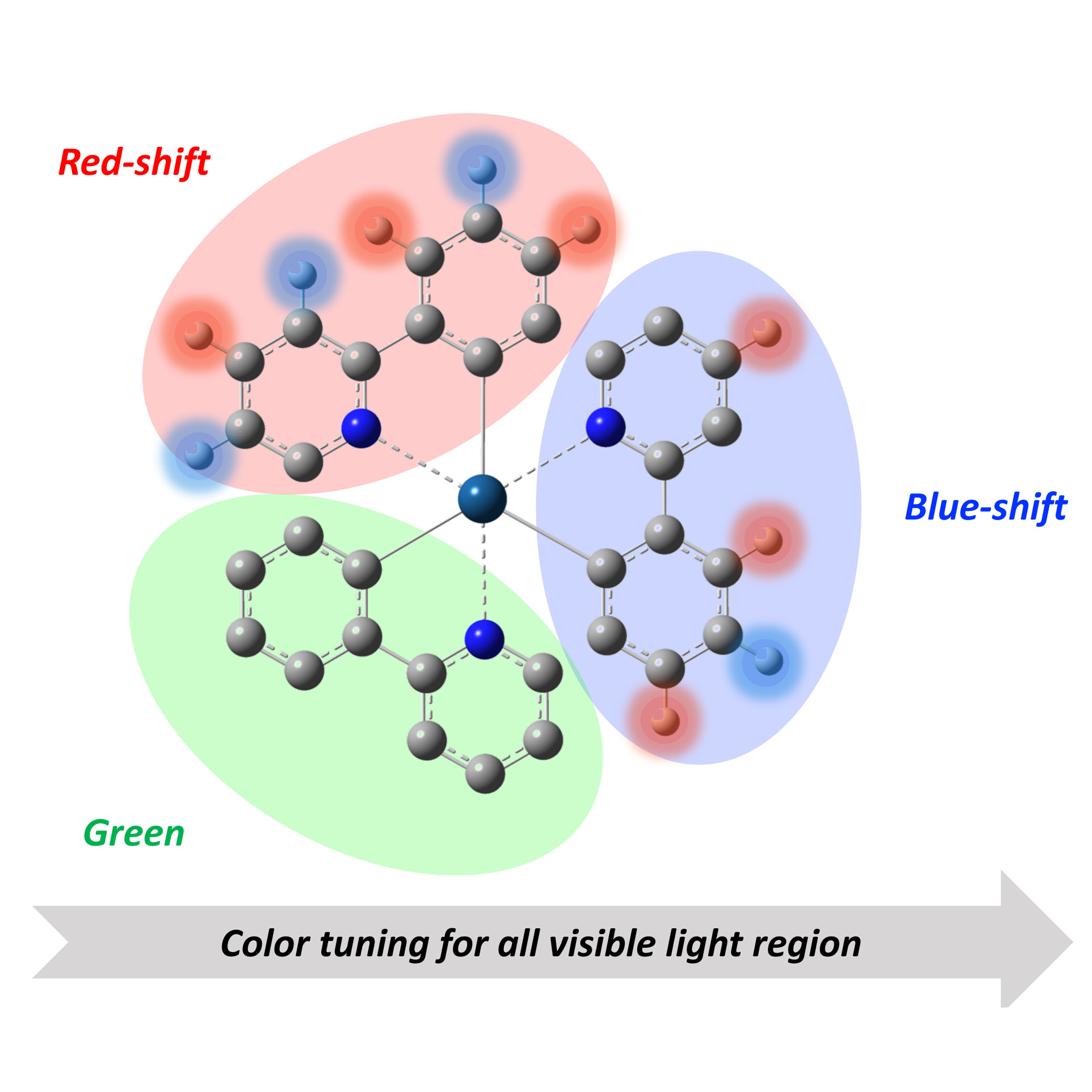
Quantum Chemical Design Guidelines for Absorption and Emission Color Tuning of fac-Ir(ppy)3 Complexes


 ,
, 
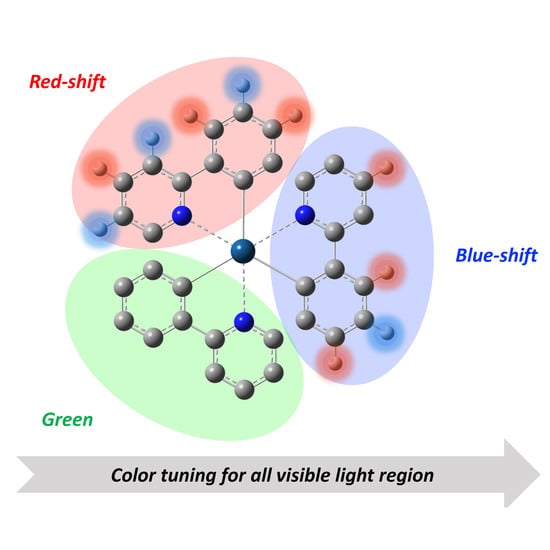
Abstract
:
1. Introduction
2. Computational Details
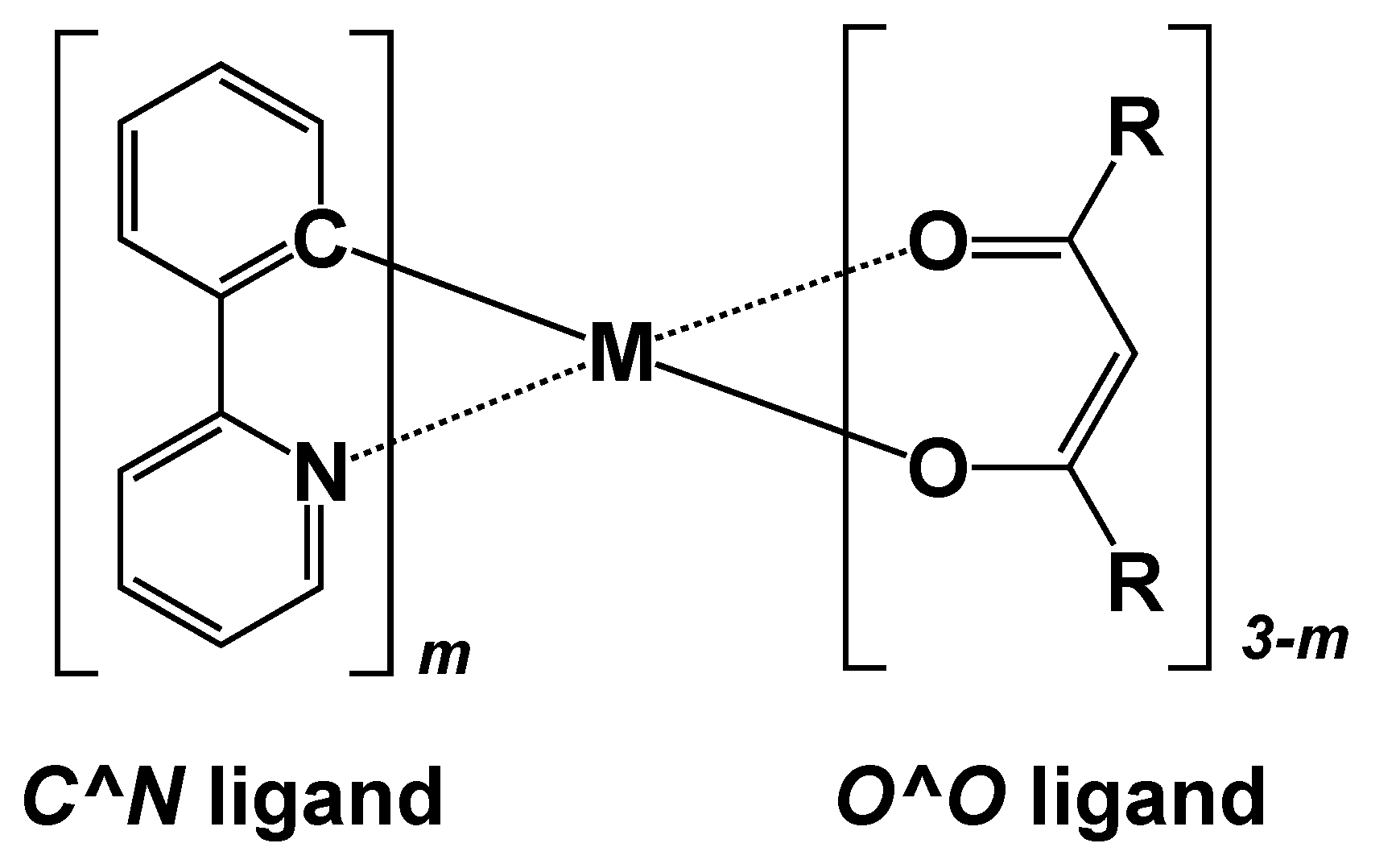
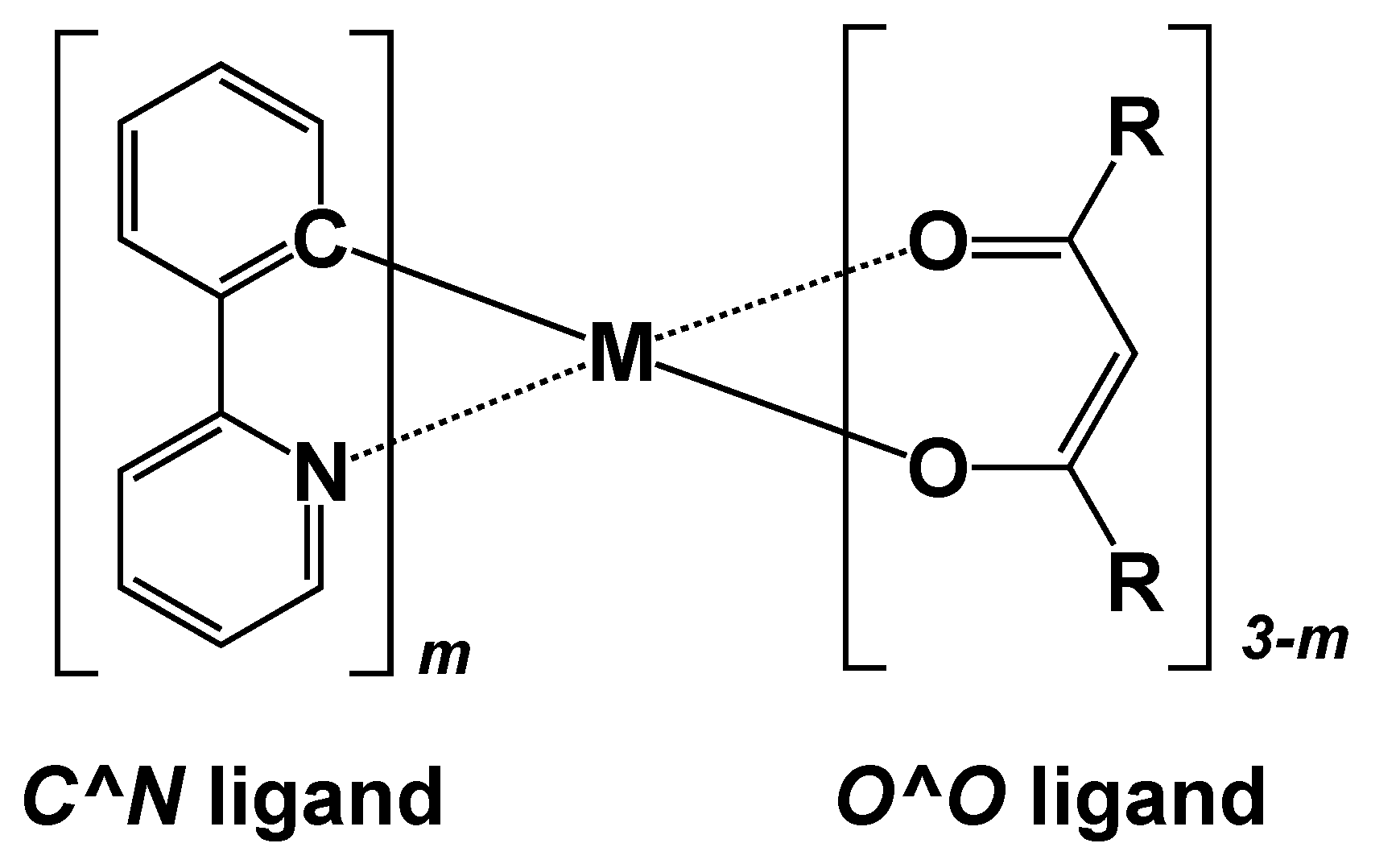
3. Results and Discussion
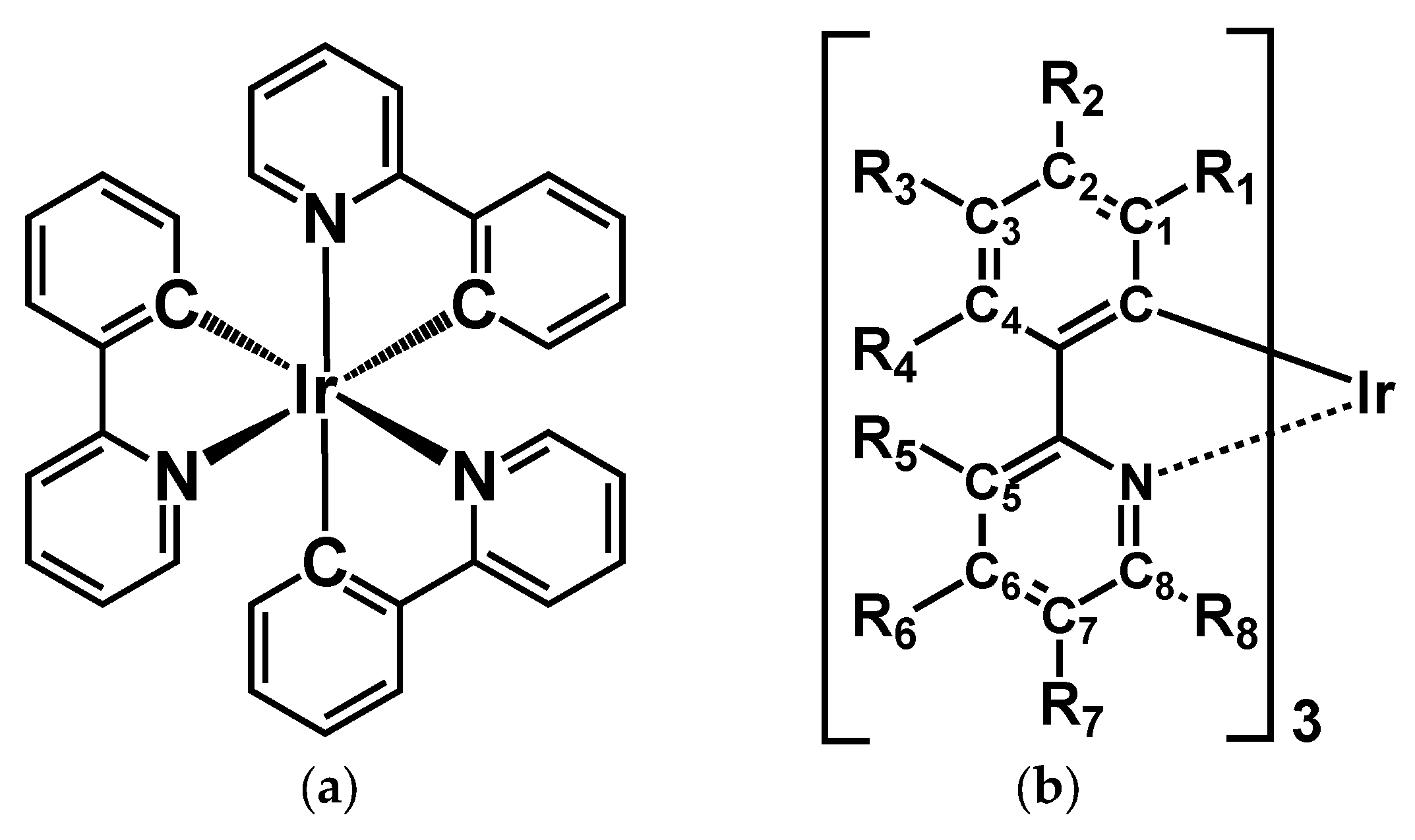
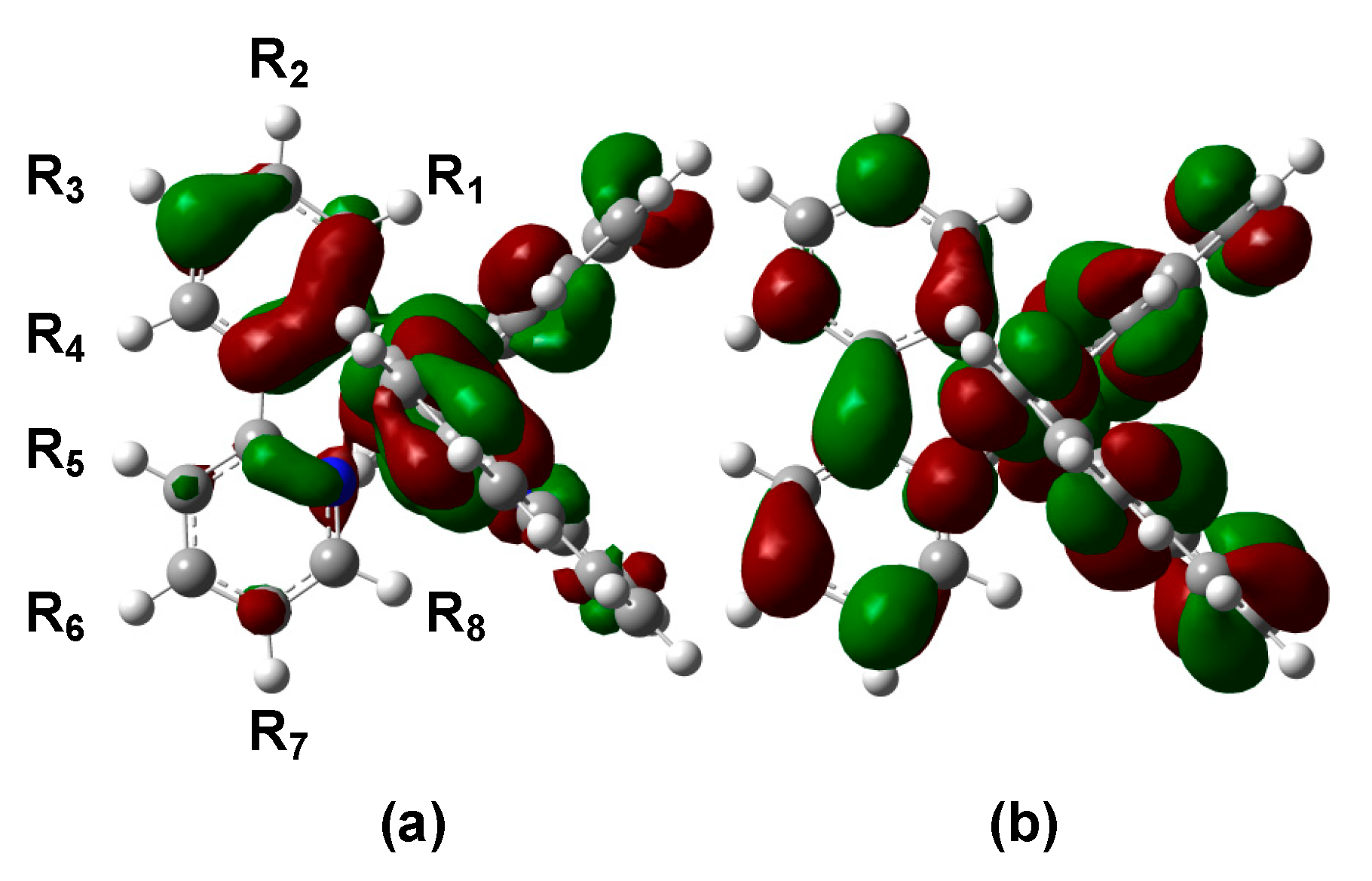
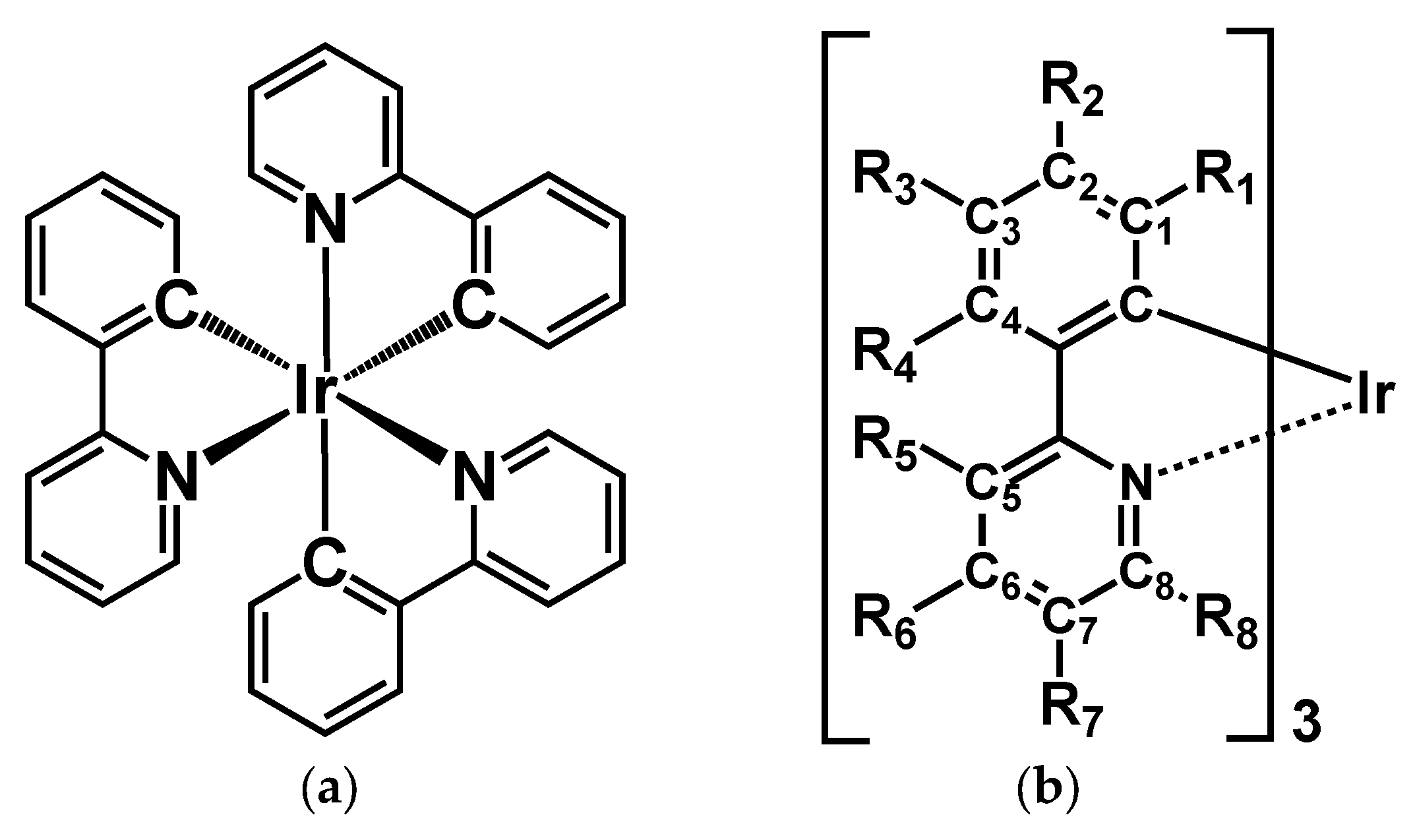
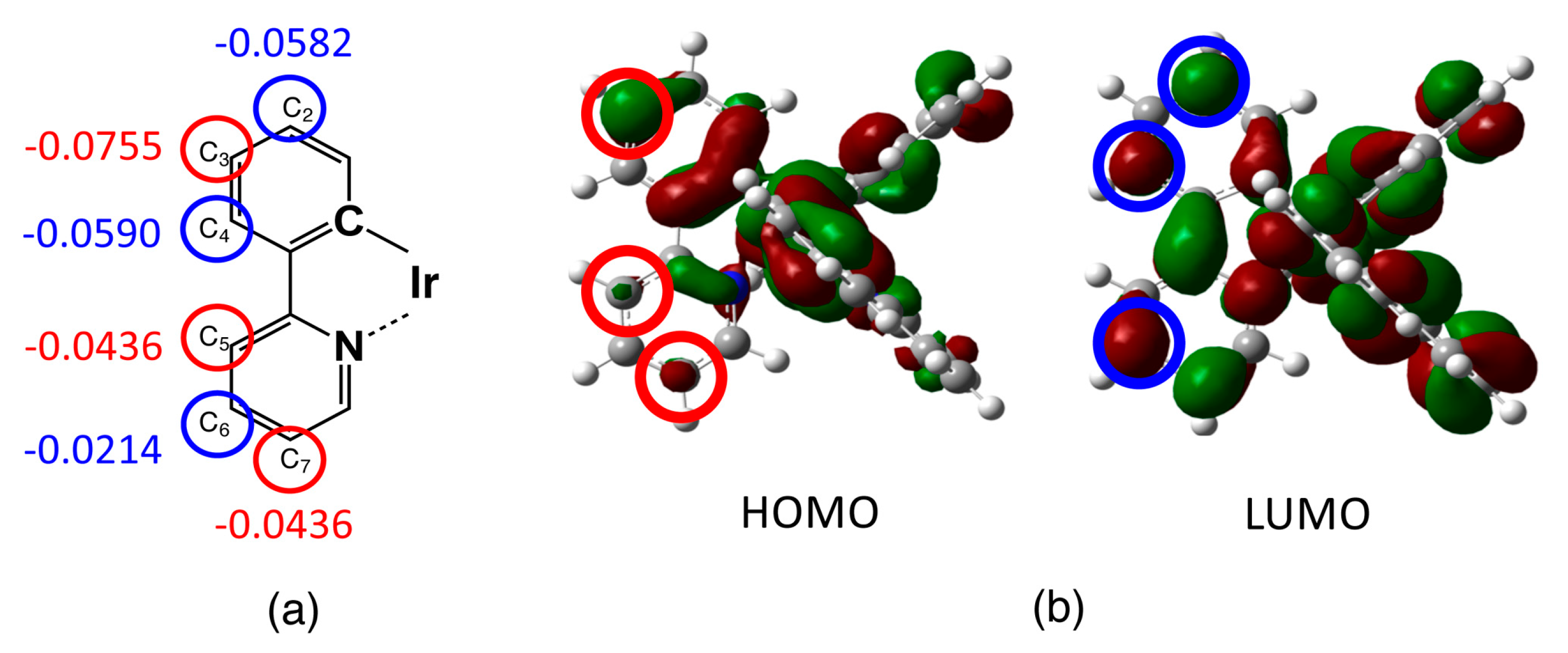
3.1. Unsubstituted Complex 1
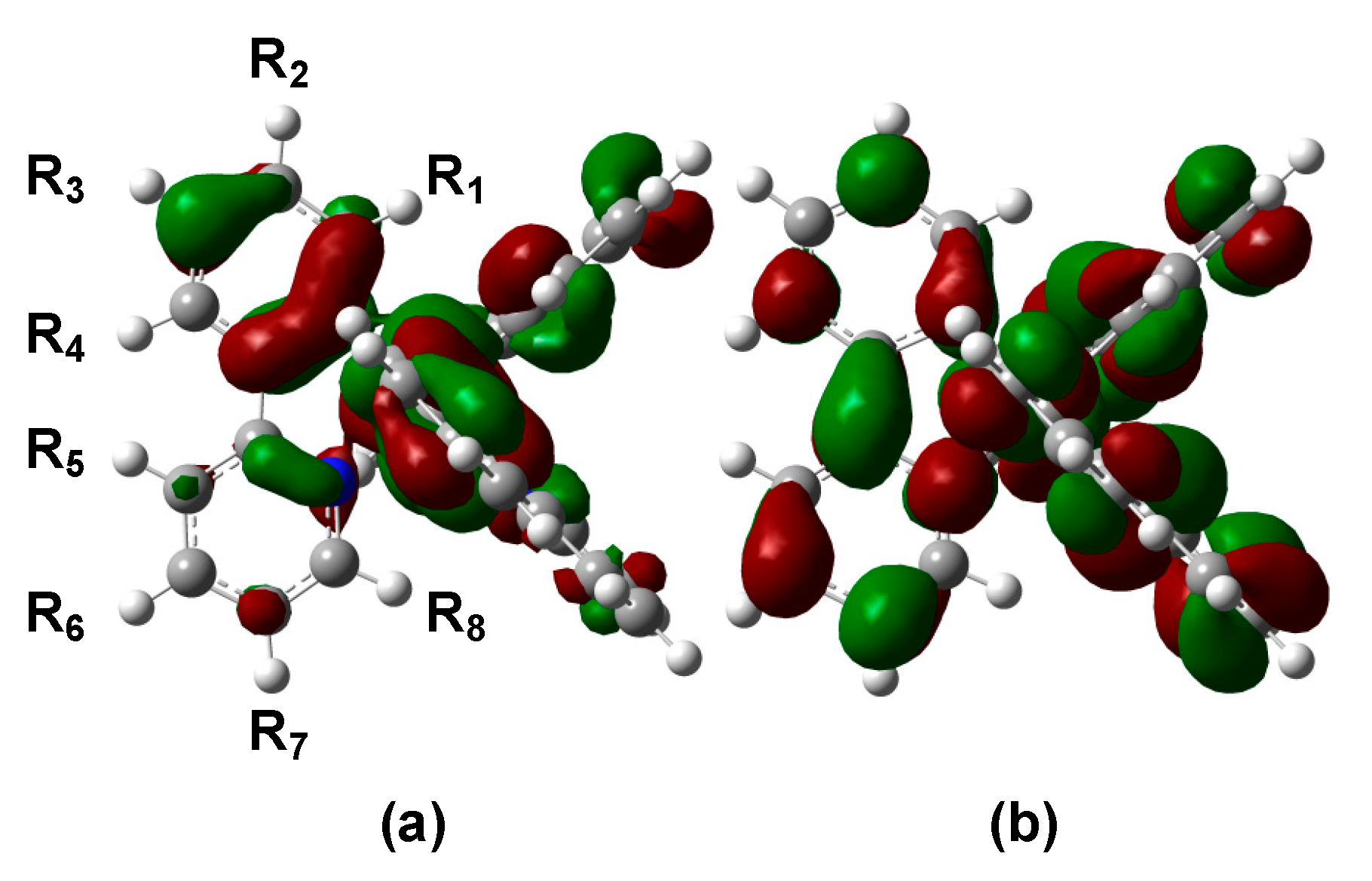
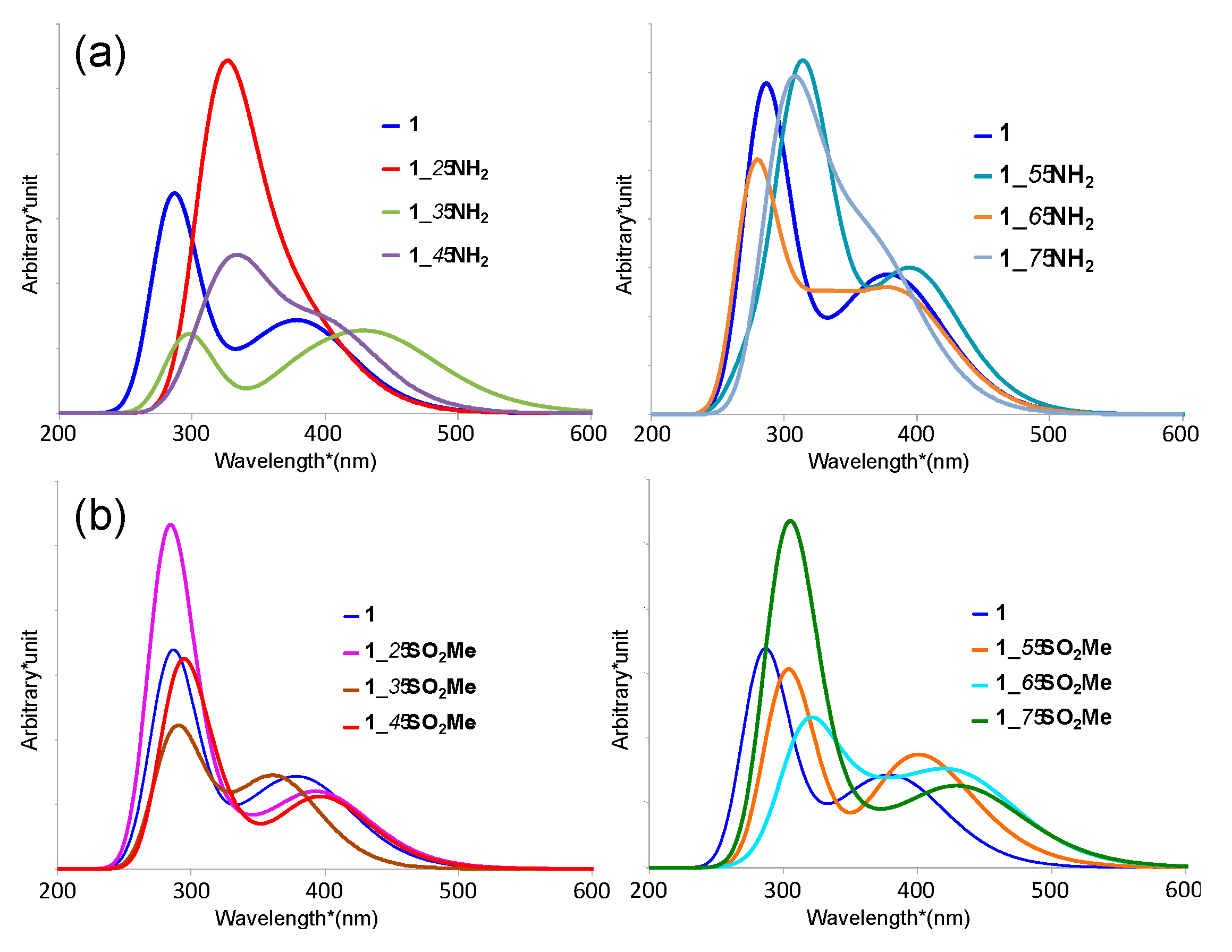
3.2. Absorption Spectra of Derivatives of Complex 1
3.3. Emission Wavelengths
4. Concluding Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baldo, M.A.; O’Brien, D.F.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M.E.; Forrest, S.R. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 1998, 395, 151–154. [Google Scholar] [CrossRef]
- Adachi, C.; Baldo, M.A.; Thompson, M.E.; Forrest, S.R. Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 2001, 90, 5048–5051. [Google Scholar] [CrossRef]
- Ikawa, S.; Yagi, S.; Maeda, T.; Nakazumi, H.; Fujiwara, H.; Sakurai, Y. Photoluminescence color tuning of phosphorescent bis-cyclometalated iridium(III) complexes by ancillary ligand replacement. Dyes Pigment. 2012, 95, 695–705. [Google Scholar] [CrossRef]
