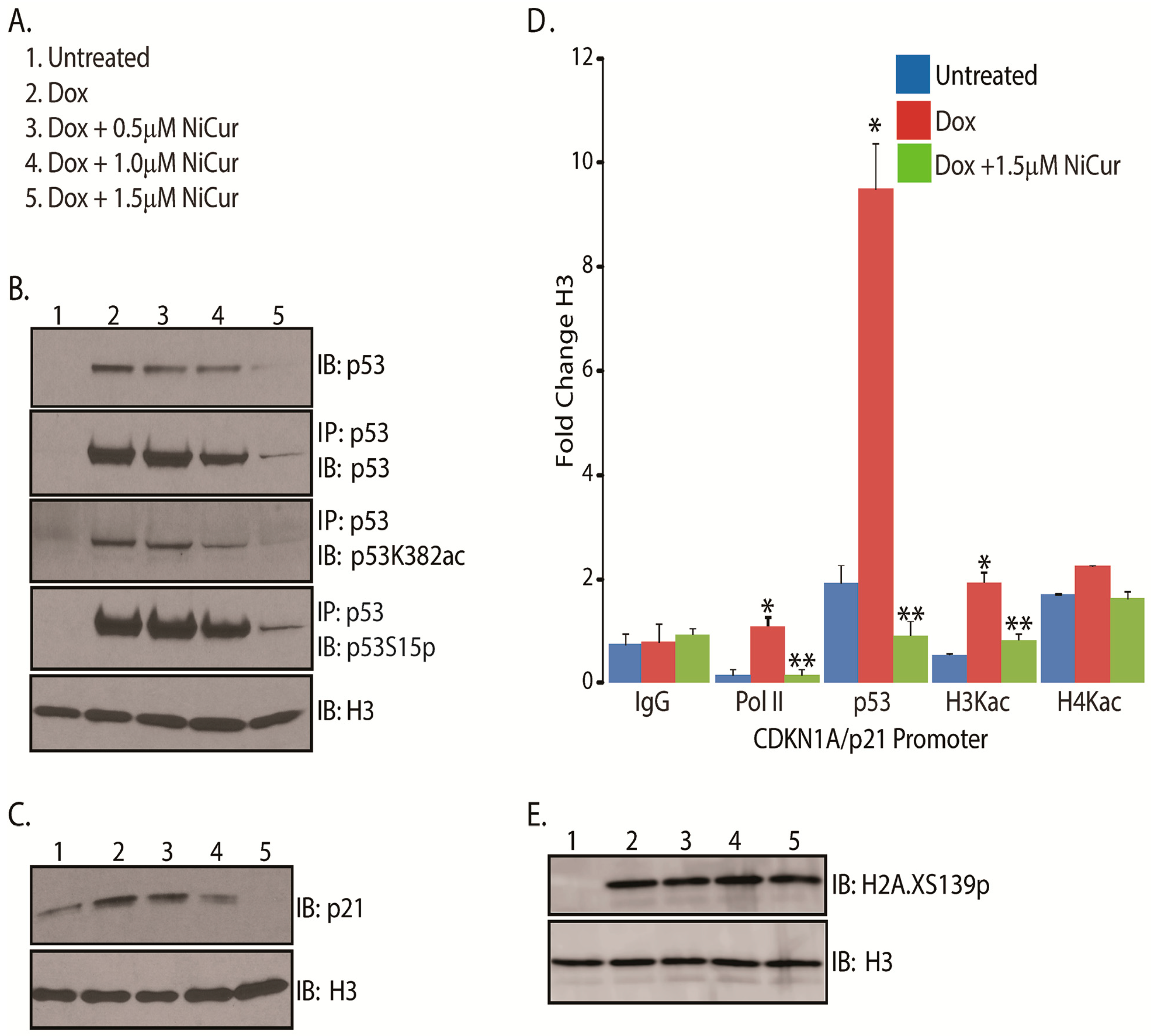
2.3. NiCur Modulates Transcription Functions of p53 during DNA Damage
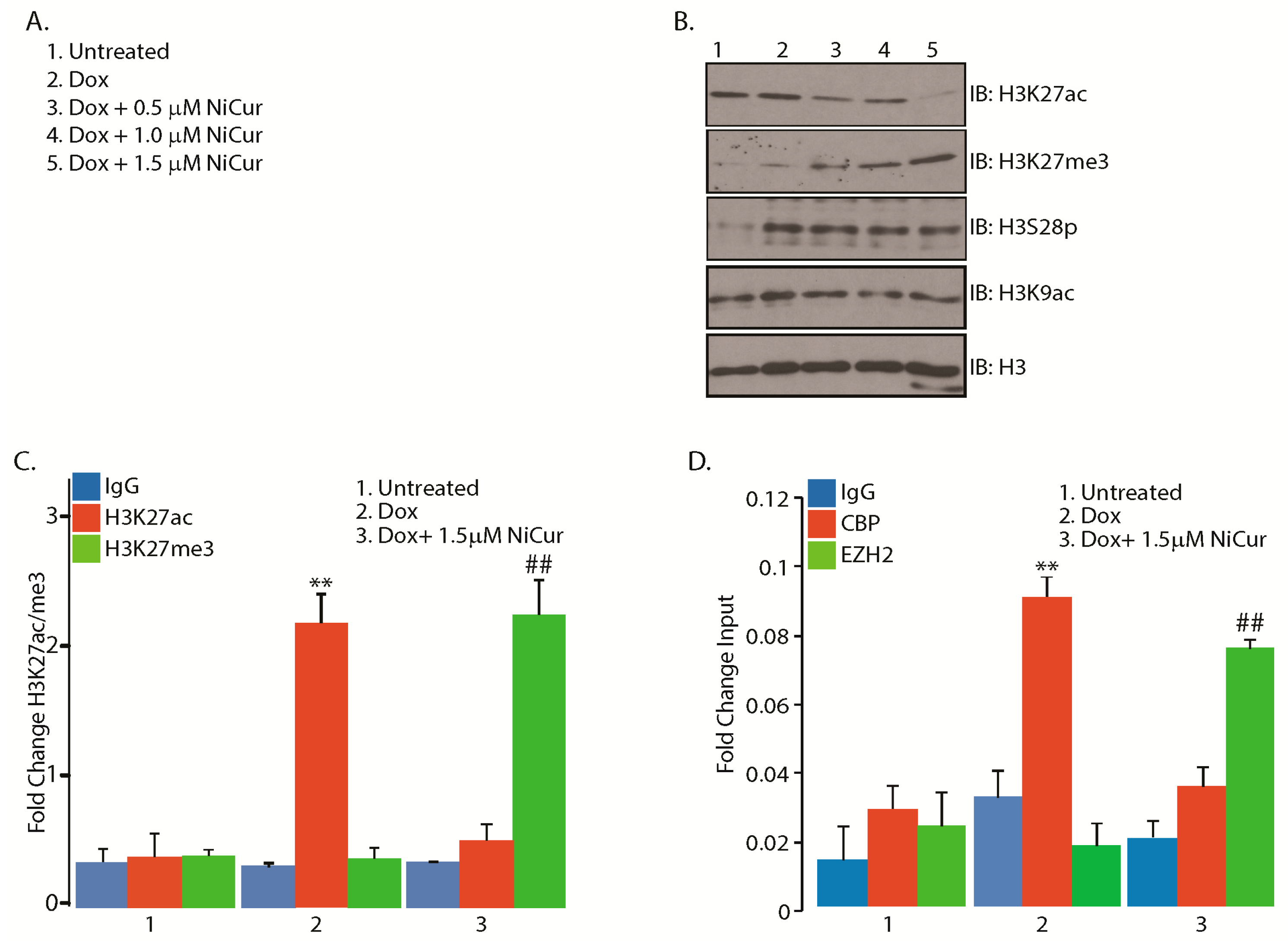
2.4. NiCur Dynamically Reprograms the Acetylation Marks on the Chromatin Landscape
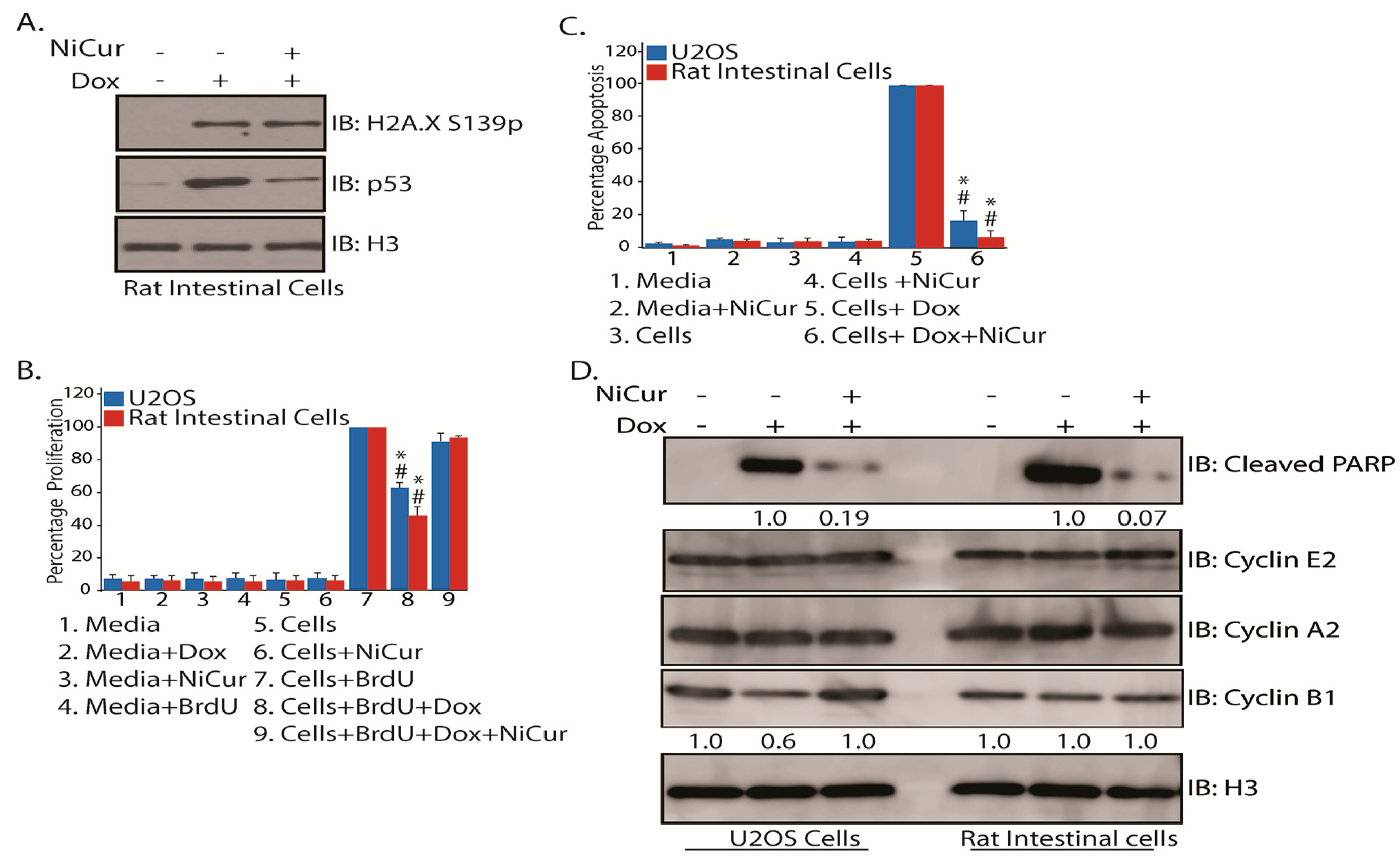
2.5. Cellular Effects of NiCur Treatment on Normal Rat Intestinal Epithelial Cells
3. Discussion
4. Conclusions
5. Materials and Methods
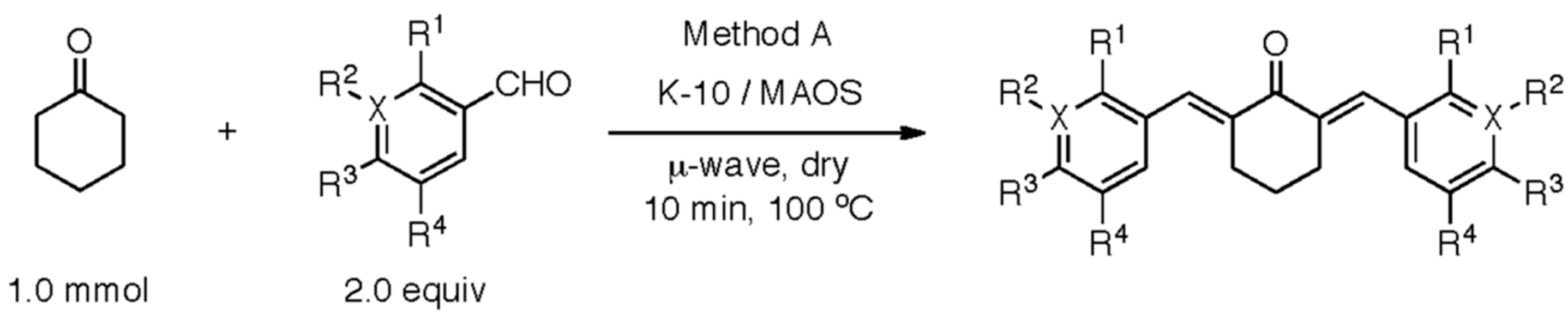
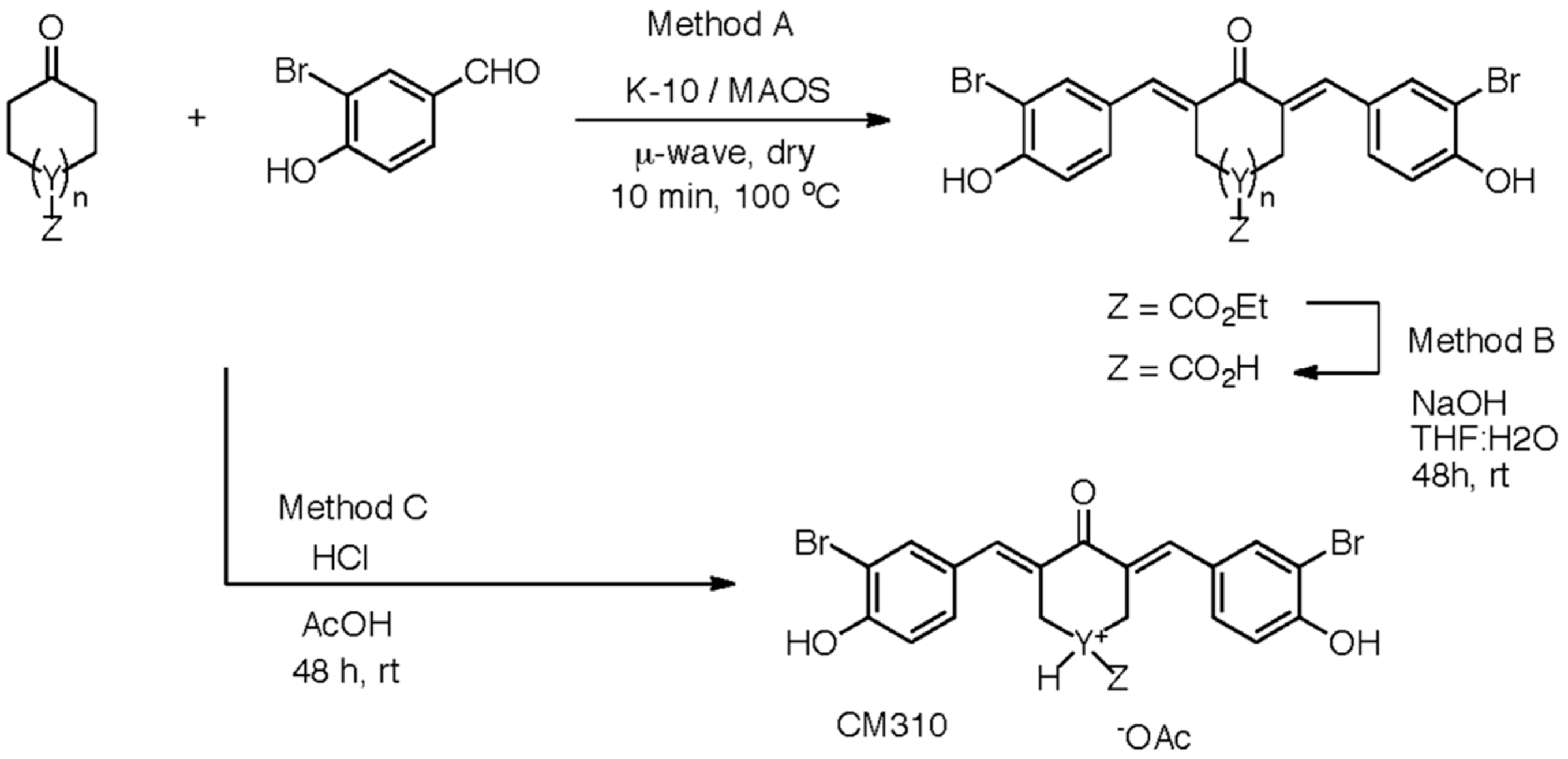
5.1. Synthetic Methods for Designing the Two-Prong Library of Potential CBP HAT Inhibitors
5.2. Chemical Synthesis and General Information
5.2.1. Claisen-Schmidt Condensation General Procedure
5.2.2. Claisen-Schmidt
5.3. Cell Lines, Plasmids, and Antibodies
5.4. Luciferase Assay for Structure Activity Relationship and IC50 Determination
5.5. In Vitro HAT Assay
5.6. Computational Analysis
5.7. Immunoblotting
5.8. Chromatin Immunoprecipitation Quantitative PCR (ChIP-qPCR)
5.9. BrdU Cell Proliferation Assay
5.10. Cleaved Caspase 3 Apoptosis Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.F.; Landauer, M.R. Protection against ionizing radiation by antioxidant nutrients and phytochemicals. Toxicology 2003, 189, 1–20. [Google Scholar] [CrossRef]
- Wang, X.; Wei, L.; Cramer, J.M.; Leibowitz, B.J.; Judge, C.; Epperly, M.; Greenberger, J.; Wang, F.; Li, L.; Stelzner, M.G.; et al. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation. Sci. Rep. 2015, 5, 8566. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.R.; Moughan, J.; Myerson, R.; Abitbol, A.; Doncals, D.E.; Johnson, D.; Schefter, T.E.; Chen, Y.; Fisher, B.; Michalski, J.; et al. Predictors of Radiation Therapy-Related Gastrointestinal Toxicity From Anal Cancer Dose-Painted Intensity Modulated Radiation Therapy: Secondary Analysis of NRG Oncology RTOG 0529. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Komarova, E.A.; Siebenhaar, F.; Botchkareva, N.V.; Sharov, A.A.; Komarov, P.G.; Marcus, M.; Gudkov, A.V.; Gilchrest, B.A. p53 Involvement in the control of murine hair follicle regression. Am. J. Pathol. 2001, 158, 1913–1919. [Google Scholar] [CrossRef]
