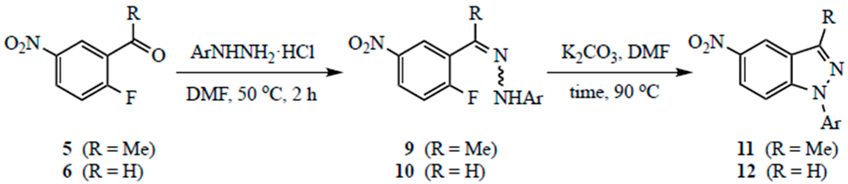
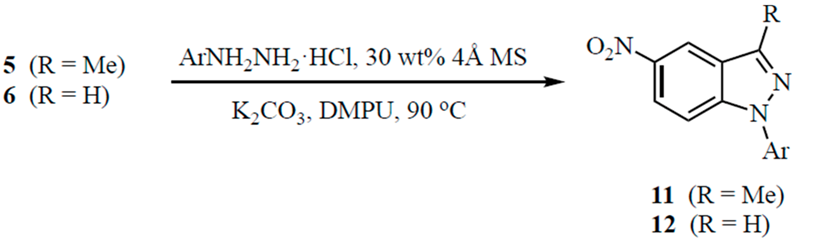
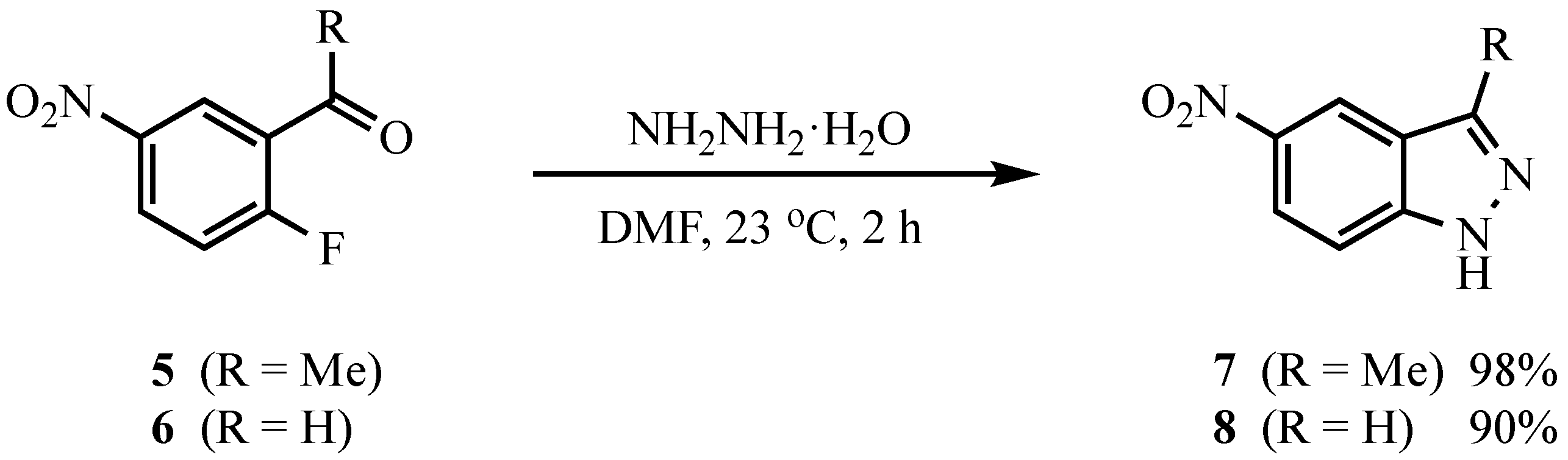
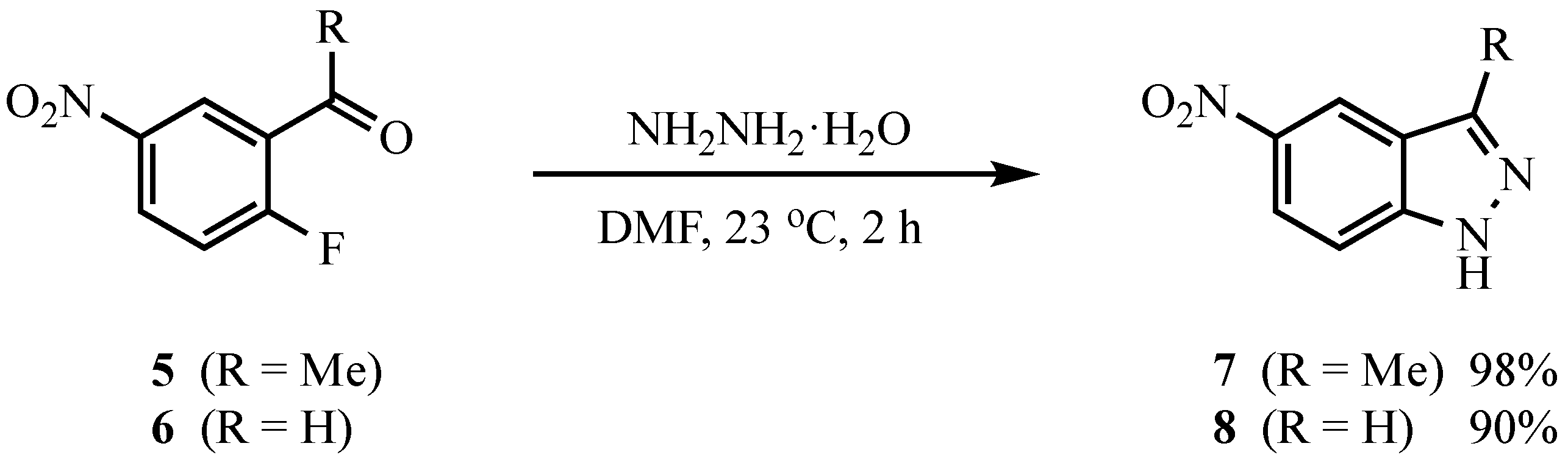
3.4. Indazoles from 5 or 6 with ArNHNH2·HCl. Method B. Representative One-Pot Procedure
To a stirred solution of the carbonyl com.p.ound (1.0 mmol) in DMPU (5 mL) were added powdered 4 Å molecular sieves (30 wt % relative to the carbonyl substrate), ArNHNH
2·HCl (3.0 mmol for
5, 2.0 mmol for
6), and K
2CO
3 (3.0 mmol for
5, 2.0 mmol for
6). For
5, all reagents were placed in the flask and heated to 90 °C; for
6, the hydrazone was allowed to form at 90 °C (1.5 h) before K
2CO
3 was added. The reaction was stirred at 90 °C for the time indicated in
Table 2. Workup was performed as described in Method A.