- Xiong, Y.; Xu, W.; Li, C.; Liang, B.; Zhao, L.; Peng, J.; Cao, Y.; Wang, J. Utilizing white OLED for full color reproduction in flat panel display. Org. Electron. 2008, 9, 533–538. [Google Scholar] [CrossRef]
- Baldo, M.A.; Lamansky, S.; Burrows, P.E.; Thompson, M.E.; Forrest, S.R. Very high-efficiency green organic light-emitting devices based on electrophosphorescence. Appl. Phys. Lett. 1999, 75, 4–6. [Google Scholar] [CrossRef]
- King, K.A.; Spellane, P.J.; Watts, R.J. Excited-state properties of a triply ortho-metalated iridium(III) complex. J. Am. Chem. Soc. 1985, 107, 1431–1432. [Google Scholar] [CrossRef]
- Hedley, G.J.; Ruseckas, A.; Samuel, I.D.W. Ultrafast luminescence in Ir(ppy)3. Chem. Phys. Lett. 2008, 450, 292–296. [Google Scholar] [CrossRef]
- Yersin, H.; Finkenzeller, W.J. Triplet Emitters for Organic Light-Emitting Diodes: Basic Properties. In Highly Efficient OLEDs with Phosphorescent Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 1–97. ISBN 9783527405947. [Google Scholar]
