Neuroprotective Effects and Mechanisms of Tea Bioactive Components in Neurodegenerative Diseases

 and
and
Abstract
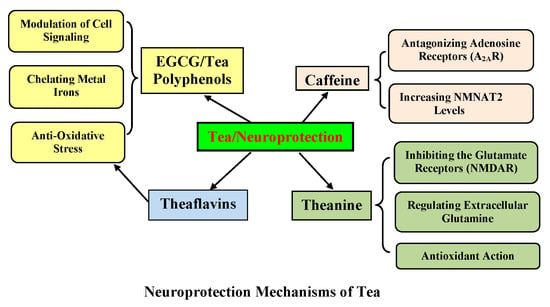
1. Introduction
2. Epidemiological Evidence
3. Neuroprotective Effects and Mechanisms of Tea Polyphenols
3.1. Anti-Oxidative Stress
3.2. Modulation of Cell Signaling Pathways
3.3. Metal Chelators Activity
4. Neuroprotective Effect and Mechanism of Theanine
5. Neuroprotective Effects and Mechanisms of Caffeine
6. Neuroprotective Effects and Mechanisms of Theaflavins
7. The Interaction between Tea Bioactive Components
8. Conclusions and Future Expectations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alzheimer’s Disease International. Disease International. World Alzheimer Report 2016: Improving Healthcare for People Living with Dementia: Coverage, Quality and Costs Now and in the Future. Available online: https://www.alz.co.uk/research/world-report-2016 (accessed on 30 September 2016).
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Prediger, R.D.S. Effects of caffeine in Parkinson's disease: from neuroprotection to the management of motor and non-motor symptoms. J. Alzheimer’s Dis. 2010, 20, S205–S220. [Google Scholar] [CrossRef] [PubMed]
- Bagga, P.; Chugani, A.N.; Patel, A.B. Neuroprotective effects of caffeine in MPTP model of Parkinson’s disease: A 13C NMR study. Neurochem. Int. 2016, 92, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry. 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Di Monte, D.A. The environment and Parkinson’s disease: Is the nigrostriatal system preferentially targeted by neurotoxins? Lancet Neurol. 2003, 2, 531–538. [Google Scholar] [CrossRef]
- Zhuo, C.; Zhu, X.; Jiang, R.; Ji, F.; Su, Z.; Xue, R.; Zhou, Y. Comparison for efficacy and tolerability among ten drugs for treatment of Parkinson’s disease: A network meta-analysis. Sci. Rep. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Schuh, C.; Schieberle, P. Characterization of the key aroma compounds in the beverage prepared from Darjeeling black tea: Quantitative differences between tea leaves and infusion. J. Agric. Food Chem. 2006, 54, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.X.; Ohata, M.; Arihara, K. Effects of odor generated from the glycine/glucose Maillard reaction on human mood and brainwaves. Food Funct. 2016, 7, 2574–2581. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Baldermann, S.; Watanabe, N. Recent studies of the volatile compounds in tea. Food Res. Int. 2013, 53, 585–599. [Google Scholar] [CrossRef]
- Kuroda, K.; Inoue, N.; Ito, Y.; Kubota, K.; Sugimoto, A.; Kakuda, T.; Fushiki, T. Sedative effects of the jasmine tea odor and (R)-(−)-linalool, one of its major odor components, on autonomic nerve activity and mood states. Eur. J. Appl. Physiol. 2005, 95, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Tomata, Y.; Kakizaki, M.; Nakaya, N.; Tsuboya, T.; Sone, T.; Kuriyama, S.; Hozawa, A.; Tsuji, I. Green tea consumption and the risk of incident functional disability in elderly Japanese: The Ohsaki Cohort 2006 Study. Am. J. Geriatr. Psychiatry. 2016, 95, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Kandinov, B.; Giladi, N.; Korczyn, A.D. Smoking and tea consumption delay onset of Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.P.; Aung, K.C.; Feng, L.; Feng, L.; Nyunt, M.S.; Yap, K.B. Tea consumption and physical function in older adults: A cross-sectional study. J. Nutr. Health Aging. 2014, 18, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gwee, X.; Kua, E.H.; Ng, T.P. Cognitive function and tea consumption in community dwelling older Chinese in Singapore. J. Nutr. Health Aging. 2010, 14, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, S.; Hozawa, A.; Ohmori, K.; Shimazu, T.; Matsui, T.; Ebihara, S.; Awata, S.; Nagatomi, R.; Arai, H.; Tsuji, I. Green tea consumption and cognitive function: A cross-sectional study from the Tsurugaya Project. Am. J. Clin. Nutr. 2006, 83, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.P.; Huang, C.; Cui, Q.Y.; Yang, D.J.; Sun, K.; Chen, X.; Li, X.H. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS ONE 2016, 11, e0165861. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatrics Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife coffee and tea drinking and the risk of late-life dementia: A population-based CAIDE study. J. Alzheimers. Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.Y.; Li, Q.S.; Lin, X.M.; Qiao, R.Y.; Yang, R.; Li, X.M.; Dong, Z.B.; Xiang, L.P.; Zheng, X.Q.; Lu, J.L.; et al. Antidiabetic effects of tea. Molecules 2017, 22, 849. [Google Scholar] [CrossRef] [PubMed]
