
Recent Developments in C–H Activation for Materials Science in the Center for Selective C–H Activation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
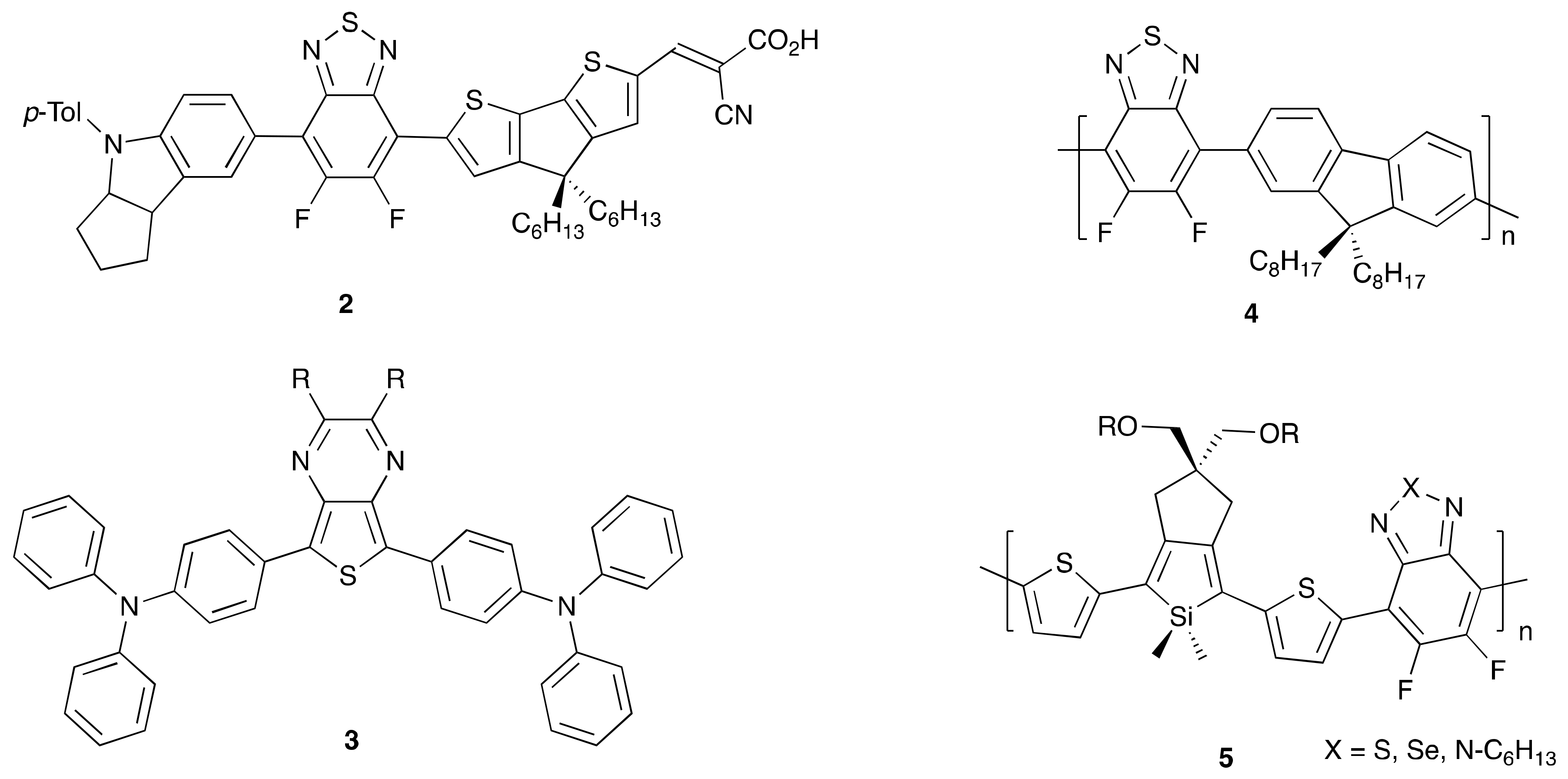
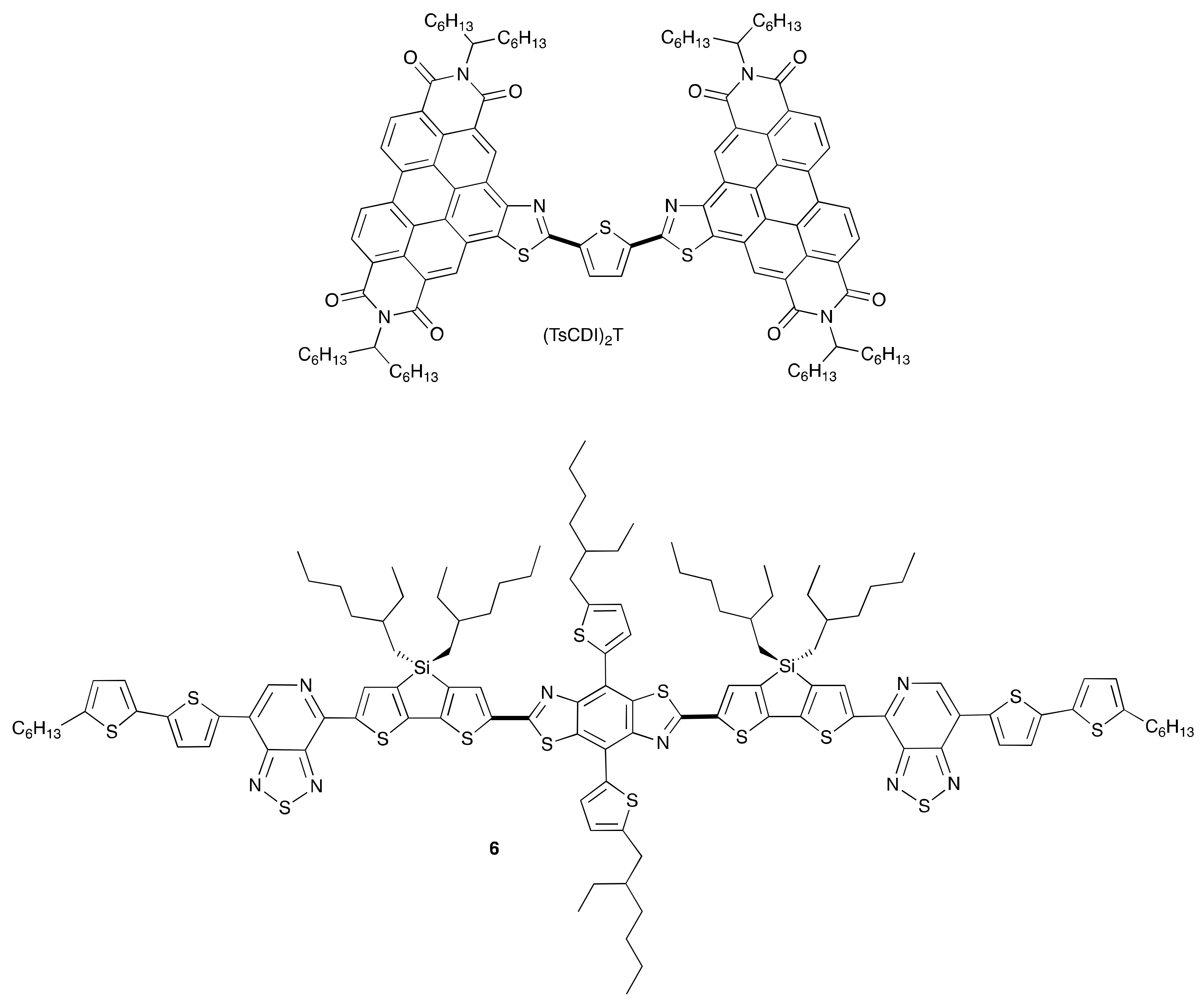
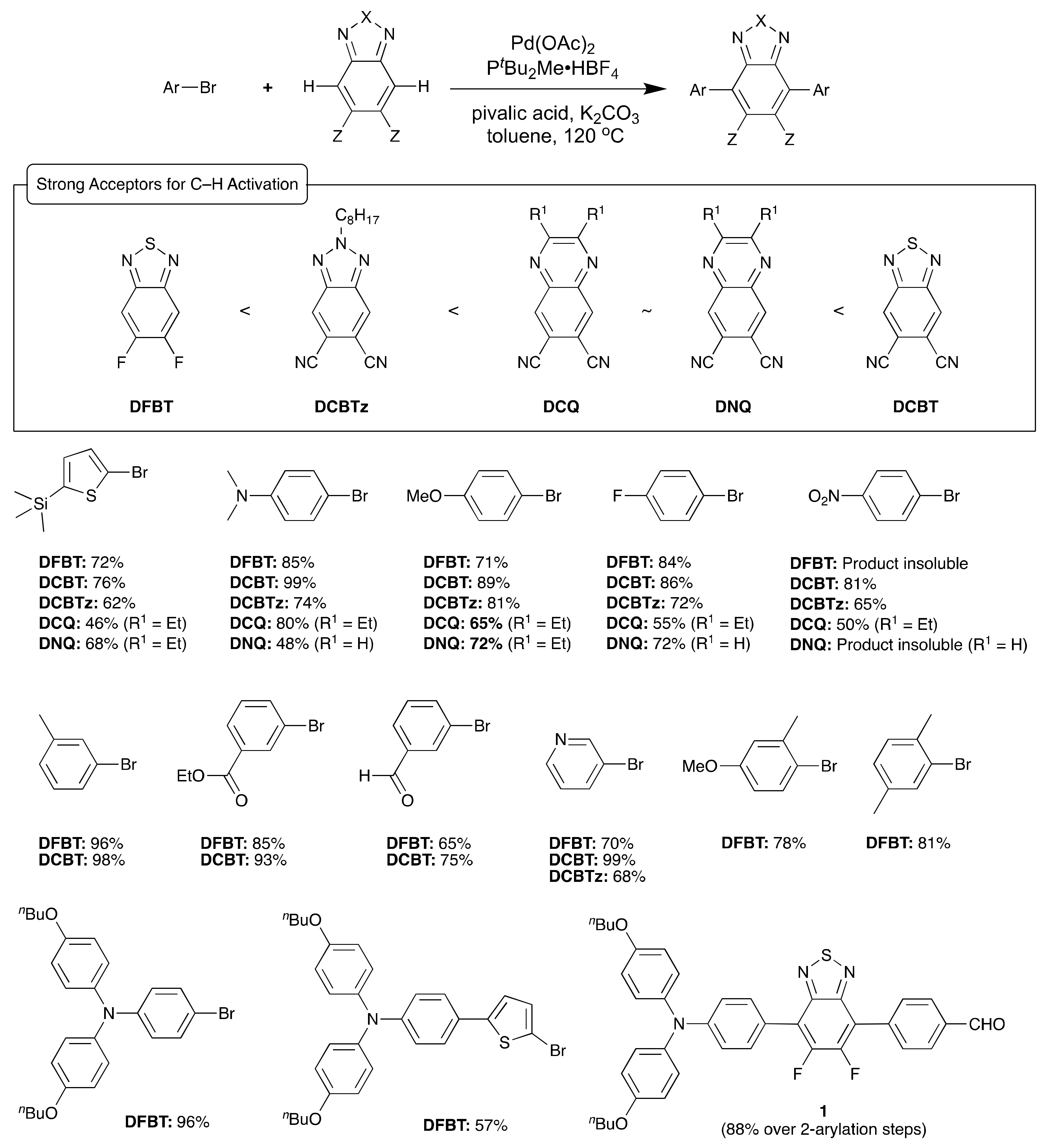
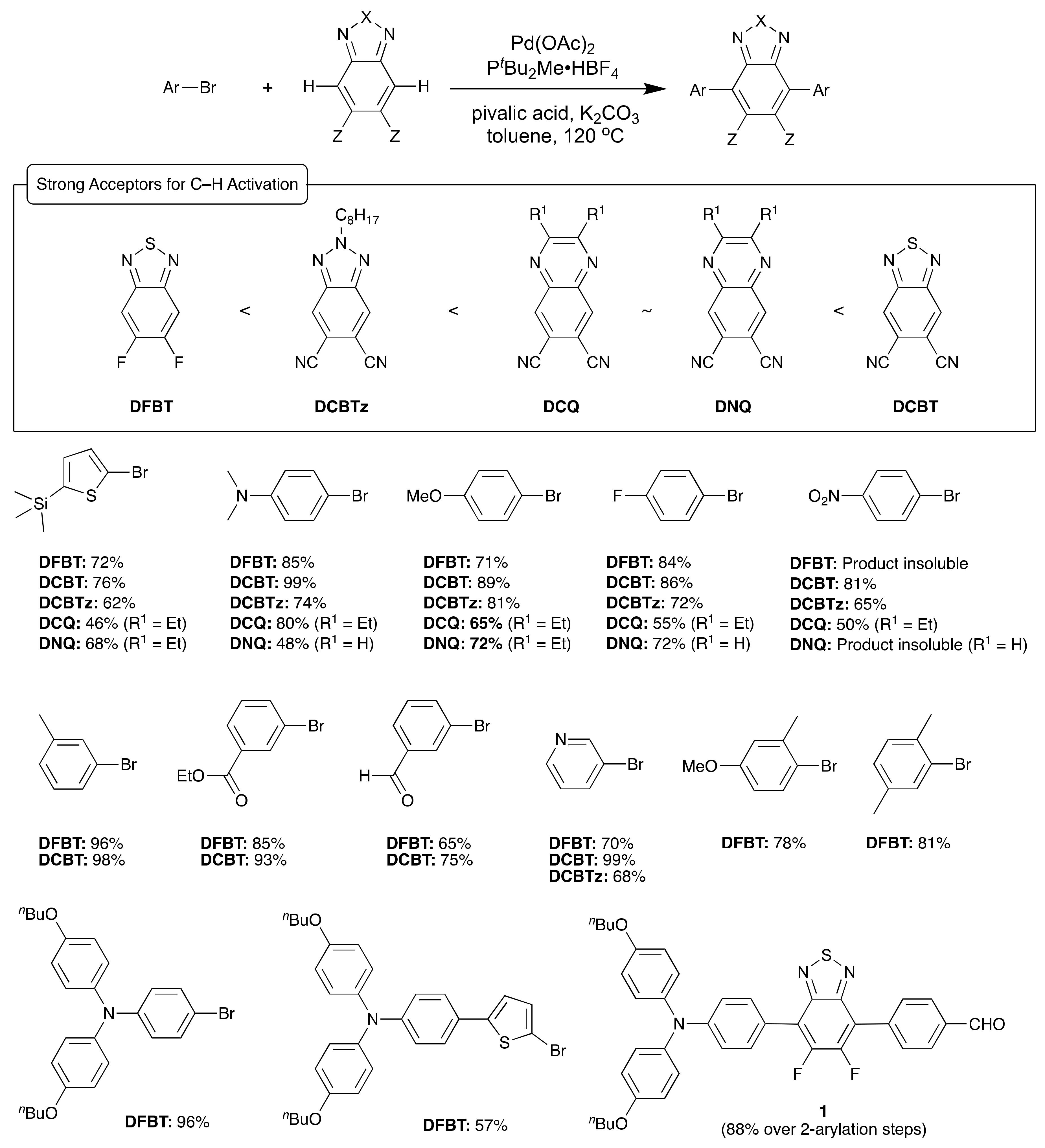
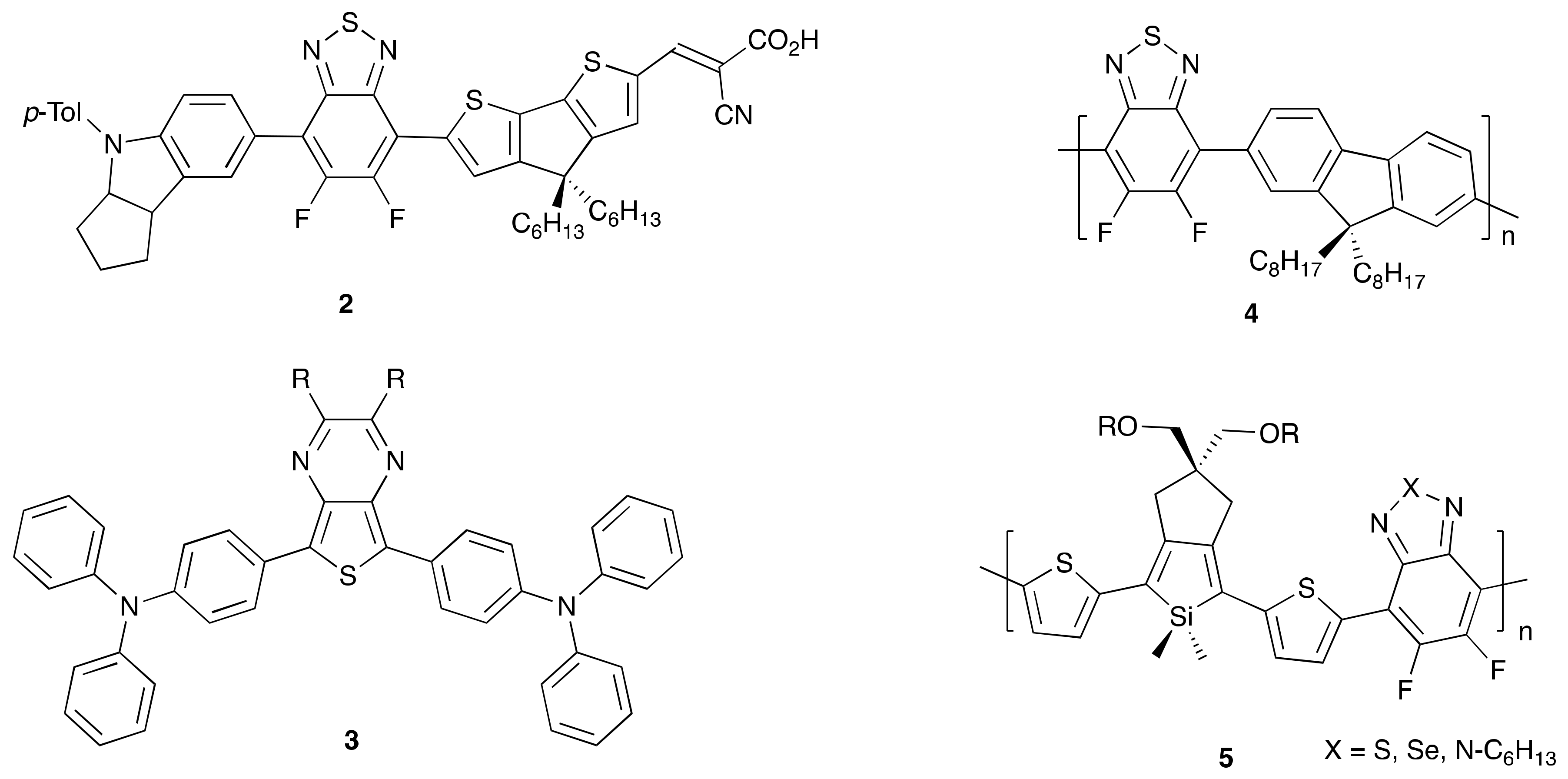
2. Direct Arylation of Electron-Poor π-Conjugated Small Molecules
Benzothiadiazole Derivatives
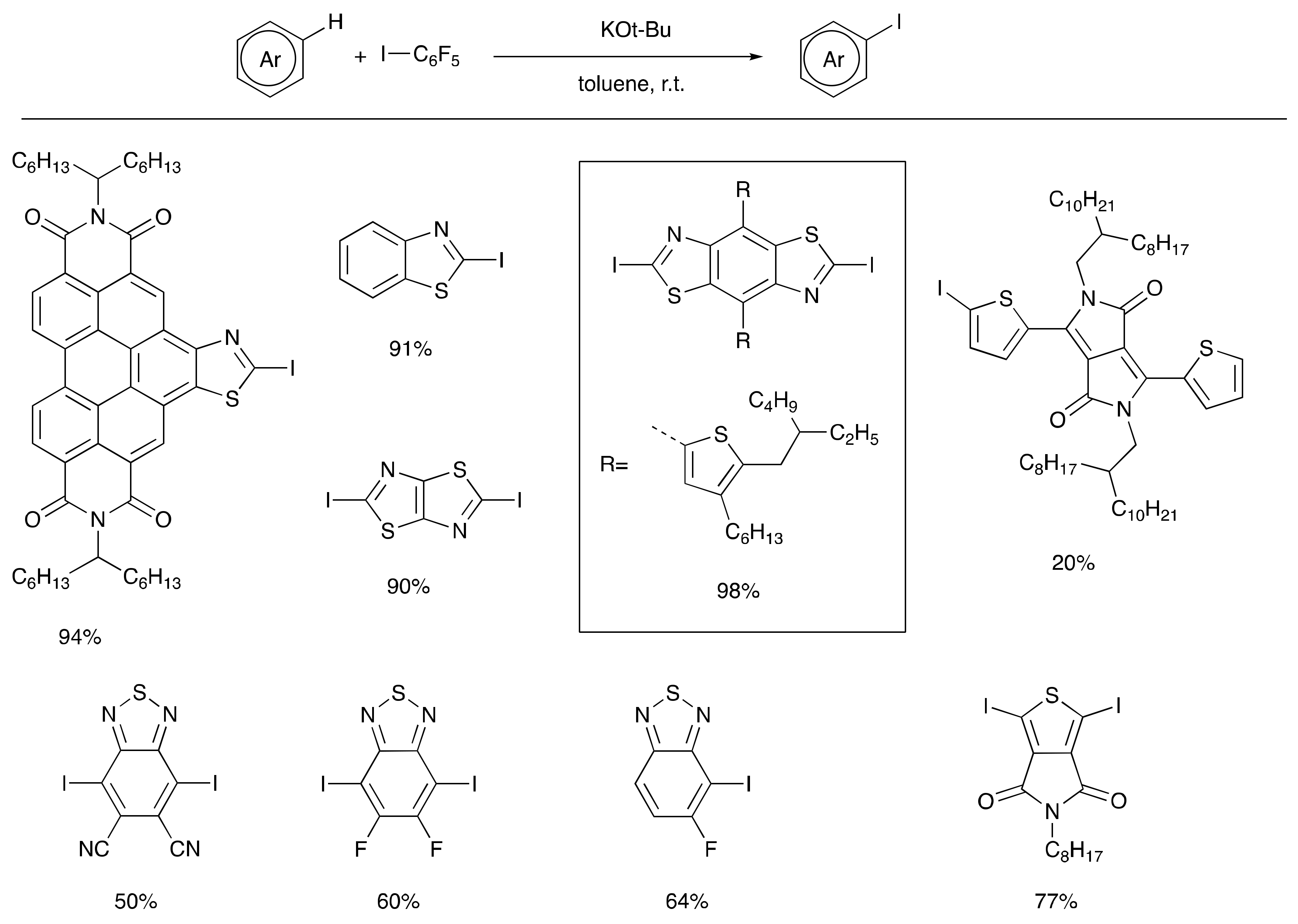
3. Earth-Abundant Metal-Initiated Iodination
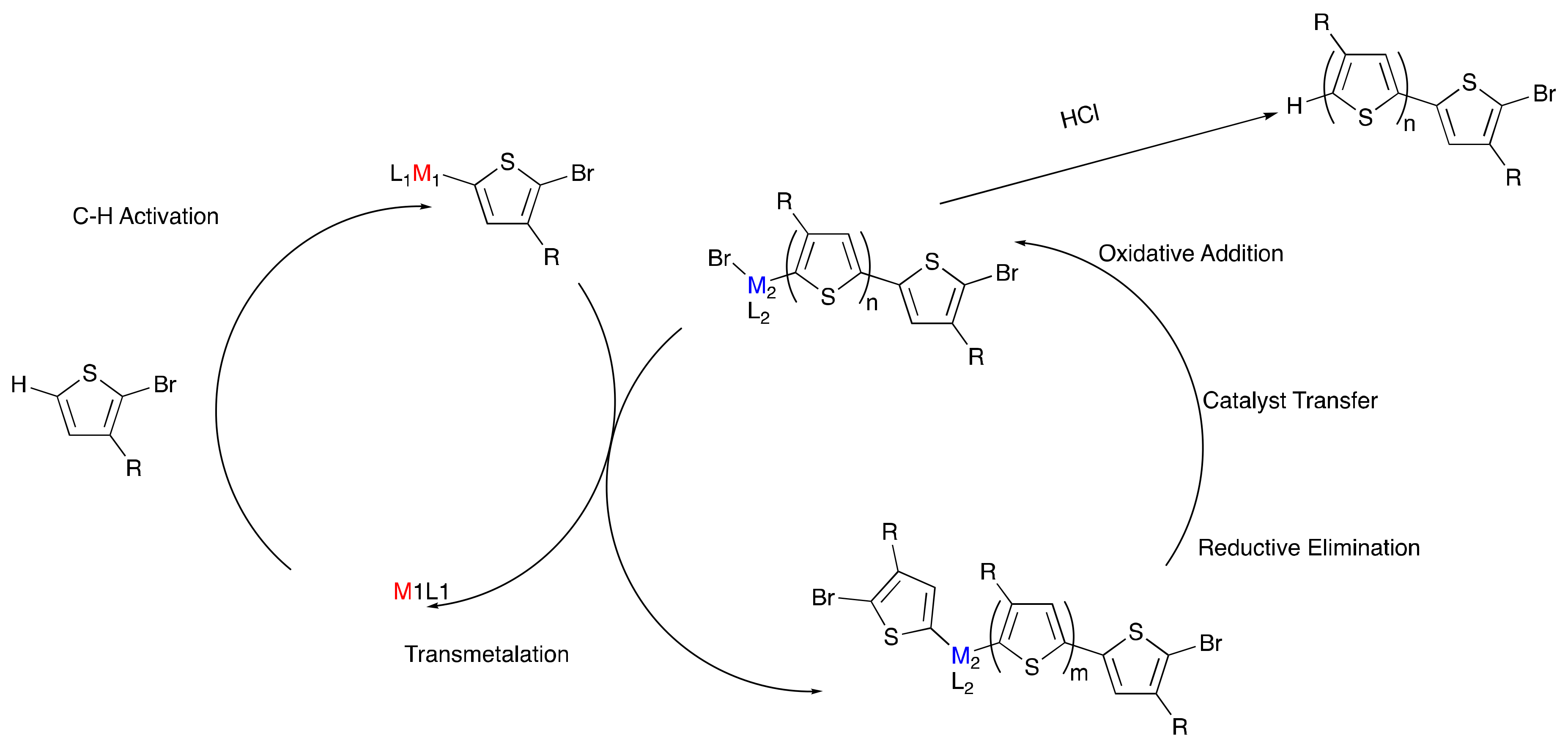
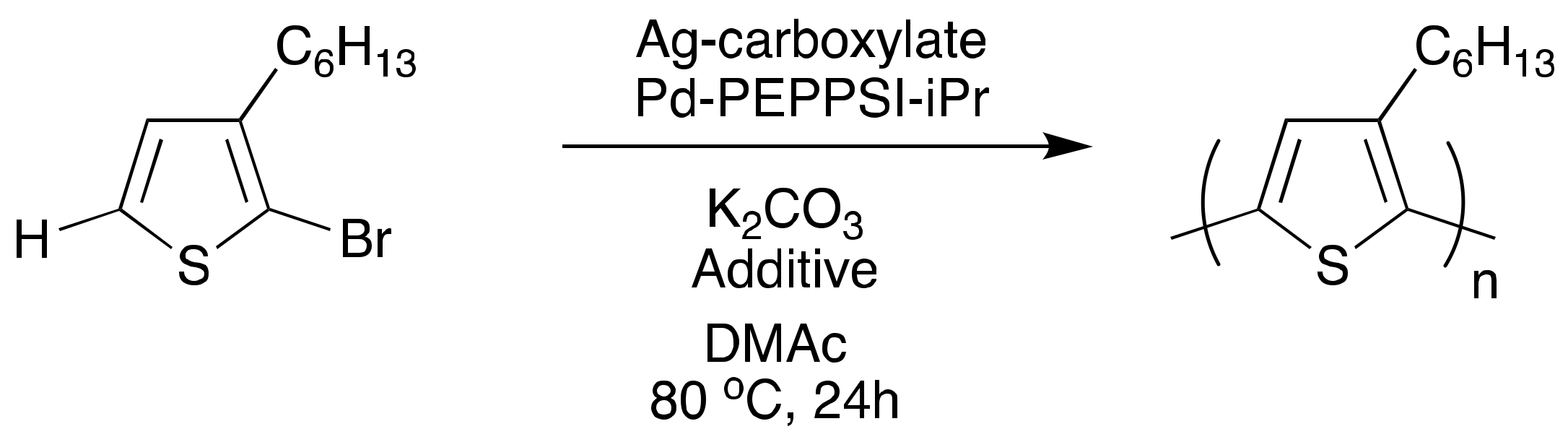
4. Toward Living Polymerizations to Synthesize Semiconducting Polymers Using Poly(3-hexylthiophene) as the Model Polymer
5. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Marzano, G.; Ciasca, C.V.; Babudri, F.; Bianchi, G.; Pellegrino, A.; Po, R.; Farinola, G.M. Organometallic Approaches to Conjugated Polymers for Plastic Solar Cells: From Laboratory Synthesis to Industrial Production. Eur. J. Org. Chem. 2014, 2014, 6583–6614. [Google Scholar] [CrossRef]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl-aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef] [PubMed]
- Schipper, D.J.; Fagnou, K. Direct Arylation as a Synthetic Tool for the Synthesis of Thiophene-Based Organic Electronic Materials. Chem. Mater. 2011, 23, 1594–1600. [Google Scholar] [CrossRef]
- Kowalski, S.; Allard, S.; Zilberberg, K.; Riedl, T.; Scherf, U. Direct arylation polycondensation as simplified alternative for the synthesis of conjugated (co)polymers. Prog. Polym. Sci. 2013, 38, 1805–1814. [Google Scholar] [CrossRef]
- Bohra, H.; Wang, M. Direct C–H arylation: A “Greener” approach towards facile synthesis of organic semiconducting molecules and polymers. J. Mater. Chem. A 2017, 5, 11550–11571. [Google Scholar] [CrossRef]
- Pouliot, J.R.; Grenier, F.; Blaskovits, J.T.; Beaupre, S.; Leclerc, M. Direct (Hetero)arylation Polymerization: Simplicity for Conjugated Polymer Synthesis. Chem. Rev. 2016, 116, 14225–14274. [Google Scholar] [CrossRef] [PubMed]