- Colombo, M.G.; Brunold, T.C.; Riedener, T.; Guedel, H.U.; Fortsch, M.; Buergi, H.-B. Facial tris cyclometalated Rh3+ and Ir3+ complexes: Their synthesis, structure, and optical spectroscopic properties. Inorg. Chem. 1994, 33, 545–550. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Kwong, R.; Tsyba, I.; Bortz, M.; Mui, B.; Bau, R.; Thompson, M.E. Synthesis and Characterization of Phosphorescent Cyclometalated Iridium Complexes. Inorg. Chem. 2001, 40, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Lenkeit, D.; Pelz, S.; Fischer, L.H.; Escudero, D.; Schiewek, R.; Klink, D.; Schmitz, O.J.; González, L.; Schäferling, M.; et al. Structure–Property Relationship of Red- and Green-Emitting Iridium(III) Complexes with Respect to Their Temperature and Oxygen Sensitivity. Eur. J. Inorg. Chem. 2010, 2010, 4875–4885. [Google Scholar] [CrossRef]
- Tamayo, A.B.; Alleyne, B.D.; Djurovich, P.I.; Lamansky, S.; Tsyba, I.; Ho, N.N.; Bau, R.; Thompson, M.E. Synthesis and Characterization of Facial and Meridional Tris-cyclometalated Iridium(III) Complexes. J. Am. Chem. Soc. 2003, 125, 7377–7387. [Google Scholar] [CrossRef] [PubMed]
- Tsuboyama, A.; Iwawaki, H.; Furugori, M.; Mukaide, T.; Kamatani, J.; Igawa, S.; Moriyama, T.; Miura, S.; Takiguchi, T.; Okada, S.; et al. Homoleptic Cyclometalated Iridium Complexes with Highly Efficient Red Phosphorescence and Application to Organic Light-Emitting Diode. J. Am. Chem. Soc. 2003, 125, 12971–12979. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; You, H.; Yang, C.; Zhang, X.; Qin, J.; Ma, D. Tuning the saturated red emission: Synthesis, electrochemistry and photophysics of 2-arylquinoline based iridium(III) complexes and their application in OLEDs. J. Mater. Chem. 2006, 16, 3332–3339. [Google Scholar] [CrossRef]
- Kusaka, S.; Sakamoto, R.; Kitagawa, Y.; Okumura, M.; Nishihara, H. meso-Alkynyl BODIPYs: Structure, Photoproperties, π-Extension, and Manipulation of Frontier Orbitals. Chem. Asian J. 2013, 8, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Sakamoto, R.; Kusaka, S.; Kitagawa, Y.; Okumura, M.; Nishihara, H. Asymmetric dinuclear bis(dipyrrinato)zinc(II) complexes: Broad absorption and unidirectional quantitative exciton transmission. Chem. Commun. 2014, 50, 5881–5883. [Google Scholar] [CrossRef] [PubMed]
- Asaoka, M.; Kitagawa, Y.; Teramoto, R.; Miyagi, K.; Natori, Y.; Sakamoto, R.; Nishihara, H.; Nakano, M. Theoretical study on S1 and T1 states of homoleptic bis(dipyrrinato)zinc(II) model complex. Polyhedron 2017, 136, 113–116. [Google Scholar] [CrossRef]
- Asaoka, M.; Kitagawa, Y.; Teramoto, R.; Miyagi, K.; Natori, Y.; Nakano, M. Origin of Solvent-independent Optical Property of Unsubstituted BODIPY Revisited. Chem. Lett. 2017, 46, 536–538. [Google Scholar] [CrossRef]