- Chakrawarti, L.; Agrawal, R.; Dang, S.; Gupta, S.; Gabrani, R. Therapeutic effects of EGCG: A patent review. Expert Opin. Ther. Patents. 2016, 26, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Mandal, A.K.A.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2015, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.P.; Bieschke, J.; Friedrich, R.P.; Zhu, D.; Wanker, E.E.; Fecht, H.J.; Mereles, D.; Hunstein, W. 670 nm laser light and EGCG complementarily reduce amyloid-β aggregates in human neuroblastoma cells: Basis for treatment of Alzheimer’s disease? Photomed. Laser Surg. 2012, 30, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.C.; Wang, M.N.; Tseng, T.Y.; Sung, J.S.; Tsai, T.H. Pharmacokinetics of (−)-epigallocatechin-3-gallate in conscious and freely moving rats and its brain regional distribution. J. Agric. Food Chem. 2007, 55, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Pogačnik, L.; Pirc, K.; Palmela, I.; Skrt, M.; Kim, K.S.; Brites, D.; Brito, M.A.; Ulrih, N.P.; Silva, R.F. Potential for brain accessibility and analysis of stability of selected flavonoids in relation to neuroprotection in vitro. Brain Res. 2016, 1651, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell. Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhao, L.; Wei, M.J.; Yao, W.F.; Zhao, H.S.; Chen, F.J. Neuroprotective effects of (−)-epigallocatechin-3-gallate on aging mice induced by D-galactose. Biol. Pharm. Bull. 2009, 32, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Mandel, S.; Youdim, M.B.H. Neuroprotective molecular mechanisms of (−)-epigallocatechin-3-gallate: A reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr. 2009, 4, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Bae, J.H.; Lee, S.R. Protective effect of green tea polyphenol EGCG against neuronal damage and brain edema after unilateral cerebral ischemia in gerbils. J. Neurosci. Res. 2004, 77, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H.; Mun, K.C.; Park, W.K.; Lee, S.R.; Suh, S.I.; Baek, W.K.; Yim, M.B.; Kwon, T.K.; Song, D.K. EGCG attenuates AMPA-induced intracellular calcium increase in hippocampal neurons. Biochem. Biophys. Res. Commun. 2002, 290, 1506–1512. [Google Scholar] [PubMed]
- Teixeira, M.D.; Souza, C.M.; Menezes, A.P.; Carmo, M.R.; Fonteles, A.A.; Gurgel, J.P.; Lima, F.A.; Viana, G.S.; Andrade, G.M. Catechin attenuates behavioral neurotoxicity induced by 6-OHDA in rats. Pharmacol. Biochem. Behav. 2013, 110, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.B.; Alexandre, B.D.S.; Neves, K.R.T.; Silva, A.H.; Leal, L.K.A.M.; Viana, G.S.B. Neuroprotective properties of the standardized extract from Camellia sinensis (green tea) and its main bioactive components, epicatechin and epigallocatechin gallate, in the 6-OHDA model of Parkinson’s disease. Evid. Based Complement. Alternat. Med. 2015, 2015, 161092. [Google Scholar] [CrossRef]
- Guo, S.H.; Bezard, E.; Zhao, B.L. Protective effect of green tea polyphenols on the SH-SY5Y cells against 6-OHDA induced apoptosis through ROS-NO pathway. Free Radical Biol. Med. 2005, 39, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, H.; Zhao, M.; Zhang, Z.; Li, Y. Chronic green tea catechins administration prevents oxidative stress-related brain aging in C57BL/6J mice. Brain Res. 2010, 1353, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.K.; Kim, J.M.; Jeong-Ja, O.; Jeon, B.S. Inhibition of inducible nitric oxide synthase expression and cell death by (−)-epigallocatechin-3-gallate, a green tea catechin, in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J. Clin. Neurosci. 2010, 17, 1165–1168. [Google Scholar]
- Sutherland, B.A.; Shaw, O.M.; Clarkson, A.N.; Jackson, D.N.; Sammut, I.A.; Appleton, I. Neuroprotective effects of (−)-epigallocatechin gallate following hypoxia-ischemia-induced brain damage: Novel mechanisms of action. FASEB 2005, 19, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.X.; Li, Y.B.; Zhao, R.P. Epigallocatechin gallate attenuates β-amyloid generation and oxidative stress involvement of PPARγ in N2a/APP695 cells. Neurochem. Res. 2017, 42, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J. Neurosci. 2001, 21, 2929–2938. [Google Scholar] [PubMed]