3-Methyl-1-phenyl-5-nitro-1H-indazole (11a). Yield: 240 mg, (0.95 mmol, 95%) as a brown solid, m.p. 115–117 °C; IR: 1516, 1336 cm−1; 1H-NMR (DMSO-d6): δ 8.89 (d, J = 2.2 Hz, 1H), 8.28 (dd, J = 9.3, 2.2 Hz, 1H), 7.93 (d, J = 9.3 Hz, 1H), 7.77 (d, J = 7.8 Hz, 2H), 7.62 (t, J = 7.7 Hz, 2H), 7.46 (t, J = 7.4 Hz, 1H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 147.3, 142.2, 141.1, 139.2, 130.3, 127.8, 124.4, 122.9, 122.8, 119.3, 111.7, 12.1; MS: m/z 253 (M+), calculated m/z 253.09. Anal. Calcd. for C14H11N3O2: C, 66.40; H, 4.38; N, 16.59. Found: C, 66.33; H, 4.34; N, 16.42.
1-(2-Methoxyphenyl)-3-methyl-5-nitro-1H-indazole (11b). Yield: 232 mg (0.82 mmol, 82%) as a light brown solid, m.p. 128–130 °C; IR: 2838, 1519, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.86 (s, 1H), 8.21 (d, J = 9.3 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.38–7.28 (overlapping signals, 2H), 7.16 (t, J = 7.7 Hz, 1H), 3.78 (s, 3H), 2.66 (s, 3H); 13C-NMR (DMSO-d6): δ 154.0, 146.6, 143.0, 141.9, 130.8, 128.6, 127.1, 123.2, 122.0, 121.4, 119.0, 113.4, 112.2, 56.2, 12.1; MS: m/z 283 (M+), calculated m/z 283.10. Anal. Calcd. for C15H13N3O3: C, 63.60; H, 4.63; N, 14.83. Found: C, 63.49; H, 4.61; N, 14.88.
1-(3-Methoxyphenyl)-3-methyl-5-nitro-1H-indazole (11c). Yield: 229 mg (0.81 mmol, 81%) as a light brown solid, m.p. 105–106 °C; IR: 2836, 1518, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.90 (s, 1H), 8.29 (d, J = 9.3 Hz, 1H), 7.96 (d, J = 9.3 Hz, 1H), 7.52 (t, J = 8.1 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.28 (s, 1H), 7.04 (d, J = 8.3 Hz, 1H); 13C-NMR (DMSO-d6): δ 160.6, 147.3, 142.3, 141.2, 140.3, 131.2, 124.4, 122.9, 119.3, 114.9, 113.7, 111.9, 108.6, 56.0, 12.1; MS: m/z 283 (M+), calculated m/z 283.10. Anal. Calcd. for C15H13N3O3: C, 63.60; H, 4.63; N, 14.83. Found: C, 63.52; H, 4.55; N, 14.71.
1-(4-Methoxyphenyl)-3-methyl-5-nitro-1H-indazole (11d). Yield: 266 mg (0.94 mmol, 94%) as a tan solid, m.p. 159–160 °C. IR: 2836, 1514, 1346 c cm−1; 1H-NMR (DMSO-d6): δ 8.88 (d, J = 2.2 Hz, 1H), 8.26 (dd, J = 9.3, 2.2 Hz, 1H), 7.79 (d, J = 9.3 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 3.85 (s, 3H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 158.9, 146.7, 142.0, 141.2, 132.2, 124.8, 123.9, 122.5, 119.3, 1153, 111.4, 56.0, 12.1; MS: m/z 283 (M+), calculated m/z 283.10. Anal. Calcd. for C15H13N3O3: C, 63.60; H, 4.63; N, 14.83. Found: C, 63.57; H, 4.60; N, 14.76.
