Synthesis of Antibacterial Nisin–Peptoid Hybrids Using Click Methodology



Abstract
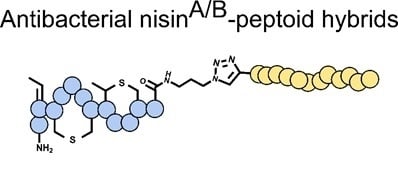
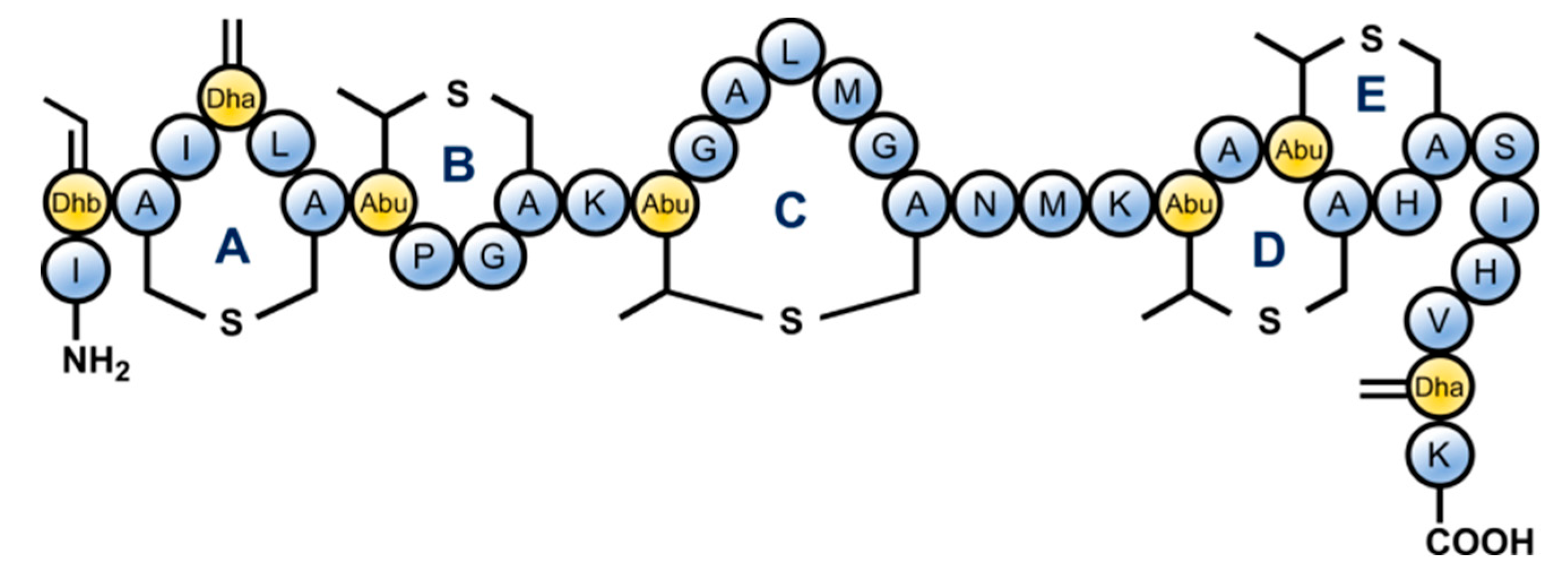
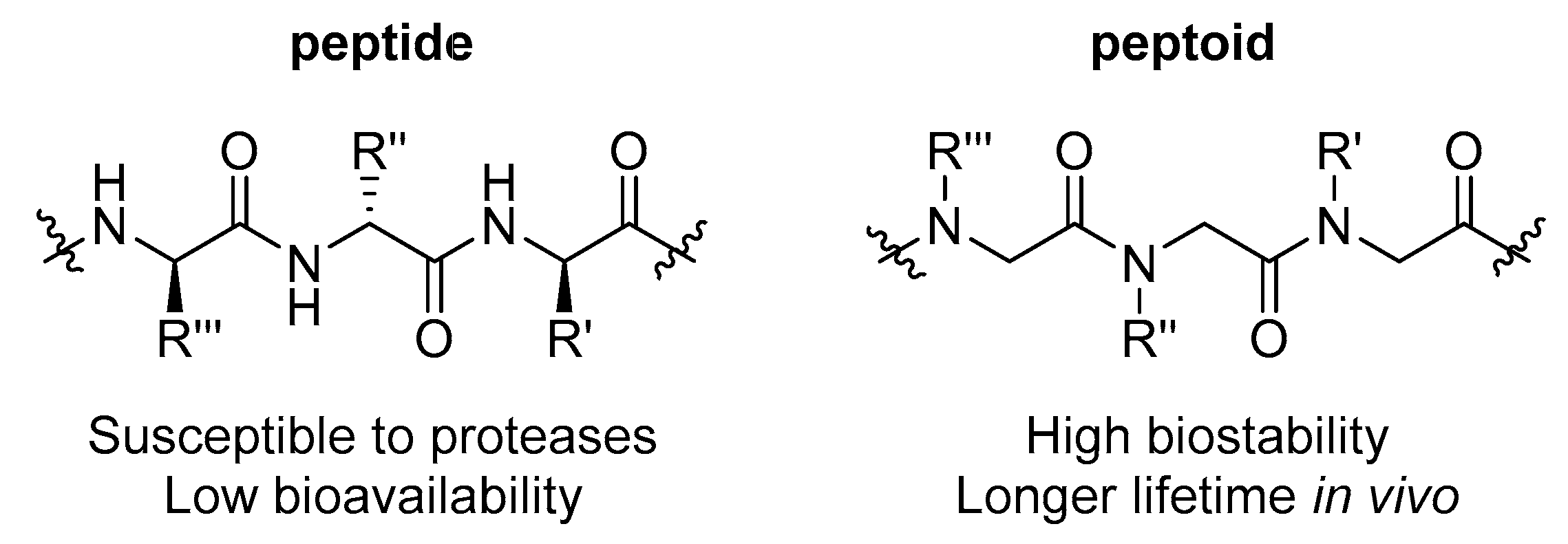
1. Introduction
2. Results and Discussion
3. Materials and Methods
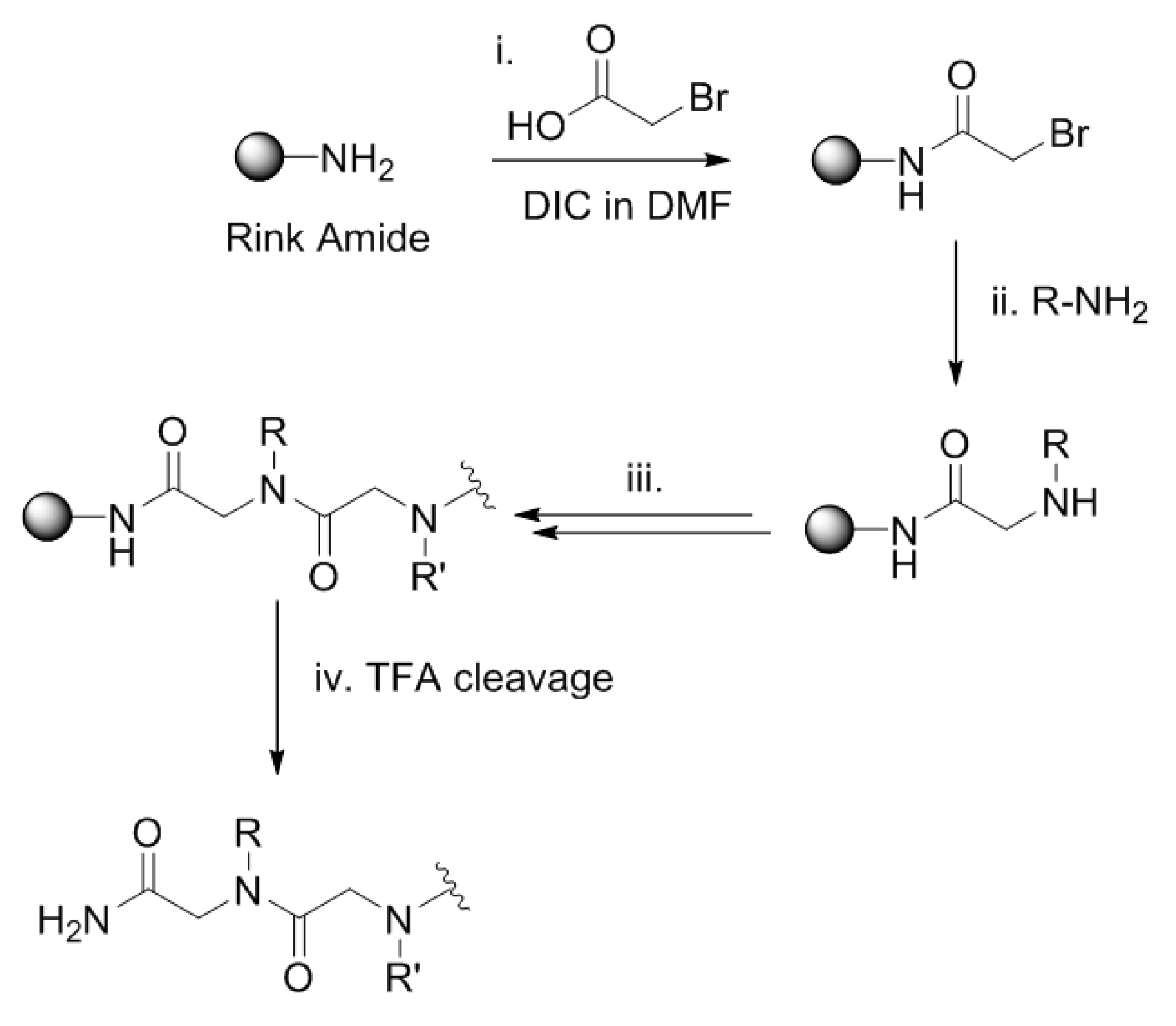
3.1. Linear Peptoid Synthesis (1a, 1b, 2a and 2b)
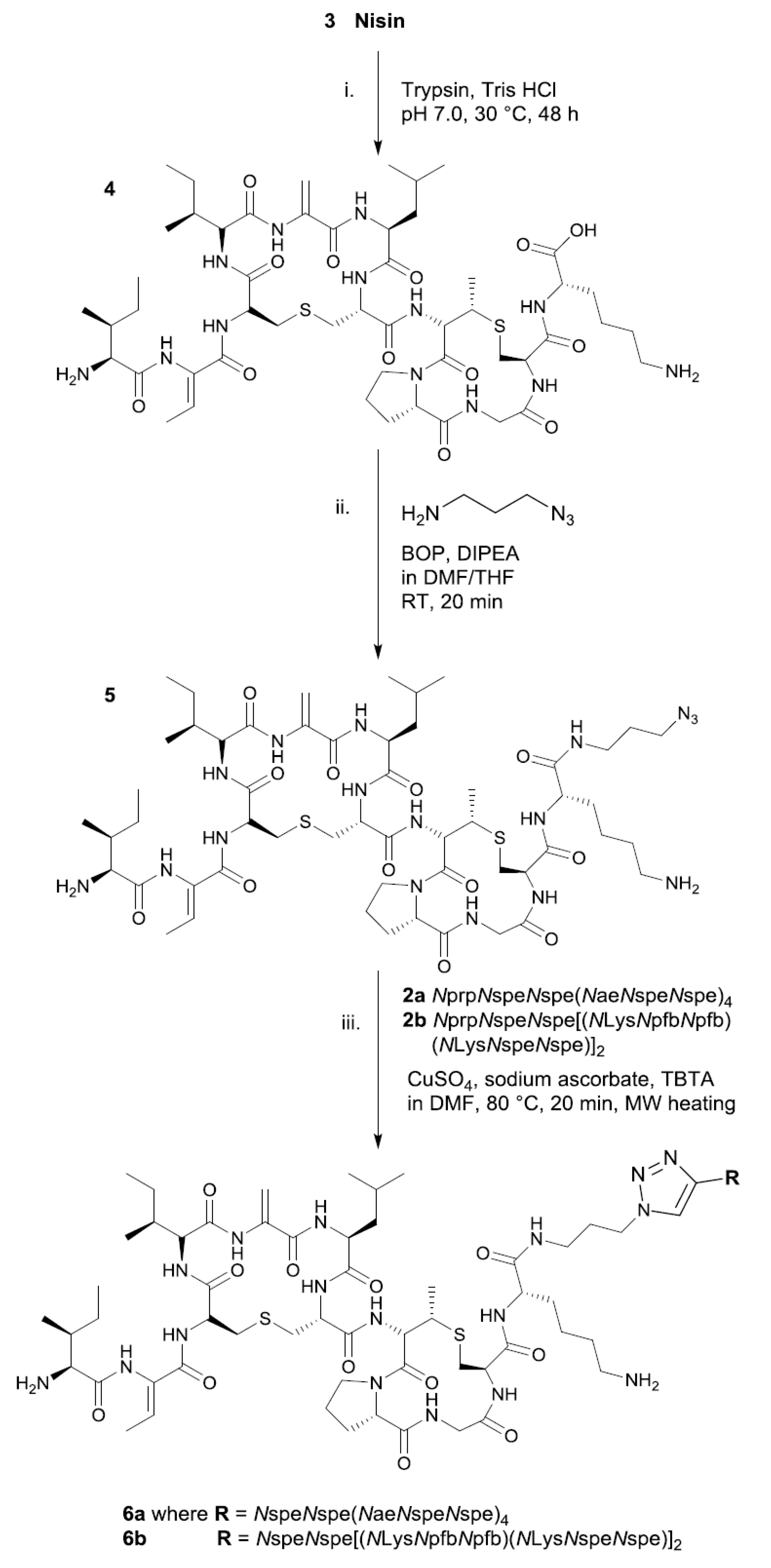
3.2. Amide-Coupled Azide-NisinA/B (5)
3.3. Click Protocol of Alkyne-Peptoids with NisinA/B-Azide (6a, 6b)
3.4. MIC Determination
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References and Note
- Department for International Development and Department of Health. United Kingdom, 2015.
- Infectious Diseases Society of America. The 10 × ‘20 Initiative: Pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin. Infect. Dis. 2010, 50, 1081–1083. [Google Scholar]
- Kramer, N.E.; Smid, E.J.; Kok, J.; de Kruijff, B.; Kuipers, O.P.; Breukink, E. Resistance of Gram-positive bacteria to nisin is not determined by lipid II levels. FEMS Microbiol. Lett. 2004, 239, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.I.; Breukink, E. The expanding role of lipid II as a target for lantibiotics. Future Microbiol. 2007, 2, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Morell, J.L. Structure of nisin. J. Am. Chem. Soc. 1971, 93, 4634–4635. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; van Heel, A.J.; Montalban-Lopez, M.; Kuipers, O.P. Potentiating the Activity of Nisin against Escherichia coli. Front. Cell Dev. Biol. 2016, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Arnusch, C.J.; Bonvin, A.M.J.J.; Verel, A.M.; Jansen, W.T.M.; Liskamp, R.M.J.; de Kruijff, D.; Pieters, R.J.; Breukink, E. The vancomycin-nisin (1–12) hybrid restores activity against vancomycin resistant Enterococci. Biochemistry 2008, 47, 12661–12663. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T.; Wood, T.M.; t’Hart, P.; Kleijn, L.H.J.; Hendrickx, A.P.A.; Willems, R.J.L.; Breukink, E.; Martin, N.I. Semisynthetic Lipopeptides Derived from Nisin Display Antibacterial Activity and Lipid II Binding on Par with That of the Parent Compound. J. Am. Chem. Soc. 2015, 137, 9382–9389. [Google Scholar] [CrossRef] [PubMed]
- Breukink, E.; Wiedemann, I.; van Kraaij, C.; Kuipers, O.P.; Sahl, H.G.; de Kruijff, B. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 1999, 286, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Breukink, E.; de Kruijff, B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 2006, 5, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Sahl, H.G. Lipid II and other bactoprenol-bound cell wall precursors as drug targets. Curr. Opin. Investig. Drug 2010, 11, 157–164. [Google Scholar]
- Van Heel, A.J.; Montalban-Lopez, M.; Kuipers, O.P. Evaluating the feasibility of lantibiotics as an alternative therapy against bacterial infections in humans. Expert Opin. Drug Metab. Toxicol. 2011, 7, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Ghalit, N.; Reichwein, J.F.; Hilbers, H.W.; Breukink, E.; Rijkers, D.T.S.; Liskamp, R.M.J. Synthesis of bicyclic alkene-/alkane-bridged nisin mimics by ring-closing metathesis and their biochemical evaluation as lipid II binders: Toward the design of potential novel antibiotics. ChemBioChem 2007, 8, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Slootweg, J.C.; Peters, N.; Quarles van Ufford, H.L.; Breukink, E.; Liskamp, R.M.J.; Rijkers, D.T. Semi-synthesis of biologically active nisin hybrids composed of the native lanthionine ABC-fragment and a cross-stapled synthetic DE-fragment. Bioorg. Med. Chem. 2014, 22, 5345–5353. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C.; McKinnie, S.M.K.; Vederas, J.C. The Synthesis of Active and Stable Diaminopimelate Analogues of the Lantibiotic Peptide Lactocin S. J. Am. Chem. Soc. 2012, 134, 2008–2011. [Google Scholar] [CrossRef] [PubMed]
- Rink, R.; Wierenga, J.; Kuipers, A.; Kluskens, L.D.; Driessen, A.J.; Kuipers, O.P.; Moll, G.N. Dissection and Modulation of the Four Distinct Activities of Nisin by Mutagenesis of Rings A and B and by C-Terminal Truncation. Appl. Environ. Microbiol. 2007, 73, 5809–5816. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K.; et al. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Patch, J.A.; Kirshenbaum, K.; Seurynck, S.L.; Zuckermann, R.N.; Barron, A.E. Pseudopeptides in Drug Development; Nielsen, P.E., Ed.; Wiley-VCH: Weinheim, Germany, 2004; p. 1. [Google Scholar]
- Zuckermann, R.N.; Kodadek, T. Peptoids as potential therapeutics. Curr. Opin. Mol. Ther. 2009, 11, 299–309. [Google Scholar] [PubMed]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.L.; Eggimann, G.A.; Jahoda, C.A.B.; Zuckermann, R.N.; Sharples, G.J.; Cobb, S.L. Exploring the links between peptoid antibacterial activity and toxicity. Med. Chem. Commun. 2017, 8, 886–896. [Google Scholar] [CrossRef]
- Kapoor, R.; Eimerman, P.R.; Hardy, J.W.; Cirillo, J.D.; Contag, C.H.; Barron, A.E. Efficacy of antimicrobial peptoids against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Wadman, M.W.; Dohm, M.T.; Czyzewski, A.M.; Spormann, A.M.; Barron, A.E. Antimicrobial Peptoids Are Effective against Pseudomonas aeruginosa Biofilms. Antimicrob. Agents Chemother. 2011, 55, 3054–3057. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Ziegler, H.L.; Nielsen, H.M.; Frimodt-Moller, N.; Jaroszewski, J.W.; Franzyk, H. Antimicrobial, hemolytic, and cytotoxic activities of beta-peptoid-peptide hybrid oligomers: Improved properties compared to natural AMPs. ChemBioChem 2010, 11, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Shin, S.B.Y.; Benson, M.A.; Torres, V.J.; Kirshenbaum, K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem 2012, 7, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Benson, M.A.; Shin, S.B.Y.; Torres, V.J.; Kirshenbaum, K. Amphiphilic cyclic peptoids that exhibit antimicrobial activity by disrupting Staphylococcus aureus membranes. Eur. J. Org. Chem. 2013, 17, 3560–3566. [Google Scholar] [CrossRef]
- Findlay, B.; Szelemej, P.; Zhanel, F.; Schweizer, F. Guanidylation and Tail Effects in Cationic Antimicrobial Lipopeptoids. PLoS ONE 2012, 7, e41141. [Google Scholar] [CrossRef] [PubMed]
- Ryge, T.S.; Frimodt-Moller, N.; Hansen, P.R. Antimicrobial activities of twenty lysine-peptoid hybrids against clinically relevant bacteria and fungi. Chemotherapy 2008, 54, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; McDermott, G.; Wetzler, M.; Le Gros, M.A.; Myllys, M.; Knoechel, C.; Barron, A.E.; Larabell, C.A. Soft X-ray tomography of phenotypic switching and the cellular response to antifungal peptoids in Candida albicans. Proc. Natl. Acad. Sci. USA 2009, 106, 19375–19380. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Bolt, H.L.; Eggimann, G.A.; Mc Auley, D.F.; Mc Mullan, R.; Curran, T.; Zhou, M.; Jahoda, C.A.B.; Cobb, S.L.; Lundy, F.T. Peptoid efficacy against polymicrobial biofilms determined using propidium monoazide-modified quantitative PCR. ChemBioChem 2017, 18, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Vedel, L.; Bonke, G.; Foged, C.; Ziegler, H.; Franzyk, H.; Jaroszewski, J.W.; Olsen, C.A. Antiplasmodial and prehemolytic activities of alpha-peptide-beta-peptoid