- Idris, I.; Gray, S.; Donnelly, R. Protein kinase C activation: isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia 2001, 44, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Musashi, M.; Ota, S.; Shiroshita, N. The role of protein kinase C isoforms in cell proliferation and apoptosis. Int. J. Hematol. 2000, 72, 12–19. [Google Scholar] [PubMed]
- Müller, J.M.; Krauss, B.; Kaltschmidt, C.; Baeuerle, P.A.; Rupec, R.A. Hypoxia induces c-fos transcription via a mitogen-activated protein kinase-dependent pathway. J. Biol. Chem. 1997, 272, 23435–23439. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, S.G.; Yuan, P.X.; Wang, M.; Vijayraghavan, S.; Bloom, A.K.; Davis, D.J.; Gobeske, K.T.; Sweatt, J.D.; Manji, H.K.; Arnsten, A.F.T. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science 2004, 306, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Vianna, M.R.M.; Barros, D.M.; Silva, T.; Choi, H.; Madche, C.; Rodrigues, C.; Medina, J.H.; Izquierdo, I. Pharmacological demonstration of the differential involvement of protein kinase c isoforms in short- and long-term memory formation and retrieval of one-trial avoidance in rats. Psychopharmacology 2000, 150, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Zheng, W.H.; Bastianetto, S.; Chabot, J.G.; Quirion, R. Neuroprotective effects of resveratrol against β-amyloid-induced neurotoxicity in rat hippocampal neurons: involvement of protein kinase C. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Pascale, A.; Jäntti, M.; Amadio, M.; Tuominen, R.K. Protein kinase C activation as a potential therapeutic strategy in Alzheimerߣs disease: Is there a role for embryonic lethal abnormal vision-like proteins? Basic Clin. Pharmacol. Toxicol. 2016, 119, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Pascale, A.; Amadio, M.; Govoni, S.; Battaini, F. The aging brain, a key target for the future: The protein kinase C involvement. Pharmacol. Res. 2007, 55, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Amit, T.S.; Mandel, S.; Youdim, M.B.H. Neuroprotection and neurorescue against a beta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (−)-epigallocatechin-3-gallate. FASEB 2003, 17, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Amit, T.; Youdim, M.B.H.; Mandel, S. Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol (−)-epigallocatechin 3-gallate neuroprotective action. J. Biol. Chem. 2002, 277, 30574–30580. [Google Scholar] [CrossRef] [PubMed]
- Kalfon, L.; Youdim, M.B.H.; Mandel, S.A. Green tea polyphenol (−)-epigallocatechin-3-gallate promotes the rapid protein kinase C- and proteasome-mediated degradation of Bad: implications for neuroprotection. J. Neurochem. 2010, 100, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Bastianetto, S.; Quirion, R. Neuroprotective effects of resveratrol and epigallocatechin gallate polyphenols are mediated by the activation of protein kinase C gamma. Front. Cell. Neurosci. 2013, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.L.; Liu, F.Q.; Jin, H.M.; Li, R.J.; Wang, Y.H.; Zhang, W.Q.; Wang, H.C.; Chen, W.Q. Involvement of PKCα and ERK1/2 signaling pathways in EGCG’s protection against stress-induced neural injuries in Wistar rats. Neuroscience 2017, 346, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, H.; Boyd, C.; Spencer, J.P.E.; Williams, R.J.; Cadenas, E.; Rice-Evans, C. MAPK signaling in neurodegeneration: influences of flavonoids and of nitric oxide. Neurobiol. Aging. 2002, 23, 861–880. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Stork, P.J.; Lazarovici, P.; Eiden, L.E. Signaling pathways for PC12 cell differentiation: Making the right connections. Science 2002, 296, 1648–1649. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. ActaMol. Basis Dis. 1802, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Barco, A.; Marie, H. Genetic approaches to investigate the role of CREB in neuronal plasticity and memory. Mol. Neurobiol. 2011, 44, 330–349. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, H.; Bahia, P.; Spencer, J.P.E.; Sheppard, O.; Rattray, M.; Cadenas, E.; Rice-Evans, C.; Williams, R.J. (−)epicatechin stimulates ERK-dependent cyclic AMP response element activity and up-regulates GluR2 in cortical neurons. J. Neurochem. 2007, 101, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, L.Y.; Zhang, H.Y.; Sun, H.Y.; Zhou, W.K. EGCG protective mitochondrial dysfunction after subarachnoid haemorrhage via inhibition p38 alpha pathway. J. Funct. Foods. 2016, 23, 115–123. [Google Scholar] [CrossRef]