1-(4-Bromophenyl)-3-methyl-5-nitro-1H-indazole (11e). Yield: 314 mg, (0.95 mmol, 95%) as a tan solid, m.p. 202–203 °C; IR: 1512, 1345 cm−1; 1H-NMR (DMSO-d6): δ 8.90 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.2, 2.2 Hz, 1H), 7.96 (d, J = 9.2 Hz, 1H), 7.80 (d, J = 8.8 Hz, 2H), 7.75 (d, J = 8.8 Hz, 2H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 147.7, 142.4, 141.1, 138.5, 133.1, 124.7, 124.6, 123.0, 120.1, 119.3, 111.8, 12.1; MS: m/z 331 (M+), calculated m/z 331.00. Anal. Calcd. for C14H10BrN3O2: C, 50.62; H, 3.03; N, 12.65. Found: C, 50.44; H, 2.99; N, 12.73.
1-(3-Chlorophenyl)-3-methyl-5-nitro-1H-indazole (11f). Yield: 250 mg, (0.87 mmol, 87%) as a tan solid, m.p. 135–136 °C; IR: 1517, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.89 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.3, 2.2 Hz, 1H), 7.98 (d, J = 9.3 Hz, 1H), 7.83 (t, J = 2.2 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.64 (t, J = 8.1 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 146.8, 141.1, 139.3, 133.4, 130.8, 126.4, 123.6, 122.0, 121.4, 120.1, 118.2, 110.9; MS: m/z 287 (M+), calculated m/z 287.05. Anal. Calcd. for C14H10ClN3O2: C, 58.45; H, 3.50; N, 14.61. Found: C, 58.58; H, 3.59; N, 14.53.
1-(4-Chlorophenyl)-3-methyl-5-nitro-1H-indazole (11g). Yield: 267 mg (0.93 mmol, 93%) as a tan solid, m.p. 217–218 °C; IR: 1511, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.91 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.3, 2.2 Hz, 1H), 7.95 (d, J = 9.3 Hz, 1H), 7.82 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 8.7 Hz, 2H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 147.7, 1424, 141.1, 138.1, 131.8, 130.2, 124.6, 124.5, 123.0, 119.4, 111.8, 12.1; MS: m/z 287 (M+), calculated m/z 287.05. Anal. Calcd. for C14H10ClN3O2: C, 58.45; H, 3.50; N, 14.61. Found: C, 58.42; H, 3.49; N, 14.48.
1-(2,4-Dichlorophenyl)-3-methyl-5-nitro-1H-indazole (11h). Yield: 257 mg (0.80 mmol, 80%), m.p. 144–145 °C; IR: 1516, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.92 (d, J = 2.1 Hz, 1H), 8.27 (dd, J = 9.2, 2.1 Hz, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.69 (dd, J = 8.5, 2.2 Hz, 1H), 7.44 (d, J = 9.2 Hz, 1H), 2.68 (s, 3H); 13C-NMR (DMSO-d6): δ 147.5, 143.1, 142.4, 135.3, 135.0, 132.0, 131.5, 130.7, 129.3, 123.5, 122.8, 119.3, 111.6, 12.1; MS: m/z 321 (M+), calculated m/z 321.01. Anal. Calcd. for C14H9Cl2N3O2: C, 52.20; H, 2.82; N, 13.04. Found: C, 52.11; H, 2.91; N, 13.09.
3-Methyl-5-nitro-1-(3-(trifluoromethyl)phenyl)-1H-indazole (11i). Yield: 282 mg (0.88 mmol, 88%) as a white solid, m.p. 112–113 °C; IR: 1520, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.90 (s, 1H), 8.32 (d, J = 9.3 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.07 (s, 1H), 7.99 (d, J = 9.3 Hz, 1H), 7.90–7.77 (complex, 2H), 2.70 (s, 3H); 13C-NMR (DMSO-d6): δ 148.0, 142.5, 141.1, 139.8, 131.6, 130.9 (q, J = 32.6 Hz), 126.2, 124.8, 124.2 (q, J = 272.6 Hz), 124.0 (q, J = 6.7 Hz), 123.1, 119.2 (2C), 111.8, 12.0; MS: m/z 321 (M+), calculated m/z 321.07. Anal. Calcd. for C15H10F3N3O2: C, 56.08; H, 3.14; N, 13.08. Found: C, 56.02; H, 3.18; N, 13.11.
3-Methyl-5-nitro-1-(4-(trifluoromethyl)phenyl)-1H-indazole (11j). Yield: 225 mg, (0.70 mmol, 70%) as a white solid, mp 167–168 °C; IR: 1525, 1331 cm−1; 1H-NMR (DMSO-d6): δ 8.89 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.3, 2.2 Hz, 1H), 8.05 (d, J = 9.3 Hz, 1H), 8.02 (d, J = 8.6 Hz, 2H), 7.95 (d, J = 8.6 Hz, 2H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 148.3, 142.6, 142.3, 141.1, 127.4 (q, J = 4.0 Hz), 127.1, 125.1, 124.5 (q, J = 269.1 Hz), 123.2, 122.6, 119.2, 112.0, 12.1; MS: m/z 321 (M+). calculated m/z 321.07. Anal. Calcd. for C15H10F3N3O2: C, 56.08; H, 3.14; N, 13.08. Found: C, 55.98; H, 3.19; N, 13.04.