- Suraru, S.-L.; Lee, J.A.; Luscombe, C.K. C–H Arylation in the Synthesis of π-Conjugated Polymers. ACS Macro Lett. 2016, 5, 724–729. [Google Scholar] [CrossRef]
- Guo, X.; Baumgarten, M.; Müllen, K. Designing π-conjugated polymers for organic electronics. Prog. Polym. Sci. 2013, 38, 1832–1908. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Klauk, H. Organic thin-film transistors. Chem. Soc. Rev. 2010, 39, 2643–2666. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, L.; You, W. Rational Design of High Performance Conjugated Polymers for Organic Solar Cells. Macromolecules 2012, 45, 607–632. [Google Scholar] [CrossRef]
- Lin, Y.; Li, Y.; Zhan, X. Small molecule semiconductors for high-efficiency organic photovoltaics. Chem. Soc. Rev. 2012, 41, 4245–4272. [Google Scholar] [CrossRef] [PubMed]
- Iino, H.; Hanna, J.-I. Liquid crystalline organic semiconductors for organic transistor applications. Polym. J. 2016, 49, 23–30. [Google Scholar] [CrossRef]
- Dalton, L.R.; Sullivan, P.A.; Bale, D.H. Electric field poled organic electro-optic materials: State of the art and future prospects. Chem. Rev. 2010, 110, 25–55. [Google Scholar] [CrossRef] [PubMed]
- Mercier, L.G.; Leclerc, M. Direct (hetero)arylation: A new tool for polymer chemists. Acc. Chem. Res. 2013, 46, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, D.; Fagnou, K. Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway. Chem. Lett. 2010, 39, 1118–1126. [Google Scholar] [CrossRef]
- Lafrance, M.; Rowley, C.N.; Woo, T.K.; Fagnou, K. Catalytic intermolecular direct arylation of perfluorobenzenes. J. Am. Chem. Soc. 2006, 128, 8754–8756. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Chen, C.-C.; Yoshimura, K.; Ohya, K.; Chang, W.-H.; Gao, J.; Liu, Y.; Richard, E.; Yang, Y. Synthesis of 5H-Dithieno[3,2-b:2′,3′-d]pyran as an Electron-Rich Building Block for Donor–Acceptor Type Low-Bandgap Polymers. Macromolecules 2013, 46, 3384–3390. [Google Scholar] [CrossRef]
- You, W. Polymers with Tunable Band Gaps for Photonic and Electronic Applications. WO Patent WO/2011/156,478, 15 December 2011. [Google Scholar]