- He, Q.Q.; Bao, L.; Zimering, J.; Zan, K.; Zhang, Z.; Shi, H.; Zu, J.; Yang, X.X.; Hua, F.; Ye, X.C. The protective role of (−)-epigallocatechin-3-gallate in thrombin-induced neuronal cell apoptosis and JNK-MAPK activation. Neuroreport 2015, 26, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Murugaiyah, V.; Mattson, M.P. Neurohormetic phytochemicals: An evolutionary–bioenergetic perspective. Neurochem. Int. 2015, 89, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discovery. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Vara, J.A.F.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/AKT signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Horwood, J.M.; Dufour, F.; Laroche, S.; Davis, S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/AKT cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 2006, 23, 3375–3384. [Google Scholar] [CrossRef] [PubMed]
- Saraceno, G.E.; Caceres, L.G.; Guelman, L.; Castilla, R.; Udovin, L.D.; Ellisman, M.H.; Brocco, M.A.; Capani, F. Consequences of excessive plasticity in the hippocampus induced by perinatal asphyxia. Exp. Neurol. 2016, 286, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wei, S.Y.; Yuan, L.; Kong, L.L.; Zhang, S.X.; Wang, Z.J.; Wu, M.N.; Qi, J.S. Davunetide improves spatial learning and memory in Alzheimer’s disease-associated rats. Physiol. Behav. 2017, 174, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Zhang, L.; Li, Y.; Cheng, Y.Q.; Zhu, X.; Zhang, F.; Yin, X.X. Activation of mTOR signaling mediates the increased expression of ache in high glucose condition: in vitro and in vivo evidences. Mol. Neurobiol. 2016, 53, 4972–4980. [Google Scholar] [CrossRef] [PubMed]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. AKT signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Cchaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.H.; Kim, S.H.; Kwon, H.; Park, Y.; Kim, K.S.; Song, C.W.; Kim, J.; Kim, M.H.; Yu, H.J.; Henkel, J.S. Epigallocatechin gallate protects nerve growth factor differentiated PC12 cells from oxidative-radical-stress-induced apoptosis through its effect on phosphoinositide 3-kinase/AKT and glycogen synthase kinase-3. Mol. Brain Res. 2003, 118, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.H.; Kim, S.H.; Kwon, H.; Kim, J.G.; Kim, J.H.; Yang, K.H.; Kim, J.; Kim, S.U.; Yu, H.J.; Do, B.R. Phosphatidylinositol-3 kinase/AKT and GSK-3 mediated cytoprotective effect of epigallocatechin gallate on oxidative stress-injured neuronal-differentiated N18D3 cells. Neurotoxicology 2004, 25, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.H.; Kwon, H.; Kim, K.S.; Kim, J.; Kim, M.H.; Yu, H.J.; Kim, M.; Lee, K.W.; Do, B.R.; Jung, H.K. Epigallocatechin gallate prevents oxidative-stress-induced death of mutant Cu/Zn-superoxide dismutase (G93A) motoneuron cells by alteration of cell survival and death signals. Toxicology 2004, 202, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-López, L.; Márquez-Valadez, B.; Gómez-Sánchez, A.; Silva-Lucero, M.D.C.; Torres-Pérez, M.; Téllez-Ballesteros, R.I.; Ichwan, M.; Meraz-Rios, M.A.; Kempermann, G.; Ramirez-Rodriguez, G.B. Green tea compound epigallo-catechin-3-gallate (EGCG) increases neuronal survival in adult hippocampal neurogenesis in vivo and in vitro. Neuroscience 2016, 322, 208–220. [Google Scholar]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Z.Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, J.; Rogers, J.; Xie, J.X. Brain iron metabolism dysfunction in Parkinson’s disease. Mol. Neurobiol. 2017, 54, 3078–3101. [Google Scholar] [CrossRef] [PubMed]
- Avramovichtirosh, Y.; Reznichenko, L.; Mit, T.; Zheng, H.; Fridkin, M.; Weinreb, O.; Mandel, S.; Youdim, M.B.H. Neurorescue activity, APP regulation and amyloid-beta peptide reduction by novel multi-functional brain permeable iron- chelating- antioxidants, m-30 and green tea polyphenol, EGCG. Curr. Alzheimer Res. 2007, 4, 403–411. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Maor, G.; Youdim, M.B.H. Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice. J. Mol. Neurosci. 2004, 24, 401–416. [Google Scholar] [CrossRef]
- Hung, V.W.S.; Bressan, L.P.; Seo, K.; Kerman, K. Electroanalysis of natural compounds as copper chelating agents for Alzheimer’s disease therapy. Electroanal 2016, 27, 2670–2678. [Google Scholar] [CrossRef]