1-(4-Cyanophenyl)-3-methyl-5-nitro-1H-indazole (11k). Yield: 222 mg (0.80 mmol, 80%) as a brown solid, m.p. ≥ 194 °C (sub), ≥ 260 °C (dec); IR: 2226, 1516, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.93 (d, J = 2.2 Hz, 1H), 8.35 (dd, J = 9.1, 2.2 Hz, 1H), 8.11 (d, J = 9.1 Hz, 1H), 8.08 (d, J = 8.9 Hz, 2H), 8.04 (d, J = 8.9 Hz, 2H), 2.71 (s, 3H); 13C-NMR (DMSO-d6): δ 148.7, 142.8, 142.7, 141.1, 134.5, 125.3, 123.3, 122.6, 119.3, 118.9, 112.3, 109.4, 12.1; MS: m/z 278 (M+), calculated m/z 278.08. Anal. Calcd. for C15H10N4O2: C, 64.74; H, 3.62; N, 20.13. Found: C, 64.69; H, 3.67; N, 20.06.
4-(3-Methyl-5-nitro-1H-indazol-1-yl)benzenesulfonamide (11l). Yield: 249 mg (0.75 mmol, 75%), m.p. 265–266 °C; IR: 3422, 1515, 1331, 1320, 1123 cm−1; 1H-NMR (DMSO-d6): δ 8.92 (d, J = 2.2 Hz, 1H), 8.34 (dd, J = 9.3, 2.2 Hz, 1H), 8.09 (d, J = 9.3 Hz, 1H), 8.04 (d, J = 8.4 Hz, 2H), 8.01 (d, J = 8.4 Hz, 2H), 7.51 (s, 2H), 2.71 (s, 3H); 13C-NMR (DMSO-d6): δ 148.2, 142.54, 145.50, 141.6, 141.0, 127.9, 125.0, 123.1, 122.3, 119.2, 112.0, 12.0; MS: m/z 332 (M+), calculated m/z 332.06. Anal. Calcd. for C14H12N4O4S: C, 50.60; H, 3.64; N, 16.86. Found: C, 50.70; H, 3.63; N, 16.78.
4-(3-Methyl-5-nitro-1H-indazol-1-yl)benzoic acid (11m). Yield: 208 mg (0.70 mmol, 70%) as a tan solid, m.p. ≥ 240 °C (sub), ≥ 320 °C (dec). IR: 3441–2352, 1697, 1514, 1346 cm−1; 1H-NMR (DMSO-d6): δ 12.8 (br s, 1H), 8.93 (d, J = 2.2 Hz, 1H), 8.34 (dd, J = 9.3, 2.2 Hz, 1H), 8.16 (d, J = 8.6 Hz, 2H), 8.09 (d, J = 9.3, Hz, 1H), 7.95 (d, J = 8.6 Hz, 2H), 2.71 (s, 3H); 13C-NMR (DMSO-d6): δ 167.1, 148.2, 142.6, 142.5, 141.1, 131.5, 125.0, 123.1, 122.0, 119.4, 119.3, 112.2, 12.1; MS: m/z 297 (M+), calculated m/z 297.07. Anal. Calcd. for C15H11N3O4: C, 60.61; H, 3.73; N, 14.14. Found: C, 60.53; H, 3.68; N, 14.22.
1-Phenyl-5-nitro-1H-indazole (
12a). Yield: 172 mg (0.72 mmol, 72%) as a tan solid, m.p. 178–180 °C (lit m.p. 178–180 °C [
18]); IR: 1534, 1341 cm
−1;
1H-NMR (DMSO-
d6): δ 8.95 (s, 1H), 8.70 (s, 1H), 8.30 (d,
J = 9.3 Hz, 1H), 7.98 (d,
J = 9.3 Hz, 1H), 7.81 (d,
J = 7.9 Hz, 2H), 7.65 (d,
J = 7.7 Hz, 2H), 7.51 (t,
J = 7.4 Hz, 1H);
13C-NMR (DMSO-
d6): δ 142.8, 140.5, 139.1, 138.9, 130.3, 128.2, 124.8, 123.3, 122.7, 119.9, 111.9; MS:
m/
z 239 (M
+), calculated
m/
z 239.07. Anal. Calcd. for C
13H
9N
3O
2: C, 65.27; H, 3.79; N, 17.56. Found: C, 65.16; H, 3.77; N, 17.57.
1-(2-Methoxyphenyl)-5-nitro-1H-indazole (12b). This reaction stopped at the hydrazone stage and did not give an indazole.