- Zhang, Y.; Chien, S.C.; Chen, K.S.; Yip, H.L.; Sun, Y.; Davies, J.A.; Chen, F.C.; Jen, A.K.-Y. Increased open circuit voltage in fluorinated benzothiadiazole-based alternating conjugated polymers. Chem. Commun. 2011, 47, 11026–11028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Parker, T.C.; Chen, W.; Williams, L.; Khrustalev, V.N.; Jucov, E.V.; Barlow, S.; Timofeeva, T.V.; Marder, S.R. C–H-Activated Direct Arylation of Strong Benzothiadiazole and Quinoxaline-Based Electron Acceptors. J. Org. Chem. 2016, 81, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Idris, I.; Tannoux, T.; Derridj, F.; Dorcet, V.; Boixel, J.; Guerchais, V.; Soulé, J.-F.; Doucet, H. Effective modulation of the photoluminescence properties of 2,1,3-benzothiadiazoles and 2,1,3-benzoselenadiazoles by Pd-catalyzed C–H bond arylations. J. Mater. Chem. C 2018, 6, 1731–1737. [Google Scholar] [CrossRef]
- He, C.Y.; Wu, C.Z.; Qing, F.L.; Zhang, X. Direct (het)arylation of fluorinated benzothiadiazoles and benzotriazole with (het)aryl iodides. J. Org. Chem. 2014, 79, 1712–1718. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.L.; Zhang, B.; He, C.Y.; Zhang, X. Direct olefination of fluorinated benzothiadiazoles: A new entry to optoelectronic materials. Chem. Eur. J. 2014, 20, 4532–4536. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Zhang, J.; O’Neil, D.; Rojas, A.J.; Chen, W.; Szymanski, P.; Marder, S.R.; El-Sayed, M.A. Effect of Molecular Structure Perturbations on the Performance of the D-A-π-A Dye Sensitized Solar Cells. Chem. Mater. 2014, 26, 4486–4493. [Google Scholar] [CrossRef]
- Kang, X.; Zhang, J.; Rojas, A.J.; O′Neil, D.; Szymanski, P.; Marder, S.R.; El-Sayed, M.A. Deposition of loosely bound organic D-A-π-A′ dyes on sensitized TiO2 film: A possible strategy to suppress charge recombination and enhance power conversion efficiency in dye-sensitized solar cells. J. Mater. Chem. A 2014, 2, 11229. [Google Scholar] [CrossRef]
- McNamara, L.E.; Liyanage, N.; Peddapuram, A.; Murphy, J.S.; Delcamp, J.H.; Hammer, N.I. Donor-Acceptor-Donor Thienopyrazines via Pd-Catalyzed C–H Activation as NIR Fluorescent Materials. J. Org. Chem. 2016, 81, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gao, Y.; Li, S.; Shi, X.; Geng, Y.; Wang, F. Synthesis of poly(5,6-difluoro-2,1,3-benzothiadiazole-alt-9,9-dioctyl-fluorene) via direct arylation polycondensation. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 2367–2374. [Google Scholar] [CrossRef]