- Mu, W.; Zhang, T.; Jiang, B. An overview of biological production of L-theanine. Biotechnol. Adv. 2015, 33, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Türközü, D.; Şanlier, N. L-theanine, unique amino acid of tea, and its metabolism, health effects, and safety. Crit. Rev. Food Sci. Nutr. 2017, 57, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Yokogoshi, H.; Kobayashi, M.; Mochizuki, M.; Terashima, T. Effect of theanine, r-glutamylethylamide, on brain monoamines and striatal dopamine release in conscious rats. Neurochem. Res. 1998, 23, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, A.; Umezawa, K.; Kobayashi, K.; Kawahara, M.; Muramoto, K.; Kakuda, T.; Kuroda, Y. Theanine, a major flavorous amino acid in green tea leaves, inhibits glutamate-induced neurotoxicity on cultured rat cerebral cortical neurons. Soc. Neurosci. Abstr. 1998, 24, 978. [Google Scholar]
- Tobaben, S.; Grohm, J.; Seiler, A.; Conrad, M.; Plesnila, N.; Culmsee, C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell. Death Differ. 2011, 18, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Deutschenbaur, L.; Beck, J.; Kiyhankhadiv, A.; Mühlhauser, M.; Borgwardt, S.; Walter, M.; Hasler, G.; Sollberger, D.; Lang, U.E. Role of calcium, glutamate and NMDA in major depression and therapeutic application. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2016, 64, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-mediated blood-brain barrier opening: Implications for neuroprotection and drug delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Myers, S.J.; Wells, G.; Nicholson, K.L.; Swanger, S.A.; Lyuboslavsky, P.; Tahirovic, Y.A.; Menaldino, D.S.; Ganesh, T.; Wilson, L.J.; et al. Context-dependent GluN2B-selective inhibitors of NMDA receptor function are neuroprotective with minimal side effects. Neuron 2015, 85, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, C.; Numakawa, T.; Ninomiya, M.; Chiba, S.; Kunugi, H. Behavioral and molecular evidence for psychotropic effects in L-theanine. Psychopharmacology 2012, 219, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, T. Neuroprotective effects of theanine and its preventive effects on cognitive dysfunction. Pharmacol. Res. 2011, 64, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, T.; Nozawa, A.; Sugimoto, A.; Niino, H. Inhibition by theanine of binding of [3H]AMPA, [3H]kainate, and [3H]MDL 105,519 to glutamate receptors. Biosci. Biotechnol. Biochem. 2002, 66, 2683–2686. [Google Scholar] [CrossRef] [PubMed]
- Zukhurova, M.; Prosvirnina, M.; Daineko, A.; Simanenkova, A.; Petrishchev, N.; Sonin, D.; Galagudza, M.; Shamtsyan, M.; Juneja, L.R.; Vlasov, T. L-theanine administration results in neuroprotection and prevents glutamate receptor agonist-mediated injury in the rat model of cerebral ischemia-reperfusion. Phytother. Res. 2013, 27, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Dulla, C.G.; Farzampour, Z.; Taylor-Weiner, A.; Huguenard, J.R.; Reimer, R.J. A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 2014, 81, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, T.; Hinoi, E.; Abe, A.; Nozawa, A.; Ogura, M.; Yoneda, Y. Theanine, an ingredient of green tea, inhibits [3H]glutamine transport in neurons and astroglia in rat brain. J. Neurosci. Res. 2008, 86, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Kakuda, T.; Takarada, T.; Nakamichi, N.; Fukumori, R.; Kim, Y.H.; Hinoi, E.; Yoneda, Y. Promotion of both proliferation and neuronal differentiation in pluripotent P19 cells with stable overexpression of the glutamine transporter slc38a1. PLoS ONE 2012, 7, e48270. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Ogura, M.; Nakamichi, N.; Kakuda, T.; Nakazato, R.; Kokubo, H.; Ikeno, S.; Nakamura, S.; Kutsukake, T.; Hinoi, E.; et al. Upregulation of slc38a1 gene along with promotion of neurosphere growth and subsequent neuronal specification in undifferentiated neural progenitor cells exposed to theanine. Neurochem. Res. 2016, 41, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Sadzuka, Y. Theanine, a specific glutamate derivative in green tea, reduces the adverse reactions of doxorubicin by changing the glutathione level. Cancer Lett. 2004, 212, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Ben, P.; Zhang, Z.P.; Xuan, X.C.; Sun, S.S.; Shen, L.; Gao, Y.H.; Cao, X.; Zhou, Y.; Lan, L.; Yin, Z.M.; et al. Protective effect of L-theanine on cadmium-induced apoptosis in PC12 cells by inhibiting the mitochondria-mediated pathway. Neurochem. Res. 2015, 40, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, M.; Miyazaki, I.; Murakami, S.; Kita, T.; Asanuma, M. L-theanine protects against excess dopamine-induced neurotoxicity in the presence of astrocytes. J. Clin. Biochem. Nutr. 2016, 59, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Kumar, P. L-theanine, a component of green tea prevents 3-nitropropionic acid (3-NP)-induced striatal toxicity by modulating nitric oxide pathway. Mol. Neurobiol. 