- Komarova, E.A.; Neznanov, N.; Komarov, P.G.; Chernov, M.V.; Wang, K.; Gudkov, A.V. p53 inhibitor pifithrin alpha can suppress heat shock and glucocorticoid signaling pathways. J. Biol. Chem. 2003, 278, 15465–15468. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; Ogryzko, V.; Kao, H.Y.; Nash, A.; Chen, H.; Nakatani, Y.; Evans, R.M. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 1999, 96, 393–403. [Google Scholar] [CrossRef]
- Ma, B.; Fey, M.; Hottiger, M.O. WNT/beta-catenin signaling inhibits CBP-mediated RelA acetylation and expression of proinflammatory NF-kappaB target genes. J. Cell Sci. 2015, 128, 2430–2436. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Pathak, R.R.; Mujtaba, S. The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutr. Metab. 2011, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Vedamurthy, B.M.; Choudhari, R.; Ostwal, Y.B.; Mantelingu, K.; Kodaganur, G.S.; Kundu, T.K. Nitric oxide-mediated histone hyperacetylation in oral cancer: Target for a water-soluble HAT inhibitor, CTK7A. Chem. Biol. 2010, 17, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Bargonetti, J.; Manfredi, J.J. Multiple roles of the tumor suppressor p53. Curr. Opin. Oncol. 2002, 14, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; Zeng, L.; Zhou, M.M. Modulating molecular functions of p53 with small molecules. Cell Cycle 2006, 5, 2575–2578. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Borah, J.C.; Mujtaba, S.; Karakikes, I.; Zeng, L.; Muller, M.; Patel, J.; Moshkina, N.; Morohashi, K.; Zhang, W.; Gerona-Navarro, G.; et al. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 2011, 18, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Arai, M.; Ferreon, J.C.; Wright, P.E. Quantitative analysis of multisite protein-ligand interactions by NMR: Binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 2012, 134, 3792–3803. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Mapping the interactions of the p53 transactivation domain with the KIX domain of CBP. Biochemistry 2009, 48, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. Prospective therapeutic applications of p53 inhibitors. Biochem. Biophys. Res. Commun. 2005, 331, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Lucia, M.S.; Hansen, K.C.; Tyler, J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009, 459, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Conrad, P.A.; Scacheri, P.C.; Harte, P.J. Histone demethylase UTX and chromatin remodeler BRM bind directly to CBP and modulate acetylation of histone H3 lysine 27. Mol. Cell Biol. 2012, 32, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Okan, N.A.; Bales, E.; Nascimento, L.; Cole, P.A.; Medrano, E.E. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002, 62, 6231–6239. [Google Scholar] [PubMed]
- Iyer, N.G.; Xian, J.; Chin, S.F.; Bannister, A.J.; Daigo, Y.; Aparicio, S.; Kouzarides, T.; Caldas, C. p300 is required for orderly G1/S transition in human cancer cells. Oncogene 2007, 26, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, T.; Yoshida, N.; Kitabayashi, I. Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. 2008, 99, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of curcumin. Curr. Drug Targets 2011, 12, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Jitoe, A.; Isobe, J.; Nakatani, N.; Yonemori, S. Anti-oxidative and anti-inflammatory curcumin-related phenolics from rhizomes of Curcuma domestica. Phytochemistry 1993, 32, 1557–1560. [Google Scholar] [CrossRef]