1-(3-Methoxyphenyl)-5-nitro-1H-indazole (
12c): Yield: 180 mg (0.67 mmol, 67%) as a yellow solid, m.p. 132–133 °C (lit [
36] m.p. 135 °C); IR: 1516, 1344 cm
−1;
1H-NMR (DMSO-
d6): δ 8.94 (s, 1H), 8.69 (s, 1H), 8.30 (d,
J = 9.2 Hz, 1H), 8.01 (d,
J = 9.3 Hz, 1H), 7.55 (t,
J = 8.0 Hz, 1H), 7.37 (d,
J = 8.0 Hz, 1H), 7.32 (s, 1H), 7.08 (d,
J = 8.2 Hz, 1H), 3.87 (s, 3H);
13C-NMR (DMSO-
d6): δ 160.6, 142.8, 140.5, 140.2, 130.8, 131.2, 124.8, 122.7, 119.8, 115.3, 114.1, 112.0, 109.0, 56.0; MS:
m/
z 269 (M
+), calculated
m/
z 269.08. Anal. Calcd. for C
14H
11N
3O
3: C, 62.45; H, 4.12; N, 15.61. Found: C, 62.37; H, 4.06; N, 15.74.
1-(4-Methoxyphenyl)-5-nitro-1H-indazole (12d). Yield: 199 mg (0.74 mmol, 74%) as a light yellow solid, m.p. 181–182 °C; IR: 1516, 1342 cm−1; 1H-NMR (DMSO-d6): δ 8.92 (d, J = 2.2 Hz, 1H), 8.64 (s, 1H), 8.27 (d, J = 9.2, 2.2 Hz, 1H), 7.84 (d, J = 9.2 Hz, 1H), 7.68 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H); 13C-NMR (DMSO-d6): δ 159.2, 142.6, 141.6, 138.3, 132.1, 125.2, 124.3, 122.4, 119.8, 115.6, 111.6, 56.2; MS: m/z 269 (M+), calculated m/z 269.08. Anal. Calcd. for C14H11N3O3: C, 62.45; H, 4.12; N, 15.61. Found: C, 62.41; H, 4.07; N, 15.56.
1-(4-Bromophenyl)-5-nitro-1H-indazole (12e). Yield: 222 mg (0.70 mmol, 70%) as a tan solid, m.p. 169–170 °C; IR: 1511, 1348 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (s, 1H), 8.71 (s, 1H), 8.32 (d, J = 9.3 Hz, 1H), 8.01 (d, J = 9.2 Hz, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H); 13C-NMR (DMSO-d6): δ 142.9, 140.4, 139.2, 138.4, 133.2, 125.2, 125.0, 122.8, 120.7, 119.9, 111.9; MS: m/z 317 (M+), calculated m/z 316.98. Anal. Calcd. for C13H8BrN3O2: C, 49.08; H, 2.53; N, 13.21. Found: C, 48.97; H, 2.59; N, 13.25.
1-(3-Chlorophenyl)-5-nitro-1H-indazole (12f). Yield: 164 mg (0.60 mmol, 60%) as an off-white solid, m.p. 143–144 °C; IR: 1514, 1346 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (s, 1H), 8.72 (s, 1H), 8.32 (d, J = 9.2 Hz, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.88 (s, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.67 (t, J = 8.1 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H); 13C-NMR (DMSO-d6): δ 142.9, 140.5, 140.3, 139.4. 134.6, 132.0, 128.0, 125.0, 123.0, 122.9, 121.7, 119.8, 112.0; MS: m/z 273 (M+), calculated m/z 273.03. Anal. Calcd. for C13H8ClN3O2: C, 57.05; H, 2.95; N, 15.35. Found: C, 56.96; H, 3.00; N, 15.29.
1-(4-Chlorophenyl)-5-nitro-1H-indazole (12g). Yield: 191 mg (0.70 mmol, 70%) as a light yellow solid, m.p. 179–180 °C; IR: 1509, 1343 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (d, J = 2.2 Hz, 1H), 8.71 (s, 1H), 8.31 (dd, J = 9.3 , 2.2 Hz, 1H), 7.99 (d, J = 9.3 Hz, 1H), 7.85 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H); 13C-NMR (DMSO-d6): δ 142.9, 140.5, 139.2, 138.0, 132.4, 130.3, 124.9, 122.8, 119.9, 111.9 (one C not observed); MS: m/z 273 (M+), calculated m/z 273.03. Anal. Calcd. for C13H8ClN3O2: C, 57.05; H, 2.95; N, 15.35. Found: C, 57.12; H, 2.97; N, 15.33.
1-(2,4-Dichlorophenyl)-5-nitro-1H-indazole (12h). Yield: 215 mg (0.70 mmol, 70%) as a light pink solid, m.p. 101–102 °C. IR: 1517, 1344 cm−1; 1H-NMR (DMSO-d6): δ 8.96 (s, 1H), 8.72 (s, 1H), 8.29 (d, J = 9.2 Hz, 1H), 8.03 (s, 1H), 7.78 (d, J = 8.3 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 7.52 (d, J = 9.2 Hz, 1H); 13C-NMR (DMSO-d6): δ 143.0, 142.3, 139.2, 135.7, 135.0, 132.2, 131.6, 130.8, 129.3, 123.9, 122.7, 119.8, 111.7; MS: m/z 307 (M+), calculated m/z 306.99. Anal. Calcd. for C13H7Cl2N3O2: C, 50.68; H, 2.29; N, 13.64. Found: C, 50.67; H, 2.34; N, 13.53.