2017, 54, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Bermejo, D.; Reglero, G.; Fornari, T. Recent advances in the processing of green tea biomolecules using ethyl lactate. A review. Trends Food Sci. Technol. 2017, 62, 1–12. [Google Scholar] [CrossRef]
- McLellan, T.M.; Caldwell, J.A.; Lieberman, H.R. A review of caffeine’s effects on cognitive, physical and occupational performance. Neurosci. Biobehav. Rev. 2016, 71, 294–312. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathologic overproduction: The bad side of adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Kolahdouzan, M.; Hamadeh, M.J. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Delic, V.; Cao, C.; Copes, N.; Lin, X.; Mamcarz, M.; Wang, L.; Arendash, G.W.; Bradshaw, P.C. Caffeine increases mitochondrial function and blocks melatonin signaling to mitochondria in Alzheimer's mice and cells. Neuropharmacology 2012, 63, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Cane, M.C.; Mukherjee, R.; Szatmary, P.; Zhang, X.; Elliott, V.; Ouyang, Y.L.; Chvanov, M.; Latawiec, D.; Wen, L.; et al. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut 2017, 66, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.A.; Sebastiao, A.M. Caffeine and adenosine. J. Alzheimer’s Dis. 2010, 20, S3–S15. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Andriolo, V.; Castorina, A. Dual blockade of the A(1) and A(2A) adenosine receptor prevents amyloid beta toxicity in neuroblastoma cells exposed to aluminum chloride. Int. J. Biochem. Cell. Biol. 2014, 54, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.F.; Chang, W.H.; Black, R.M.; Liu, J.R.; Sompol, P.; Chen, Y.M.; Wei, H.L.; Zhao, Q.Y.; Cheng, I.H. Crude caffeine reduces memory impairment and amyloid β(1–42) levels in an Alzheimer’s mouse model. Food Chem. 2012, 135, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Santosrodrigues, A.D.; Schwarzschild, M.A.; Chen, J.F.; Cunha, R.A.; Agostinho, P. Adenosine A(2A) receptors modulate glutamate uptake in cultured astrocytes and gliosomes. GLIA 2012, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.A.; Zhao, Y.; Ning, Y.L.; Yang, N.; Peng, Y.; Li, P.; Chen, X.Y.; Liu, D.; Wang, H.; Chen, X. Adenosine A(2A) receptor inactivation alleviates early-onset cognitive dysfunction after traumatic brain injury involving an inhibition of tau hyperphosphorylation. Transl. Psychiatry. 2017, 7, e1123. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.V.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Goncalves, F.Q.; Grosjean, N. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A(2A) receptors. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van der Jeugd, A. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry. 2016, 21, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Muller, C.E.; Buee, L. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging. 2014, 35, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.D.; Goldberg, J.A.; Surmeier, D.J. Adenosine A2a receptor antagonists attenuate striatal adaptations following dopamine depletion. Neurobiol. Dis. 2012, 45, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.G.; Batalha, V.L.; Vicente, M.H.; Coelho, J.E.; Gomes, R.; Gonçalves, F.Q.; Real, J.I.; Rino, J.; Albino-Teixeira, A.; Cunha, R.A. Adenosine A(2A) receptors modulate alpha-synuclein aggregation and toxicity. Cereb. Cortex 2015, 27, 718–730. [Google Scholar]