chimeras. ChemBioChem 2007, 8, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Eggimann, G.A.; Bolt, H.L.; Denny, P.W.; Cobb, S.L. Investigating the anti-leishmanial effects of linear peptoids. ChemMedChem 2015, 10, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.L.; Denny, P.W.; Cobb, S.L. An Efficient Method for the Synthesis of Peptoids with Mixed Lysine-type/Arginine-type Monomers and Evaluation of Their Anti-leishmanial Activity. J. Vis. Exp. 2016, 117, e54750. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.L.; Eggimann, G.A.; Denny, P.W.; Cobb, S.L. Enlarging the chemical space of anti-leishmanials: A structure–activity relationship study of peptoids against Leishmania mexicana, a causative agent of cutaneous leishmaniasis. MedChemComm 2016, 7, 799–805. [Google Scholar] [CrossRef]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Comparison of the Proteolytic Susceptibilities of Homologous L-Amino Acid, D-Amino Acid, and N-Substituted Glycine Peptide and Peptoid Oligomers. Drug Dev. Res. 1995, 35, 20–32. [Google Scholar] [CrossRef]
- Mojsoska, B.; Jenssen, H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals 2015, 8, 366–415. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; Leyland, M.; Clark, J.; Dodd, H.M.; Lian, L.Y.; Gasson, M.J.; Bycroft, B.W.; Roberts, G.C. Structure-activity relationships in the peptide antibiotic nisin: Antibacterial activity of fragments of nisin. FEBS Lett. 1996, 390, 129–132. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1–3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Bolt, H.L.; Cobb, S.L. A practical method for the synthesis of peptoids containing both lysine-type and arginine-type monomers. Org. Biomol. Chem. 2016, 14, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids [oligo (N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef] [PubMed]