1-(3-(Trifluoromethyl)phenyl)-5-nitro-1H-indazole (12i). Yield: 190 mg (0.62 mmol, 62%) as a light yellow solid, m.p. 118–119 °C. IR: 1513, 1347 cm−1; 1H-NMR (DMSO-d6): δ 8.97 (s, 1H), 8.75 (s, 1H), 8.35 (d, J = 9.2 Hz, 1H), 8.18 (d, J = 7.2 Hz, 1H), 8.13 (s, 1H), 8.05 (d, J = 9.3 Hz, 1H), 7.94–7.85 (complex, 2H); 13C-NMR (DMSO-d6): δ 143.0, 140.6, 139.8, 139.6, 131.7, 131.0 (q, J = 32.7 Hz), 126.9, 125.2, 124.6 (q, J = 4.3 Hz), 124.1 (q, J = 272.6 Hz), 123.0, 119.8 (q, J = 4.9 Hz), 112.0 (one C not observed); MS: m/z 307 (M+), calculated m/z 307.06. Anal. Calcd. for C14H8F3N3O2: C, 54.73; H, 2.62; N, 13.68. Found: C, 54.66; H, 2.64; N, 13.57.
1-(4-(Trifluoromethyl)phenyl)-5-nitro-1H-indazole (12j). Yield: 209 mg (0.68 mmol, 68%) as a light yellow solid, m.p. 151–152 °C. IR: 1519, 1327 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (s, 1H), 8.76 (s, 1H), 8.34 (d, J = 9.5 Hz, 1H), 8.11 (obscured d, J = 9.5 Hz, 1H), 8.08 (d, J = 8.5 Hz, 2H), 8.00 (d, J = 8.5 Hz, 2H); 13C-NMR (DMSO-d6): δ 143.1, 142.3, 140.5, 139.8, 127.9, (q, J = 32.3 Hz), 127.5 (q, J = 3.5 Hz), 125.4, 124.4 (q, J = 273.7 Hz), 123.3, 123.1, 119.9, 112.1; MS: m/z 307 (M+), calculated m/z 307.06. Anal. Calcd. for C14H8F3N3O2: C, 54.73; H, 2.62; N, 13.68. Found: C, 54.68; H, 2.61; N, 13.64.
1-(4-Cyanophenyl)-5-nitro-1H-indazole (12k). Yield: 158 mg (0.60 mmol, 60%) as a light yellow solid, m.p. 250–251 °C. IR: 2227, 1512, 1344 cm−1; 1H-NMR (DMSO-d6): δ 8.97 (d, J = 2.2 Hz, 1H), 8.78 (s, 1H), 8.36 (dd, J = 9.3, 2.2 Hz, 1H), 8.15 (d, J = 9.3 Hz, 1H), 8.12 (d, J = 8.8 Hz, 2H), 8.07 (d, J = 8.8 Hz, 2H); 13C-NMR (DMSO-d6): δ 142.1, 141.6, 139.4, 139.1, 133.6, 124.5, 122.1, 120.0, 118.9, 117.7, 111.3, 109.0; MS: m/z 264 (M+), calculated m/z 264.06. Anal. Calcd. for C14H8N4O2: C, 63.64; H, 3.05; N, 21.20. Found: C, 63.55; H, 3.09; N, 21.08.
4-(5-Nitro-1H-indazol-1-yl)benzenesulfonamide (12l). Yield: 159 mg (0.50 mmol, 50%), m.p. 237–238 °C; IR: 3329, 3241, 1512, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.97 (d, J = 2.2 Hz, 1H), 8.76 (s, 1H), 8.35 (dd, J = 9.3, 2.2 Hz, 1H), 8.13 (d, J = 9.3 Hz, 1H), 8.06 (s, 4H), 7.53 (br s, 2H); 13C-NMR (DMSO-d6): δ 143.1, 141.60, 141.55, 140.5, 139.8, 128.0, 125.3,123.1, 122.2, 119.9, 112.2; MS: m/z 318 (M+), calculated m/z 318.04. Anal. Calcd. for C13H10N4O4S: C, 49.05; H, 3.17; N, 17.60. Found: C, 49.11; H, 3.16; N, 17.66.
4-(5-Nitro-1H-indol-1yl)benzoic acid (12m). This product was formed only using the two-step procedure. Yield: 142 mg (0.50 mmol, 50%) as a brown product, m.p. 158–159 °C; IR: 3395–2372, 1694, 1518, 1345 cm−1; 1H-NMR (DMSO-d6): δ 13.3 (br s, 1H), 8.97 (d, J = 2.2 Hz, 1H), 8.76 (s, 1H), 8.35 (dd, J = 9.3, 2.2 Hz , 1H), 8.18 (d, J = 8.4 Hz, 2H), 8.13 (d, J = 9.3 Hz, 1H), 7.98 (d, J = 8.4 Hz, 2H); 13C-NMR (DMSO-d6): δ 167.0, 143.0, 142.5, 140.4, 139.7, 131.5, 129.8, 125.3, 123.0, 122.5, 119.9, 112.2; MS: m/z 283 (M+), calculated m/z 283.06. Anal. Calcd. for C14H9N3O4: C, 59.37; H, 3.20; N, 14.84. Found: C, 59.32; H, 3.23; N, 14.75.