- Scott, C.N.; Bisen, M.D.; Stemer, D.M.; McKinnon, S.; Luscombe, C.K. Direct Arylation Polycondensation of 2,5-Dithienylsilole with a Series of Difluorobenzodiimine-Based Electron Acceptors. Macromolecules 2017, 50, 4623–4628. [Google Scholar] [CrossRef]
- Mkhalid, I.A.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C–H activation for the construction of C-B bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Hartwig, J.F. Catalytic Silylation of Unactivated C–H Bonds. Chem. Rev. 2015, 115, 8946–8975. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Hiyama, T. Cross-coupling of organosilanes with organic halides mediated by a palladium catalyst and tris(diethylamino)sulfonium difluorotrimethylsilicate. J. Org. Chem. 1988, 53, 918–920. [Google Scholar] [CrossRef]
- Denmark, S.E.; Regens, C.S. Palladium-catalyzed cross-coupling reactions of organosilanols and their salts: Practical alternatives to boron- and tin-based methods. Acc. Chem. Res. 2008, 41, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Toutov, A.A.; Liu, W.B.; Betz, K.N.; Fedorov, A.; Stoltz, B.M.; Grubbs, R.H. Silylation of C–H bonds in aromatic heterocycles by an Earth-abundant metal catalyst. Nature 2015, 518, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.B.; Schuman, D.P.; Yang, Y.F.; Toutov, A.A.; Liang, Y.; Klare, H.F.T.; Nesnas, N.; Oestreich, M.; Blackmond, D.G.; Virgil, S.C.; et al. Potassium tert-Butoxide-Catalyzed Dehydrogenative C–H Silylation of Heteroaromatics: A Combined Experimental and Computational Mechanistic Study. J. Am. Chem. Soc. 2017, 139, 6867–6879. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Yang, Y.F.; Jenkins, I.D.; Liang, Y.; Toutov, A.A.; Liu, W.B.; Schuman, D.P.; Grubbs, R.H.; Stoltz, B.M.; Krenske, E.H.; et al. Ionic and Neutral Mechanisms for C–H Bond Silylation of Aromatic Heterocycles Catalyzed by Potassium tert-Butoxide. J. Am. Chem. Soc. 2017, 139, 6880–6887. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.Q.; Zhang, S.Y.; Zhang, J.X.; Oswald, V.F.; Amassian, A.; Marder, S.R.; Blakey, S.B. (KOBu)-Bu-t-Initiated Aryl C–H Iodination: A Powerful Tool for the Synthesis of High Electron Affinity Compounds. J. Am. Chem. Soc. 2016, 138, 3946–3949. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, X.; Liang, F.; Ren, B. t-BuONa-mediated direct C–H halogenation of electron-deficient (hetero)arenes. Org. Biomol. Chem. 2018, 16, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.Q.; Andreansky, E.S.; Marder, S.R.; Blakey, S.B. Synthesis and C–H Functionalization Chemistry of Thiazole-Semicoronenediimide (TsCDIs) and -Coronenediimides (TCDIs). J. Org. Chem. 2017, 82, 10139–10148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Zhang, J.X.; Abdelsamie, M.; Shi, Q.Q.; Zhang, Y.D.; Parker, T.C.; Jucov, E.V.; Timofeeva, T.V.; Amassian, A.; Bazan, G.C.; et al. Intermediate-Sized Conjugated Donor Molecules for Organic Solar Cells: Comparison of Benzodithiophene and Benzobisthiazole-Based Cores. Chem. Mater. 2017, 29, 7880–7887. [Google Scholar] [CrossRef]
- Tamba, S.; Shono, K.; Sugie, A.; Mori, A. C–H functionalization polycondensation of chlorothiophenes in the presence of nickel catalyst with stoichiometric or catalytically generated magnesium amide. J. Am. Chem. Soc. 2011, 133, 9700–9703. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, A.; Vaccaro, L.; Marrocchi, A. Semiconducting polymers prepared by direct arylation polycondensation. Angew. Chem. Int. Ed. 2012, 51, 3520–3523. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Zhang, J.; Housekeeper, J.B.; Marder, S.R.; Luscombe, C.K. C–H Arylation Reaction: Atom Efficient and Greener Syntheses of π-Conjugated Small Molecules and Macromolecules for Organic Electronic Materials. Macromolecules 2013, 46, 8059–8078. [Google Scholar] [CrossRef]
- Segawa, Y.; Maekawa, T.; Itami, K. Synthesis of extended π-systems through C–H activation. Angew. Chem. Int. Ed. 2015, 54, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Perez-Temprano, M.H.; Casares, J.A.; Espinet, P. Bimetallic catalysis using transition and Group 11 metals: An emerging tool for C-C coupling and other reactions. Chem. Eur. J. 2012, 18, 1864–1884. [Google Scholar] [CrossRef] [PubMed]
