Enantiomeric Mixtures in Natural Product Chemistry: Separation and Absolute Configuration Assignment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
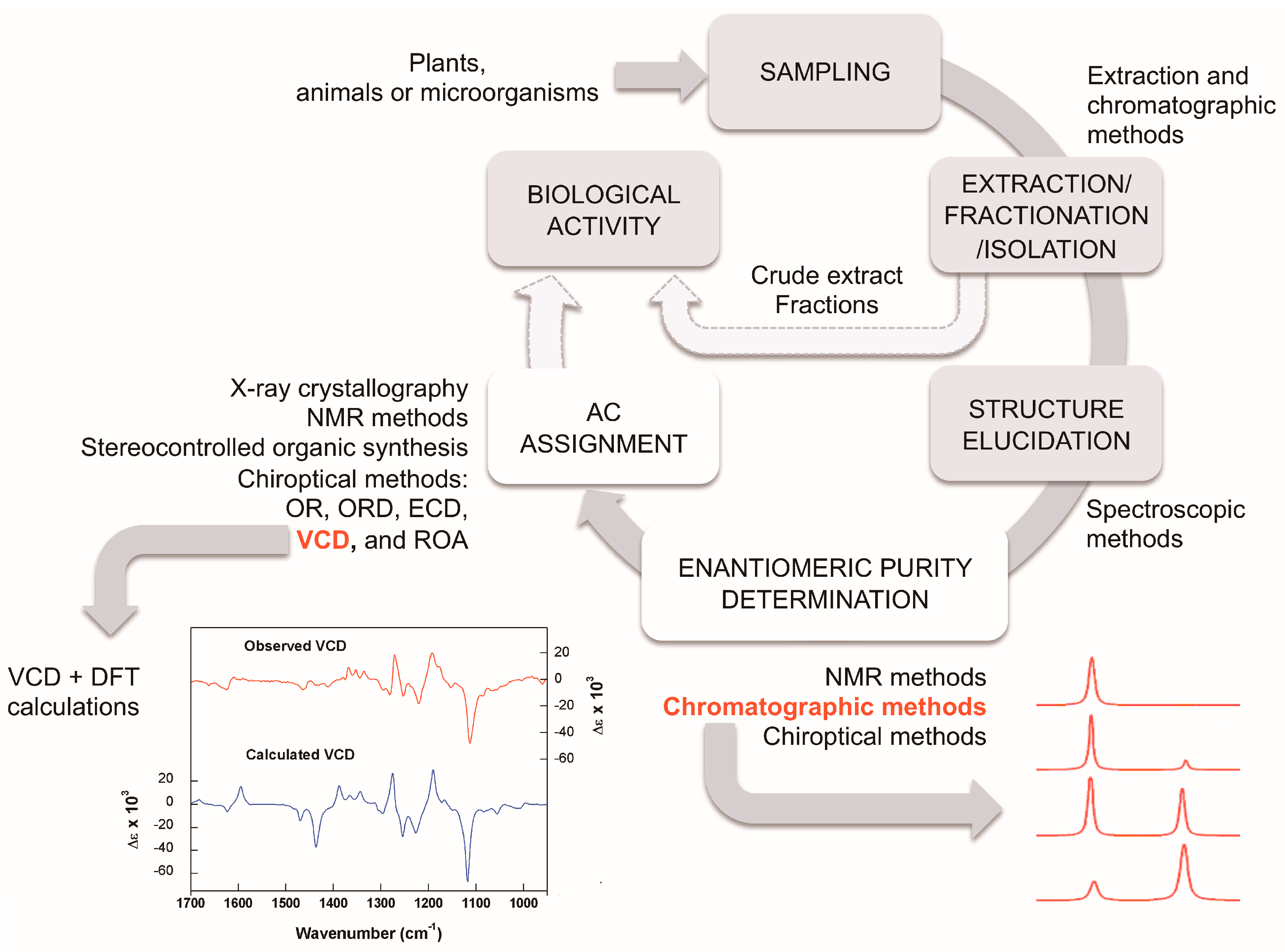
1. Introduction
2. Tutorial
2.1. Enantioselective Chromatography
2.1.1. The most used CSPs
- Polysaccharide-based CSPs
- Macrocyclic antibiotics CSPs
- Pirkle type CSPs
- Cyclodextrin-based CSPs
2.1.2. Screening Development
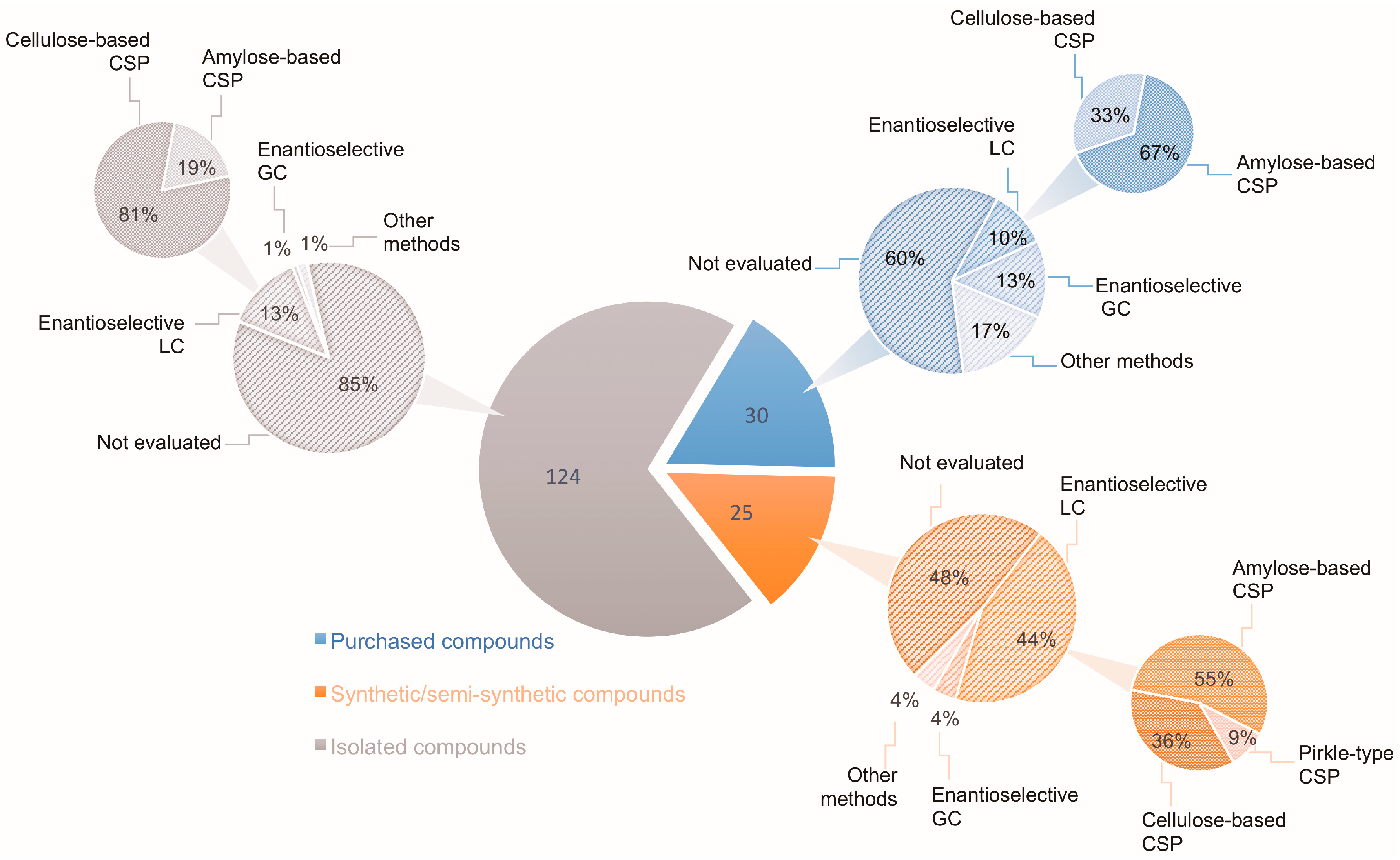
- Polysaccharide-based CSPs are the ones with the broadest enantiomeric discrimination abilities and are complementary to the macrocyclic antibiotics CSPs. For their screening, it is important to explore multimodal elution, thus, one must be aware of solvent miscibility. 100% ethanol (EtOH) is an excellent choice for changing from normal to polar or reverse-phase modes. As a rule, one should mind the instructions provided by the manufacturer.
- For the screening of polysaccharide-based CSPs, it is better to start by exploring the normal elution mode with different proportions of alkane (hexane or heptane) to EtOH or 2-propanol. The amylose- and cellulose-tris-3,5-dimethylphenylcarbamate derivative CSPs are the ones with the highest success rates [67]. Initially, the polysaccharide coated CSPs were commercialized only by the Daicel; currently, they are commercialized also by other companies with different brand names [71]. With the immobilized CSPs one can explore eluents such as mixtures of alkane with ethyl acetate and tetrahydrofuran (THF) before going to the polar organic mode.
- The polar organic mode also gives high selectivity and it is usually a good choice for multi-milligram separations. It is important to give time for equilibration, especially for the coated polysaccharide-based CSPs. To avoid immiscibility problems with the eluents, one should go first to 100% EtOH. Methanol (MeOH) or acetonitrile (ACN) are the solvents of choice, or mixtures of them [55,84].