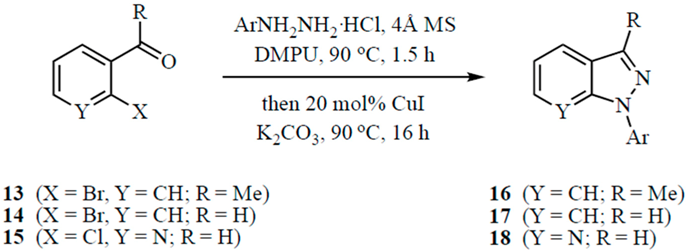
3.5. Representative Procedure for the General Indazole Synthesis
To a stirred solution of the carbonyl compound (13, 14 or 15, 1.0 mmol) in DMPU (5 mL) were added powdered 4 Å molecular sieves (30 wt% relative to the carbonyl substrate) and ArNHNH2·HCl (1.5 mmol). The mixture was heated at 90 °C (oil bath) for 1.5 h at which time CuI (0.2 mmol) and K2CO3 (2.5 mmol) were added and heating was continued at this temperature for 16 h. The crude reaction mixture was cooled to 23 °C and filtered through a Celite® pad (Fisher Scientific, Pittsburgh, PA, USA). The pad was rinsed with ether (2 × 20 mL) and the combined filtrate was washed with water (25 mL), saturated NaCl (25 mL), dried (MgSO4), filtered, and concentrated under vacuum. The products were purified by silica gel chromatography using increasing concentrations of EtOAc in hexanes. Yields as well as physical and spectral data are given below.
3-Methyl-1-phenyl-1H-indazole (
16a). Yield: 169 mg (0.81 mmol, 81%) as tan solid, m.p. 73–74 °C (lit [
18] m.p. 73–74 °C); IR: 1597, 1505 cm
−1;
1H-NMR (CDCl
3): δ 7.75–7.96 (complex, 4H), 7.52 (t,
J = 7.3 Hz, 2H), 7.42 (t,
J = 7.5 Hz, 1H), 7.32 (t,
J = 7.3 Hz, 1H), 7.21 (t,
J = 7.5 Hz, 1H), 2.65 (s, 3H);
13C-NMR (CDCl
3): δ 144.0, 140.3, 139.5, 129.4, 127.1, 126.1, 124.9, 122.4, 120.8, 120.6, 110.3, 12.0; MS:
m/
z 208 (M
+), calculated
m/
z 208.10. Anal. Calcd. for C
14H
12N
2: C, 80.74; H, 5.81; N, 13.45. Found: C, 81.01; H, 6.08; N, 13.23.
1-(4-Methoxyphenyl)-3-methyl-1H-indazole (16d). Yield: 207 mg (0.87 mmol, 87%) as a yellow oil; IR: 2845, 1517 cm−1; 1H-NMR (CDCl3): δ 7.72 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.59 (d, J = 8.9 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H), 2.65 (s, 3H); 13C-NMR (CDCl3): δ 158.1, 143.8, 139.7, 133.5, 126.9, 124.4, 124.3, 120.50, 120.47, 114.6, 110.1, 56.6, 11.9; MS: m/z 238 (M+), calculated m/z 238.11. Anal. Calcd. for C15H14N2O: C, 75.61; H, 5.92; N, 11.76. Found: C, 75.35; H, 6.18; N, 11.25.
4-(3-Methyl-1H-indazol-1-yl)benzonitrile (16k). Yield: 198 mg (0.85 mmol, 85%) as tan solid, m.p. 124–126 °C; IR: 2224, 1604, 1517 cm−1; 1H-NMR (CDCl3): δ 7.91 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 8.6 Hz, 2H), 7.78 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 7.9 Hz, 1H), 7.50 (t, J = 8.3 Hz, 1H), 7.28 (t, J = 7.4 Hz, 1H), 2.65 (s, 3H); 13C-NMR (CDCl3): δ 146.1, 143.9, 139.1, 133.5, 128.0, 125.9, 121.9, 121.3, 121.1, 118.7, 110.5, 108.5, 12.0; MS: m/z 233 (M+), calculated m/z 233.10. Anal. Calcd. for C15H11N3: C, 77.23; H, 4.75; N, 18.01. Found: C, 77.35; H, 4.31; N, 18.32.
1-Phenyl-1H-indazole (
17a). Yield: 149 mg (0.77 mmol, 77%) as off-white solid, m.p. 77–78 °C (lit [
18] m.p. 76–78 °C); IR: 1595, 1500 cm
−1;
1H-NMR (CDCl
3): δ 8.21 (s, 1H), 7.81 (d,
J = 8.1 Hz, 1H), 7.77 (d,
J = 8.4 Hz, 1H), 7.75 (d,
J = 8.7 Hz, 2H), 7.54 (t,
J = 8.4 Hz, 2H), 7.43 (t,
J = 8.3 Hz, 1H), 7.37 (t,
J = 8.3 Hz, 1H), 7.23 (t,
J = 8.1 Hz, 1H);
13C-NMR (CDCl
3): δ 140.2, 138.8, 135.4, 129.5, 127.1, 126.6, 125.3, 122.8, 121.5, 121.3, 110.4; MS:
m/
z 194 (M
+), calculated
m/
z 194.08. Anal. Calcd. for C
13H
10N
2: C, 80.39; H, 5.19; N, 14.42. Found: C, 80.15; H, 5.31; N, 14.69.