- Vilekar, P.; Awasthi, S.; Natarajan, A.; Anant, S.; Awasthi, V. EF24 suppresses maturation and inflammatory response in dendritic cells. Int. Immunol. 2012, 24, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Du, Z.Y.; Zheng, X.; Cui, X.X.; Conney, A.H.; Zhang, K. Synthesis and evaluation of curcumin-related compounds for anticancer activity. Eur. J. Med. Chem. 2012, 53, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Valente, S.; Castellano, S.; Sbardella, G.; Di Santo, R.; Costi, R.; Bedford, M.T.; Mai, A. Novel 3,5-bis(bromohydroxybenzylidene)piperidin-4-ones as coactivator-associated arginine methyltransferase 1 inhibitors: Enzyme selectivity and cellular activity. J. Med. Chem. 2011, 54, 4928–4932. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Cheng, D.; Bedford, M.T.; Valente, S.; Nebbioso, A.; Perrone, A.; Brosch, G.; Sbardella, G.; de Bellis, F.; Miceli, M.; et al. Epigenetic multiple ligands: Mixed Histone/Protein methyltransferase, acetyltransferase, and class III deacetylase (Sirtuin) inhibitors. J. Med. Chem. 2008, 51, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [PubMed]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2008, 457, 57. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.L.; Briggs, S.D.; Allis, C.D. Acetylation and chromosomal functions. Curr. Opin. Cell Biol. 2000, 12, 326–333. [Google Scholar] [CrossRef]
- Mujtaba, S.; He, Y.; Zeng, L.; Farooq, A.; Carlson, J.E.; Ott, M.; Verdin, E.; Zhou, M. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell 2002, 9, 575–586. [Google Scholar] [CrossRef]
- Komarova, E.A.; Gudkov, A.V. Suppression of p53: A new approach to overcome side effects of antitumor therapy. Biochemistry 2000, 65, 41–48. [Google Scholar] [PubMed]