- Nozaki, K. Theoretical Studies on Photophysical Properties and Mechanism of Phosphorescence in [fac-Ir(2-phenylpyridine)3]. J. Chin. Chem. Soc. 2006, 53, 101–112. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Rev.C01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Younker, J.M.; Dobbs, K.D. Correlating Experimental Photophysical Properties of Iridium(III) Complexes to Spin–Orbit Coupled TDDFT Predictions. J. Phys. Chem. C 2013, 117, 25714–25723. [Google Scholar] [CrossRef]
- Mehata, M.S.; Yang, Y.; Qu, Z.-J.; Chen, J.-S.; Zhao, F.-J.; Han, K.-L. Spin mixed charge transfer states of iridium complex Ir(ppy)3: Transient absorption and time-resolved photoluminescence. RSC Adv. 2015, 5, 34094–34099. [Google Scholar] [CrossRef]
- Wang, P.; Wang, F.-F.; Chen, Y.; Niu, Q.; Lu, L.; Wang, H.-M.; Gao, X.-C.; Wei, B.; Wu, H.-W.; Cai, X.; et al. Synthesis of all-deuterated tris(2-phenylpyridine)iridium for highly stable electrophosphorescence: The “deuterium effect”. J. Mater. Chem. C 2013, 1, 4821–4825. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Yagi, S.; Asuka, H.; Inui, Y.; Ikawa, S.; Maeda, T.; Nakazumi, H.; Sakurai, Y. Pure red electrophosphorescence from polymer light-emitting diodes doped with highly emissive bis-cyclometalated iridium(III) complexes. J. Organomet. Chem. 2010, 695, 1972–1978. [Google Scholar] [CrossRef]