1-(4-Methoxyphenyl)-1H-indazole (17d). Yield: 177 mg (0.79 mmol, 79%) as a yellow oil; IR: 2836, 1515 cm−1; 1H-NMR (CDCl3): δ 8.17 (s, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.9 Hz, 2H), 7.41 (t, J = 8.3 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 7.06 (d, J = 8.9 Hz, 2H), 3.88 (s, 3H); 13C-NMR (CDCl3): δ 158.4, 139.0, 134.8, 133.4, 126.9, 124.9, 124.5, 121.23, 121.21, 114.6, 110.2, 55.6; MS: m/z 224 (M+), calculated m/z 224.09. Anal. Calcd. for C14H12N2O: C, 74.98; H, 5.39; N, 12.49. Found: C, 74.63; H, 5.52; N, 12.63.
4-(1H-Indazol-1-yl)benzonitrile (
17k). Yield: 164 mg (0.75 mmol, 75%) as white solid, m.p. 104–106 °C (lit m.p. [
18] 103–106 °C); IR: 2225, 1604, 1510 cm
−1;
1H-NMR (CDCl
3): δ 8.26 (s, 1H), 7.94 (d,
J = 8.6 Hz, 2H), 7.84 (d,
J = 8.6 Hz, 2H), 7.86–7.81 (complex, 2H), 7.51 (t,
J = 8.4 Hz, 1H), 7.31 (t,
J = 8.0 Hz, 1H);
13C-NMR (CDCl
3): δ 143.8, 138.5, 137.3, 133.6, 128.1, 126.1, 122.5, 121.9, 121.8, 118.5, 110.4, 109.3; MS:
m/
z 219 (M
+), calculated
m/
z 219.08. Anal. Calcd. for C
14H
9N
3: C, 76.70; H, 4.14; N, 19.17. Found: C, 76.85; H, 4.31; N, 19.32.
1-Phenyl-1H-pyrazolo[3,4-b]pyridine (
18a). Yield: 133 mg (0.68 mmol, 68%) as a white solid, m.p. 52–54 °C (lit [
10] 53–55 °C); IR: 1595, 1499 cm
−1;
1H-NMR (CDCl
3): δ 8.65 (dd,
J = 4.5, 1.7 Hz, 1H), 8.28 (dd,
J = 7.7, 1.2 Hz, 2H), 8.21 (s, 1H), 8.14 (dd,
J = 8.0, 1.7 Hz, 1H), 7.54 (t,
J = 7.7 Hz, 1H), 7.33 (t,
J = 7.5 Hz, 1H), 7.22 (dd,
J = 8.0, 4.5 Hz, 1H);
13C-NMR (CDCl
3): δ 150.1, 149.2, 139.5, 133.8, 130.2, 129.1, 126.1, 121.4, 117.6, 117.2; MS:
m/
z 195 (M
+), calculated
m/
z 195.08. Anal. Calcd. for C
12H
9N
3: C, 73.83; H, 4.65; N, 21.52. Found: C, 73.95; H, 4.87; N, 21.78.
1-(4-Methoxyphenyl)-1H-pyrazolo[3,4-b]pyridine (18d). Yield: 140 mg (0.62 mmol, 62%) as a purple solid, m.p. 204–205 °C; IR: 2833, 1513 cm−1; 1H-NMR (CDCl3): δ 9.00 (dd, J = 8.0, 1.7 Hz, 1H), 8.70 (dd, J = 4.5, 1.7 Hz, 1H), 8.28 (d, J = 9.0 Hz, 2H), 7.35 (dd, J = 8.0, 4.5 Hz, 1H), 7.26 (s, 1H), 7.14 (d, J = 9.0 Hz, 2H), 3.91 (s, 3H); 13C-NMR (CDCl3): δ 158.0, 150.6, 149.6, 138.7, 132.9, 132.6, 123.0, 118.3, 115.3, 114.4, 55.6; MS: m/z 225 (M+), calculated m/z 225.09. Anal. Calcd. for C13H11N3O: C, 69.32; H, 4.92; N, 18.66. Found: C, 69.68; H, 5.11; N, 19.82.
4-(1H-Pyrazolo[3,4-b]pyridine-1-yl)benzonitrile (18k). Yield: 172 mg (0.78 mmol, 78%) as a white solid, m.p. 125–127 °C; IR: 2222, 1603, 1450 cm−1; 1H-NMR (CDCl3): δ 8.68 (dd, J = 4.5, 1.6 Hz, 1H), 8.66 (d, J = 8.8 Hz, 2H), 8.25 (s, 1H), 8.17 (dd. J = 8.0, 1.6 Hz, 1H), 7.82 (d, J = 8.8 Hz, 2H), 7.30 (dd, J = 8.0, 4.5 Hz, 1H); 13C-NMR (CDCl3): δ 150.7, 149.4, 143.1, 135.4, 133.2, 130.6, 120.3, 11.89, 118.4, 117.9, 108.7; MS: m/z 220 (M+), calculated m/z 220.07. Anal. Calcd. for C13H8N4: C, 70.90; H, 3.66; N, 25.44. Found: C, 71.23; H, 3.82; N, 25.67.




{kind=link}
{kind=link}
{kind=link}