- Boija, A.; Mahat, D.B.; Zare, A.; Holmqvist, P.H.; Philip, P.; Meyers, D.J.; Cole, P.A.; Lis, J.T.; Stenberg, P.; Mannervik, M. CBP Regulates Recruitment and Release of Promoter-Proximal RNA Polymerase II. Mol. Cell 2017, 68, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Innocente, S.A.; Abrahamson, J.L.; Cogswell, J.P.; Lee, J.M. p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. USA. 1999, 96, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Heger, M.; van Golen, R.F.; Broekgaarden, M.; Michel, M.C. The molecular basis for the pharmacokinetics and pharmacodynamics of curcumin and its metabolites in relation to cancer. Pharmacol. Rev. 2014, 66, 222–307. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.C.; Chen, L.F. Regulation of NF-kappaB action by reversible acetylation. Novartis Found Symp. 2004, 259, 208–217. [Google Scholar] [PubMed]
- Debes, J.D.; Schmidt, L.J.; Huang, H.; Tindall, D.J. p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002, 62, 5632–5636. [Google Scholar] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.T.; Wagenblast, E.; Davis, C.A.; Moon, S.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Balasubramanyam, K.; Cebrat, M.; Buck, D.; Guidez, F.; Zelent, A.; Alani, R.M.; Cole, P.A. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. J. Am. Chem. Soc. 2005, 127, 17182–17183. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Pradhan, S.K.; Thanuja, G.R.; Vedamurthy, B.M.; Agrawal, S.; Dasgupta, D.; Kundu, T.K. Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. J. Med. Chem. 2009, 52, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Quang, P.; Torok, B. Microwave-Assisted Solid-Acid-Catalyzed Friedel-Crafts Alkylation and Electrophilic Annulation of Indoles Using Alcohols as Alkylating Agents. Synthesis 2009, 23, 4010–4014. [Google Scholar] [CrossRef]
- Kulkarni, A.; Torok, B. Microwave-assisted multicomponent domino cyclization-aromatization: An efficient approach for the synthesis of substituted quinolines. Green Chem. 2010, 12, 875–878. [Google Scholar] [CrossRef]