- Aoki., S.; Matsuo, Y.; Ogura., S.; Ohwada, H.; Hisamatsu, Y.; Moromizato, S.; Shiro, M.; Kitamura, M. Regioselective Aromatic Substitution Reactions of Cyclometalated Ir(III) Complexes: Synthesis and Photochemical Properties of Substituted Ir(III) Complexes That Exhibit Blue, Green, and Red Color Luminescence Emission. Inorg. Chem. 2011, 50, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Rausch, A.F.; Thompson, M.E.; Yersin, H. Matrix Effects on the Triplet State of the OLED Emitter Ir(4,6-dFppy)2(pic) (FIrpic): Investigations by High-Resolution Optical Spectroscopy. Inorg. Chem. 2009, 48, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms 2 | Phenyl Ring | Pyridine Ring | ||||
|---|---|---|---|---|---|---|
| C2 | C3 | C4 | C5 | C6 | C7 | |
| Hirshfeld charges 3 | −0.0582 | −0.0755 | −0.0590 | −0.0436 | −0.0214 | −0.0472 |
| (↗) | (↘) | (↗) | (↘) | (↗) | (↘) | |
| Complex | Absorption Wavelength (λab) (nm) | ∆λab (nm) 1 | |||
|---|---|---|---|---|---|
| LLCT Peak | MLCT Peak | LLCT Peak | MLCT Peak | ||
| 1 | 287 | (a) 362, (b) 387 | - | (a) | (b) |
| 1_2-NH2 | 320 | 386, 415 | 33 | 24 | 28 |