- Margaret, C.; Eli, N.P.; Michael, Z.D. USA300 Methicillin-Resistant Staphylococcus aureus, United States, 2000–2013. Emerg. Infect. Dis. 2015, 21, 1973–1980. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | MIC (µM) | ED50 (μM) | ||
|---|---|---|---|---|
| S. aureus (USA300) | HaCaT | HepG2 | ||
| 7 | Vancomycin | 1 | ||
| 3 | Nisin | 5 | ||
| 4 | Nisin[1,2,3,4,5,6,7,8,9,10,11,12] | >100 | ||
| 5 | Nisin[1,2,3,4,5,6,7,8,9,10,11,12]-azide | 26 | ||
| 1a | (NaeNspeNspe)4 | 2 | 26 | 15 |
| 1b | [(NLysNpfbNpfb)(NLysNspeNspe)]2 | 2–4 | 53 | 18 |
| 2a | NprpNspeNspe(NaeNspeNspe)4 | 4 | 20 | 12 |
| 2b | NprpNspeNspe[(NLysNpfbNpfb)(NLysNspeNspe)]2 | 4–7 | 15 | 17 |
| 6a | Nisin[1,2,3,4,5,6,7,8,9,10,11,12]-NspeNspe(NaeNspeNspe)4 | 5–10 | 24 | 15 |
| 6b | Nisin[1,2,3,4,5,6,7,8,9,10,11,12]-NspeNspe[(NLysNpfbNpfb)(NLysNspeNspe)]2 | 9–18 | 22 | 23 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolt, H.L.; Kleijn, L.H.J.; Martin, N.I.; Cobb, S.L. Synthesis of Antibacterial Nisin–Peptoid Hybrids Using Click Methodology. Molecules 2018, 23, 1566. https://doi.org/10.3390/molecules23071566
Bolt HL, Kleijn LHJ, Martin NI, Cobb SL. Synthesis of Antibacterial Nisin–Peptoid Hybrids Using Click Methodology. Molecules. 2018; 23(7):1566. https://doi.org/10.3390/molecules23071566
Chicago/Turabian StyleBolt, Hannah L., Laurens H. J. Kleijn, Nathaniel I. Martin, and Steven L. Cobb. 2018. "Synthesis of Antibacterial Nisin–Peptoid Hybrids Using Click Methodology" Molecules 23, no. 7: 1566. https://doi.org/10.3390/molecules23071566
APA StyleBolt, H. L., Kleijn, L. H. J., Martin, N. I., & Cobb, S. L. (2018). Synthesis of Antibacterial Nisin–Peptoid Hybrids Using Click Methodology. Molecules, 23(7), 1566. https://doi.org/10.3390/molecules23071566