- Xu, K.; Di, L.D.; Orrú, M.; Xu, Y.; Chen, J.F.; Schwarzschild, M.A. Neuroprotection by caffeine in the MPTP model of Parkinson’s disease and its dependence on adenosine A(2A) receptors. Neuroscience 2016, 322, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Machado, J.A.; Correia, A.O.; Montenegro, A.B.A.; Nobre, M.E.P.; Cerqueira, G.S.; Neves, K.R.T.; Naffah-Mazzacoratti, M.D.; Cavalheiro, E.A.; Brito, G.A.D.; Viana, G.S.D. Caffeine neuroprotective effects on 6-OHDA-lesioned rats are mediated by several factors, including pro-inflammatory cytokines and histone deacetylase inhibitions. Behav. Brain Res. 2014, 264, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Nobre, H.V.; Cunha, G.M.D.; de Vasconcelos, L.M.; Magalhaes, H.I.F.; Neto, R.N.O.; Maia, F.D.; de Moraes, M.O.; Leal, L.K.A.M.; Viana, G.S.D. Caffeine and CSC, adenosine A(2A) antagonists, offer neuroprotection against 6-OHDA-induced neurotoxicity in rat mesencephalic cells. Neurochem. Int. 2010, 56, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.S.; Bjorklund, N.; Taglialatela, G. NMNAT2: HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016, 14, e1002472. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Bradley, G.; Lu, H.C. Corrigendum: Screening with an NMNAT2-MSD platform identifies small molecules that modulate NMNAT2 levels in cortical neurons. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.L.; Su, Y.; Chen, R.; Zhang, Z.; Huang, Y.; Chen, Z.Y. Theaflavins in black tea and catechins in green tea are equally effective antioxidants. J. Nutr. 2001, 131, 2248–2251. [Google Scholar]
- Miller, N.J.; Castelluccio, C.; Tijburg, L.; Rice-Evans, C. The antioxidant properties of theaflavins and their gallate esters—Radical scavengers or metal chelators? FEBS Lett. 1996, 392, 40–44. [Google Scholar] [CrossRef]
- Ben Lagha, A.; Grenier, D. Black tea theaflavins attenuate Porphyromonas gingivalis virulence properties, modulate gingival keratinocyte tight junction integrity and exert anti-inflammatory activity. J. Periodontal Res. 2017, 52, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Greaves, P.; Cooke, D.N.; Edwards, R.; Steward, W.P.; Gescher, A.J.; Marczylo, T.H. Breast cancer prevention by green tea catechins and black tea theaflavins in the C3(1) SV40 T,t antigen transgenic mouse model is accompanied by increased apoptosis and a decrease in oxidative DNA adducts. J. Agric. Food Chem. 2007, 55, 3378–3385. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Yue, Q.; Zhang, D.D.; Li, H.; Yang, S.Q.; Gao, W.Y. Antimicrobial mechanism of theaflavins: They target 1-deoxy-D-xylulose 5-phosphate reductoisomerase, the key enzyme of the MEP terpenoid biosynthetic pathway. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Lu, H.; Lu, H.; He, Y.X.; Niu, J.K.; Debnath, A.K.; Wu, S.G.; Jiang, S.B. Theaflavin derivatives in black tea and catechin derivatives in green tea inhibit HIV-1 entry by targeting gp41. Biochim. Biophys. Acta 2005, 1723, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Grelle, G.; Otto, A.; Lorenz, M.; Frank, R.F.; Wanker, E.E.; Bieschke, J. Black tea theaflavins inhibit formation of toxic amyloid-beta and alpha-synuclein fibrils. Biochemistry 2011, 50, 10624–10636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, S.X.; Li, J.; Xiong, L.G.; Tian, L.L.; Liu, J.J.; Huang, J.N.; Liu, Z.H. Neuroprotective effects of theaflavins against oxidative stress-induced apoptosis in PC12 cells. Neurochem. Res. 2016, 41, 3364–3372. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zhao, Y.; Wang, Y.; Yang, X.; Zhao, B. Protective effect of theaflavins on neuron against 6-hydroxydopamine-induced apoptosis in SH-SY5Y cells. J. Clin. Biochem. Nutr. 2012, 50, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Tamilselvam, K.; Radhiga, T.; Rao, S.; Essa, M.M.; Manivasagam, T. Theaflavin, a black tea polyphenol, protects nigral dopaminergic neurons against chronic MPTP/probenecid induced Parkinson’s disease. Brain Res. 2012, 1433, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Dodd, F.L.; Kennedy, D.O.; Riby, L.M.; Haskellramsay, C.F. A double-blind, placebo-controlled study evaluating the effects of caffeine and l-theanine both alone and in combination on cerebral blood flow, cognition and mood. Psychopharmacology 2015, 232, 2563–2576. [Google Scholar] [CrossRef] [PubMed]
- Haskell, C.F.; Kennedy, D.O.; Milne, A.L.; Wesnes, K.A.; Scholey, A.B. The effects of L-theanine, caffeine and their combination on cognition and mood. Biol. Psychology. 2008, 77, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Oh, J.H.; Yoo, H.S.; Lee, Y.M.; Lee, M.K.; Hong, J.T.; Oh, K.W. (−)-Epigallocatechin-3-O-gallate (EGCG) reverses caffeine-induced anxiogenic-like effects. Neurosci. Lett. 2010, 481, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (−)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J. Neurochem. 2004, 88, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Utsunomiya, K.; Kimbara, A.; Fukushima, K.; Mori, T.; Shiba, J. Preventive effect of green tea containing theanine at a high concentration on dementia in aged volunteers. J. Jpn. Mibyo. Syst. Assoc. 2009, 15, 17–23. [Google Scholar]