| 1_3-NH2 | 296 | 405, 439 | 9 | 43 | 52 |
| 1_4-NH2 | 322 | 393, 413 | 35 | 31 | 26 |
| 1_5-NH2 | 319 | 383, 398 | 32 | 21 | 11 |
| 1_6-NH2 | 276 | 381, 402 | –11 | 19 | 15 |
| 1_7-NH2 | 307, 360 | 380, 391 | 20, 73 | 18 | 4 |
| 1_2-SO2Me | 284 | 391, 409 | –3 | 29 | 21 |
| 1_3-SO2Me | 294 | 364, 376 | 7 | 2 | –11 |
| 1_4-SO2Me | 291 | 397, 420 | 4 | 35 | 33 |
| 1_5-SO2Me | 304 | 393, 419 | 14 | 29 | 32 |
| 1_6-SO2Me | 318 | 386, 431 | 31 | 24 | 44 |
| 1_7-SO2Me | 304 | 427 | 17 | 40 | - |
| Complexes | Emission Wavelength (λem, nm) | ∆λem (nm) 1 |
|---|---|---|
| 1 | 532 | |
| 1_2-NH2 | 533 | 1 |
| 1_3-NH2 | 698 | 166 |
| 1_4-NH2 | 533 | 1 |
| 1_5-NH2 | 591 | 59 |
| 1_6-NH2 | 520 | −12 |
| 1_7-NH2 | 575 | 43 |
| 1_2-SO2Me | 566 | 34 |
| 1_3-SO2Me | 514 | −18 |
| 1_4-SO2Me | 592 | 60 |
| 1_5-SO2Me | 575 | 43 |
| 1_6-SO2Me | 605 | 73 |
| 1_7-SO2Me | 574 | 42 |
| Complexes | HOMO (eV) | LUMO (eV) | HOMO–LUMO Gap (eV) |
|---|---|---|---|
| 1 | −5.343 | −1.685 | 3.658 |
| 1_2-NH2 | −5.130 | −1.435 | 3.695 |
| 1_3-NH2 | −4.903 | −1.635 | 3.267 |
| 1_4-NH2 | −5.218 | −1.654 | 3.564 |
| 1_5-NH2 | −5.225 | −1.564 | 3.661 |
| 1_6-NH2 | −5.113 | −1.438 | 3.675 |
| 1_7-NH2 | −5.132 | −1.425 | 3.706 |
| 1_2-SO2Me | −5.776 | −2.185 | 3.590 |
| 1_3-SO2Me | −5.857 | −2.001 | 3.856 |