- Lohr, T.L.; Marks, T.J. Orthogonal tandem catalysis. Nat. Chem. 2015, 7, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Furstner, A.; Davies, P.W. Catalytic carbophilic activation: Catalysis by platinum and gold π acids. Angew. Chem. Int. Ed. 2007, 46, 3410–3449. [Google Scholar] [CrossRef] [PubMed]
- Wegner, H.A.; Auzias, M. Gold for C-C coupling reactions: A Swiss-Army-knife catalyst? Angew. Chem. Int. Ed. 2011, 50, 8236–8247. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, T.; Livendahl, M.; Rosellon, A.; Espinet, P.; Echavarren, A.M. Unlikeliness of Pd-free gold(I)-catalyzed Sonogashira coupling reactions. Org. Lett. 2010, 12, 3006–3009. [Google Scholar] [CrossRef] [PubMed]
- Livendahl, M.; Goehry, C.; Maseras, F.; Echavarren, A.M. Rationale for the sluggish oxidative addition of aryl halides to Au(I). Chem. Commun. 2014, 50, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Joost, M.; Zeineddine, A.; Estevez, L.; Mallet-Ladeira, S.; Miqueu, K.; Amgoune, A.; Bourissou, D. Facile oxidative addition of aryl iodides to gold(I) by ligand design: Bending turns on reactivity. J. Am. Chem. Soc. 2014, 136, 14654–14657. [Google Scholar] [CrossRef] [PubMed]
- Guenther, J.; Mallet-Ladeira, S.; Estevez, L.; Miqueu, K.; Amgoune, A.; Bourissou, D. Activation of aryl halides at gold(I): Practical synthesis of (P,C) cyclometalated gold(III) complexes. J. Am. Chem. Soc. 2014, 136, 1778–1781. [Google Scholar] [CrossRef] [PubMed]
- Cambeiro, X.C.; Boorman, T.C.; Lu, P.; Larrosa, I. Redox-controlled selectivity of C–H activation in the oxidative cross-coupling of arenes. Angew. Chem. Int. Ed. 2013, 52, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Boorman, T.C.; Larrosa, I. Gold-mediated C–H bond functionalisation. Chem. Soc. Rev. 2011, 40, 1910–1925. [Google Scholar] [CrossRef] [PubMed]