- Pool, H.; Quintanar, D.; Figueroa, J.D.D.; Mano, C.M.; Bechara, J.E.H.; Mendoza, S. Antioxidant effects of quercetin and catechin encapsulated into PLGA nanoparticles. J. Nanomater. 2012, 2012. [Google Scholar] [CrossRef]
- Forester, S.C.; Lambert, J.D. The catechol-O-methyltransferase inhibitor, tolcapone, increases the bioavailability of unmethylated (-)-epigallocatechin-3-gallate in mice. J. Funct. Foods. 2015, 17, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hamagami, H.; Kumazoe, M.; Yamaguchi, Y.; Fuse, S.; Tachibana, H.; Tanaka, H. 6-Azido-6-deoxy-l-idose as a hetero-bifunctional spacer for the synthesis of azido-containing chemical probes. Chemistry 2016, 22, 12884–12890. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; De, R.; Begun, J.; Popat, A. Cancer therapeutics with epigallocatechin-3-gallate encapsulated in biopolymeric nanoparticles. Int. J. Pharm. 2017, 518, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.Q.; Li, Q.S.; Xiang, L.P.; Liang, Y.R. Recent Advances in Volatiles of Teas. Molecules 2016, 21, 338. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Reference | Study Design | Location | Study Sample | Main Results |
|---|---|---|---|---|
| Tomata et al. [14] | Cohort study | Japan | 5.7-year follow-up (13,645 participants aged 65 years or more) | Green tea consumption related to reduced dementia incidence, with interval (CI) (0.61–0.87) |
| Eskelinen et al. [21] | Population-based cohort study | North Karelia | 1409 participants (71%) aged 65 to 79 years | Midlife tea consumption was not related to AD or dementia, while midlife coffee related to decreased AD or dementia risk |
| Kandinov et al. [15] | Population Cardiovascular Risk Factors, Aging, and Dementia (CAIDE) study | Tel Aviv Sourasky Medical Center, UK | 278 patients who noticed first motor symptoms of PD(4 groups, aged <50, 51–60, 61–70, >70 years) | Tea consumption >3 cups/day delayed PD onset by 7.7 years |
| Ng et al. [16] | Cross-sectional study | Singapore | 2398 adults aged ≥ 55 years | Regular drinking of tea, especially oolong tea or black tea, was related to better functional and physical performance |
| Feng et al. [17] | Cross-sectional study | Singapore | 716 Chinese adults aged ≥ 55 years | Tea consumption was conductive to cognitive performance |
| Kuriyama et al. [18] | Cross-sectional study | Tsurugaya, Japan | 1003 Japanese aged ≥70 years | Higher level green tea consumption was related to lower cognitive impairment |
| Ma et al. [19] | Meta-analysis | Asia, Europe, Australia, and USA | 52, 503 participants aged ≥50 years | Tea drinking could decrease the attack of cognitive decline, cognitive impairment, and mild cognitive impairment in elderly |
| Qi and Li [20] | Meta-analysis | Asia, USA | Tea consumption(34,4895 participants), caffeine consumption (49, 2 724 participants)aged 30–85 years | Tea consumption could decrease PD risk with a linear relation, and the consumption increment of caffeine (200 mg/day) and tea (2 cups/day) decreased PD risk by 17% and 26%, respectively |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-Q.; Wang, Z.-S.; Ma, Y.-X.; Zhang, W.; Lu, J.-L.; Liang, Y.-R.; Zheng, X.-Q. Neuroprotective Effects and Mechanisms of Tea Bioactive Components in Neurodegenerative Diseases. Molecules 2018, 23, 512. https://doi.org/10.3390/molecules23030512
Chen S-Q, Wang Z-S, Ma Y-X, Zhang W, Lu J-L, Liang Y-R, Zheng X-Q. Neuroprotective Effects and Mechanisms of Tea Bioactive Components in Neurodegenerative Diseases. Molecules. 2018; 23(3):512. https://doi.org/10.3390/molecules23030512
Chicago/Turabian StyleChen, Shu-Qing, Ze-Shi Wang, Yi-Xiao Ma, Wei Zhang, Jian-Liang Lu, Yue-Rong Liang, and Xin-Qiang Zheng. 2018. "Neuroprotective Effects and Mechanisms of Tea Bioactive Components in Neurodegenerative Diseases" Molecules 23, no. 3: 512. https://doi.org/10.3390/molecules23030512
APA StyleChen, S.-Q., Wang, Z.-S., Ma, Y.-X., Zhang, W., Lu, J.-L., Liang, Y.-R., & Zheng, X.-Q. (2018). Neuroprotective Effects and Mechanisms of Tea Bioactive Components in Neurodegenerative Diseases. Molecules, 23(3), 512. https://doi.org/10.3390/molecules23030512