| 1_4-SO2Me | −5.766 | −2.094 | 3.672 |
| 1_5-SO2Me | −5.620 | −2.104 | 3.516 |
| 1_6-SO2Me | −5.647 | −2.342 | 3.304 |
| 1_7-SO2Me | −5.687 | −2.320 | 3.367 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natori, Y.; Kitagawa, Y.; Aoki, S.; Teramoto, R.; Tada, H.; Era, I.; Nakano, M. Quantum Chemical Design Guidelines for Absorption and Emission Color Tuning of fac-Ir(ppy)3 Complexes. Molecules 2018, 23, 577. https://doi.org/10.3390/molecules23030577
Natori Y, Kitagawa Y, Aoki S, Teramoto R, Tada H, Era I, Nakano M. Quantum Chemical Design Guidelines for Absorption and Emission Color Tuning of fac-Ir(ppy)3 Complexes. Molecules. 2018; 23(3):577. https://doi.org/10.3390/molecules23030577
Chicago/Turabian StyleNatori, Yoshiki, Yasutaka Kitagawa, Shogo Aoki, Rena Teramoto, Hayato Tada, Iori Era, and Masayoshi Nakano. 2018. "Quantum Chemical Design Guidelines for Absorption and Emission Color Tuning of fac-Ir(ppy)3 Complexes" Molecules 23, no. 3: 577. https://doi.org/10.3390/molecules23030577
APA StyleNatori, Y., Kitagawa, Y., Aoki, S., Teramoto, R., Tada, H., Era, I., & Nakano, M. (2018). Quantum Chemical Design Guidelines for Absorption and Emission Color Tuning of fac-Ir(ppy)3 Complexes. Molecules, 23(3), 577. https://doi.org/10.3390/molecules23030577