- Boogaerts, I.I.F.; Nolan, S.P. Carboxylation of C–H bonds using N-heterocyclic carbene gold(I) complexes. J. Am. Chem. Soc. 2010, 132, 8858–8859. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, S.; Cazin, C.S.; Nolan, S.P. N-heterocyclic carbene gold(I) and copper(I) complexes in C–H bond activation. Acc. Chem. Res. 2012, 45, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Ahlsten, N.; Perry, G.J.P.; Cambeiro, X.C.; Boorman, T.C.; Larrosa, I. A silver-free system for the direct C–H auration of arenes and heteroarenes from gold chloride complexes. Catal. Sci. Technol. 2013, 3, 2892. [Google Scholar] [CrossRef]
- Hansmann, M.M.; Pernpointner, M.; Dopp, R.; Hashmi, A.S. A theoretical DFT-based and experimental study of the transmetalation step in Au/Pd-mediated cross-coupling reactions. Chem. Eur. J. 2013, 19, 15290–15303. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Döpp, R.; Lothschütz, C.; Rudolph, M.; Riedel, D.; Rominger, F. Scope and Limitations of Palladium-Catalyzed Cross-Coupling Reactions with Organogold Compounds. Adv. Synth. Catal. 2010, 352, 1307–1314. [Google Scholar] [CrossRef]
- Del Pozo, J.; Casares, J.A.; Espinet, P. The decisive role of ligand metathesis in Au/Pd bimetallic catalysis. Chem. Commun. 2013, 49, 7246–7248. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Roth, K.E.; Ramgren, S.D.; Blum, S.A. Catalyzed catalysis using carbophilic Lewis acidic gold and Lewis basic palladium: Synthesis of substituted butenolides and isocoumarins. J. Am. Chem. Soc. 2009, 131, 18022–18023. [Google Scholar] [CrossRef] [PubMed]
- Perez-Temprano, M.H.; Casares, J.A.; de Lera, A.R.; Alvarez, R.; Espinet, P. Strong metallophilic interactions in the palladium arylation by gold aryls. Angew. Chem. Int. Ed. 2012, 51, 4917–4920. [Google Scholar] [CrossRef] [PubMed]
- Suraru, S.-L.; Lee, J.A.; Luscombe, C.K. Preparation of an Aurylated Alkylthiophene Monomer via C–H Activation for Use in Pd-PEPPSI-iPr Catalyzed-Controlled Chain Growth Polymerization. ACS Macro Lett. 2016, 5, 533–536. [Google Scholar] [CrossRef]
- Lotz, M.D.; Camasso, N.M.; Canty, A.J.; Sanford, M.S. Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C–H Functionalization Reactions. Organometallics 2016, 36, 165–171. [Google Scholar] [CrossRef]