- Yadav, B.; Taurin, S.; Rosengren, R.J.; Schumacher, M.; Diederich, M.; Somers-Edgar, T.J.; Larsenc, L. Synthesis and cytotoxic potential of heterocyclic cyclohexanone analogues of curcumin. Bioorg. Med. Chem. 2010, 18, 6701–6707. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, S.; Sharma, R.; Chakraborti, A.K. Dual-activation protocol for tandem cross-aldol condensation: An easy and highly efficient synthesis of alpha,alpha’-bis(aryl/alkylmethylidene)ketones. J. Mol. Catal. A Chem. 2006, 260, 235–240. [Google Scholar] [CrossRef]
- Costi, R.; Di Santo, R.; Artico, M.; Miele, G.; Valentini, P.; Novellino, E.; Cereseto, A. Cinnamoyl compounds as simple molecules that inhibit p300 histone acetyltransferase. J. Med. Chem. 2007, 50, 1973–1977. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Vieweg, H.; Horn, H. Synthesis of Alpha,Alpha’-Bis-[Amidinobenzylidene] and Alpha,Alpha’-Bis-[Amidinobenzyl]-Cycloalkanones. Pharmazie 1977, 32, 141–145. [Google Scholar] [PubMed]
- Kumar, R.R.; Loganayaki, B.; Perumal, S. Sequential 1,3-Dipolar Cycloadditions in the Synthesis of Novel Tri-spiro Cyclohexanones and Piperidin-4-ones. Synth. Commun. 2009, 39, 3197–3216. [Google Scholar] [CrossRef]
- Appiah-Opong, R.; de Esch, I.; Commandeur, J.N.M.; Andarini, M.; Vermeulen, N.P.E. Structure-activity relationships for the inhibition of recombinant human cytochromes P450 by curcumin analogues. European. J. Med. Chem. 2008, 43, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.M.; Lu, Y.J.; Hu, H.P.; Shoji, M.; Liotta, D.C.; Snyder, J.P. Curcumin analog cytotoxicity against breast cancer cells: Exploitation of a redox-dependent mechanism. Bioorg. Med. Chem. Lett. 2009, 19, 6627–6631. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.X.; Liang, G.; Chu, Y.; Li, X.; Lian, Q.Q.; Lin, He, H.Y.; Huang, Y.; Hardy, D.O.; Ge, R. Curcumin derivatives inhibit testicular 17beta-hydroxysteroid dehydrogenase 3. Bioorg. Med. Chem. Lett. 2010, 20, 2549–2551. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Yang, S.L.; Jiang, L.J.; Zhao, Y.; Shao, L.L.; Xiao, J.; Ye, F.; Li, Y.; Li, X. Synthesis and anti-bacterial properties of mono-carbonyl analogues of curcumin. Chem. Pharm. Bull. 2008, 56, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Di Santo, R.; Costi, R.; Novellino, E.; Greco, G.; Massa, S.; Tramontano, E.; Marongiu, M.E.; de Montis, A.; La Colla, P. Geometrically and conformationally restrained cinnamoyl compounds as inhibitors of HIV-1 integrase: Synthesis, biological evaluation, and molecular modeling. J. Med. Chem. 1998, 41, 3948–3960. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanyam, K.; Swaminathan, V.; Ranganathan, A.; Kundu, T.K. Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 2003, 278, 19134–19140. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mizzen, C.; Ying, C.; Candau, R.; Barlev, N.; Brownell, J.; Allis, C.D.; Berger, S.L. Histone acetyltransferase activity is conserved between yeast and human GCN5 and is required for complementation of growth and transcriptional activation. Mol. Cell Biol. 1997, 17, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | X | R1 | R2 | R3 | R4 | % Inhibition |
|---|---|---|---|---|---|---|
| DMSO | - | - | - | - | - | 0 |
| Curcumin | - | - | - | - | - | 61 |
| 1 | C | H | Br | OH | H | 31 |
| 2 | C | H | CN | H | H | >99 |
| 3 | C | H | Br | OCH3 | H | 53 |
| 4 | C | CH3 | H | H | H | 6 |
| 5 | C | H | CH3 | H | CH3 | 25 |
| 6 | C | H | CH3 | OH | CH3 | 40 |
| 7 | C | H | H | OH | H | 38 |
| 8 | C | F | H | H | H | 13 |
| 9 | C | Br | H | H | H | 17 |
| 10 | C | H | Br | H | H | 88 |
| 11 | C | H | H | Br | H | 4 |