- Wang, Q.; Takita, R.; Kikuzaki, Y.; Ozawa, F. Palladium-catalyzed dehydrohalogenative polycondensation of 2-bromo-3-hexylthiophene: An efficient approach to head-to-tail poly(3-hexylthiophene). J. Am. Chem. Soc. 2010, 132, 11420–11421. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-Y.; Tung, T.-C.; Liang, W.-W.; Cheng, Y.-J. Synthesis of Poly(3-hexylthiophene), Poly(3-hexylselenophene), and Poly(3-hexylselenophene-alt-3-hexylthiophene) by Direct C–H Arylation Polymerization via N-Heterocyclic Carbene Palladium Catalysts. Macromolecules 2015, 48, 2978–2988. [Google Scholar] [CrossRef]
- Rudenko, A.E.; Thompson, B.C. Influence of the Carboxylic Acid Additive Structure on the Properties of Poly(3-hexylthiophene) Prepared via Direct Arylation Polymerization (DArP). Macromolecules 2015, 48, 569–575. [Google Scholar] [CrossRef]
- Lee, J.A.; Luscombe, C.K. Dual-catalytic Ag-Pd System for a Chain-Growth Direct Arylation Polymerization to Synthesize Poly(3-hexylthiophene). ACS Macro Lett. Manuscript Submitted, 28 March 2018.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Kang, L.J.; Parker, T.C.; Blakey, S.B.; Luscombe, C.K.; Marder, S.R. Recent Developments in C–H Activation for Materials Science in the Center for Selective C–H Activation. Molecules 2018, 23, 922. https://doi.org/10.3390/molecules23040922
Zhang J, Kang LJ, Parker TC, Blakey SB, Luscombe CK, Marder SR. Recent Developments in C–H Activation for Materials Science in the Center for Selective C–H Activation. Molecules. 2018; 23(4):922. https://doi.org/10.3390/molecules23040922
Chicago/Turabian StyleZhang, Junxiang, Lauren J. Kang, Timothy C. Parker, Simon B. Blakey, Christine K. Luscombe, and Seth R. Marder. 2018. "Recent Developments in C–H Activation for Materials Science in the Center for Selective C–H Activation" Molecules 23, no. 4: 922. https://doi.org/10.3390/molecules23040922
APA StyleZhang, J., Kang, L. J., Parker, T. C., Blakey, S. B., Luscombe, C. K., & Marder, S. R. (2018). Recent Developments in C–H Activation for Materials Science in the Center for Selective C–H Activation. Molecules, 23(4), 922. https://doi.org/10.3390/molecules23040922