| 12 | N | H | - a | H | H | >99 |
| Compound ID | n | Y | Z | (%) Inhibition |
|---|---|---|---|---|
| DMSO | - | - | - | 0 |
| Curcumin | - | - | - | 61 |
| 1 | 1 | CH | H | 31 |
| 13 | 0 | - | - | 39 |
| 14 | 1 | CH | CH3 | 53 |
| 15 | 1 | CH | CO2Et | 82 |
| 16 | 1 | CH | CO2H | 26 |
| 17 | 1 | CH | OH | 26 |
| 18 | 1 | O | - a | 22 |
| 19 | 1 | N b | H | >99 |
| 20 | 1 | N | CH3 | 29 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincek, A.S.; Patel, J.; Jaganathan, A.; Green, A.; Pierre-Louis, V.; Arora, V.; Rehmann, J.; Mezei, M.; Zhou, M.-M.; Ohlmeyer, M.; et al. Inhibitor of CBP Histone Acetyltransferase Downregulates p53 Activation and Facilitates Methylation at Lysine 27 on Histone H3. Molecules 2018, 23, 1930. https://doi.org/10.3390/molecules23081930
Vincek AS, Patel J, Jaganathan A, Green A, Pierre-Louis V, Arora V, Rehmann J, Mezei M, Zhou M-M, Ohlmeyer M, et al. Inhibitor of CBP Histone Acetyltransferase Downregulates p53 Activation and Facilitates Methylation at Lysine 27 on Histone H3. Molecules. 2018; 23(8):1930. https://doi.org/10.3390/molecules23081930
Chicago/Turabian StyleVincek, Adam S., Jigneshkumar Patel, Anbalagan Jaganathan, Antonia Green, Valerie Pierre-Louis, Vimal Arora, Jill Rehmann, Mihaly Mezei, Ming-Ming Zhou, Michael Ohlmeyer, and et al. 2018. "Inhibitor of CBP Histone Acetyltransferase Downregulates p53 Activation and Facilitates Methylation at Lysine 27 on Histone H3" Molecules 23, no. 8: 1930. https://doi.org/10.3390/molecules23081930
APA StyleVincek, A. S., Patel, J., Jaganathan, A., Green, A., Pierre-Louis, V., Arora, V., Rehmann, J., Mezei, M., Zhou, M.-M., Ohlmeyer, M., & Mujtaba, S. (2018). Inhibitor of CBP Histone Acetyltransferase Downregulates p53 Activation and Facilitates Methylation at Lysine 27 on Histone H3. Molecules, 23(8), 1930. https://doi.org/10.3390/molecules23081930