- Reversed elution mode should also be explored. This is usually done by using acetonitrile or methanol as modifiers in water. The use of buffer solutions is necessary only if the analytes require [19].
- It is recommended to start the screening with the Chirobiotic® CSPs at polar organic mode for nonionizable enantiomers, or at the polar ionic mode for the ionizable ones. In the first case, ACN is usually used with a small amount of a protic organic solvent, while in the polar ionic mode acid or base is added to MeOH [42]. The manufacturer recommends MeOH with acid and base in proportions 4/1 to 1/4, respectively.
- The Chirobiotic® CSPs operate well under the reversed mode with MeOH, ACN, and THF as modifiers. The use of an aqueous buffer solution, such as ammonium acetate, is a good selection. The pH can affect drastically the enantioselectivity and should therefore be explored.
- The normal elution mode is carried out with alkane and 2-propanol or ethanol. The use of acidic or basic additives for Chirobiotic® CSPs under normal elution is also important [57].
2.2. Chiroptical Methods and Absolute Configuration Assignment
2.2.1. VCD Measurements
2.2.2. Calculations of Theoretical VCD Spectra
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Glossary
- Enantiomer excess (enantiomeric excess): For a mixture of (+)- and (−)-enantiomers, with composition given as the mole or weight fractions F(+) and F(−) (where F(+) + F(−) = 1) the enantiomer excess is defined as | F(+) − F(−) | (and the percent enantiomer excess by 100 | F(+) − F(−) |). Frequently this term is abbreviated as e.e.
- Enantiomeric purity: See: enantiomer excess
- Enantiomeric ratio: The ratio of the percentage of one enantiomer in a mixture to that of the other e.g., 70 (+):30 (−).
- Enantiomerically enriched: A sample of a chiral substance whose enantiomeric ratio is greater than 50:50 but less than 100:0.
- Enantiomerically pure: A sample for which all of its molecules have (within the limits of detection) the same chirality sense. Use of homochiral as a synonym is strongly discouraged.
- Racemate: An equimolar mixture of a pair of enantiomers. It does not exhibit optical activity. The chemical name or formula of a racemate is distinguished from those of the enantiomers by the prefix (±)- or rac- (or racem-) or by the symbols RS and SR.
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Moore, B.S.; Yoon, Y.J. Reinvigorating natural product combinatorial biosynthesis with synthetic biology. Nat. Chem. Biol. 2015, 11, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Gardner, D.R.; Chang, C.W.; Panter, K.E.; Molyneux, R.J. Separation and measurement of plant alkaloid enantiomers by RP-HPLC analysis of their Fmoc-Alanine analogs. Phytochem. Anal. 2008, 19, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Colegate, S.M.; Molyneux, R.J. Bioactive Natural Products: Detection, Isolation, and Structural Determination, 2nd ed.; Taylor and Francis: Boca Raton, FL, USA, 2008; pp. 1–605, ISBN-13 978-0849372582. [Google Scholar]
- Miller, K.A.; Tsukamoto, S.; Williams, R.M. Asymmetric total syntheses of (+)- and (−)-versicolamide b and biosynthetic implications. Nat. Chem. 2009, 1, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Bioactive natural products and chirality. Chirality 2011, 23, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Mane, S. Racemic drug resolution: A comprehensive guide. Anal. Methods 2016, 8, 87567–87586. [Google Scholar] [CrossRef]
- Zask, A.; Ellestad, G. Biomimetic syntheses of racemic natural products. Chirality 2017. [Google Scholar] [CrossRef] [PubMed]
- Marín-Sáez, J.; Romero-González, R.; Frenich, A.G. Enantiomeric determination and evaluation of the racemization process of atropine in Solanaceae seeds and contaminated samples by high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2016, 1474, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Vistoli, G.; Pedretti, A. Mechanisms and pharmaceutical consequences of processes of stereoisomerisation—A didactic excursion. Eur. J. Pharm. Sci. 2016, 88, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Batista, J.M., Jr.; Blanch, E.W.; Bolzani, V.S. Recent advances in the use of vibrational chiroptical spectroscopic methods for stereochemical characterization of natural products. Nat. Prod. Rep. 2015, 32, 1280–1302. [Google Scholar] [CrossRef] [PubMed]
- Finefield, J.M.; Sherman, D.H.; Kreitman, M.; Williams, R.M. Enantiomeric natural products: Occurrence and biogenesis. Angew. Chem. Int. Ed. Engl. 2012, 51, 4802–4836. [Google Scholar] [CrossRef] [PubMed]
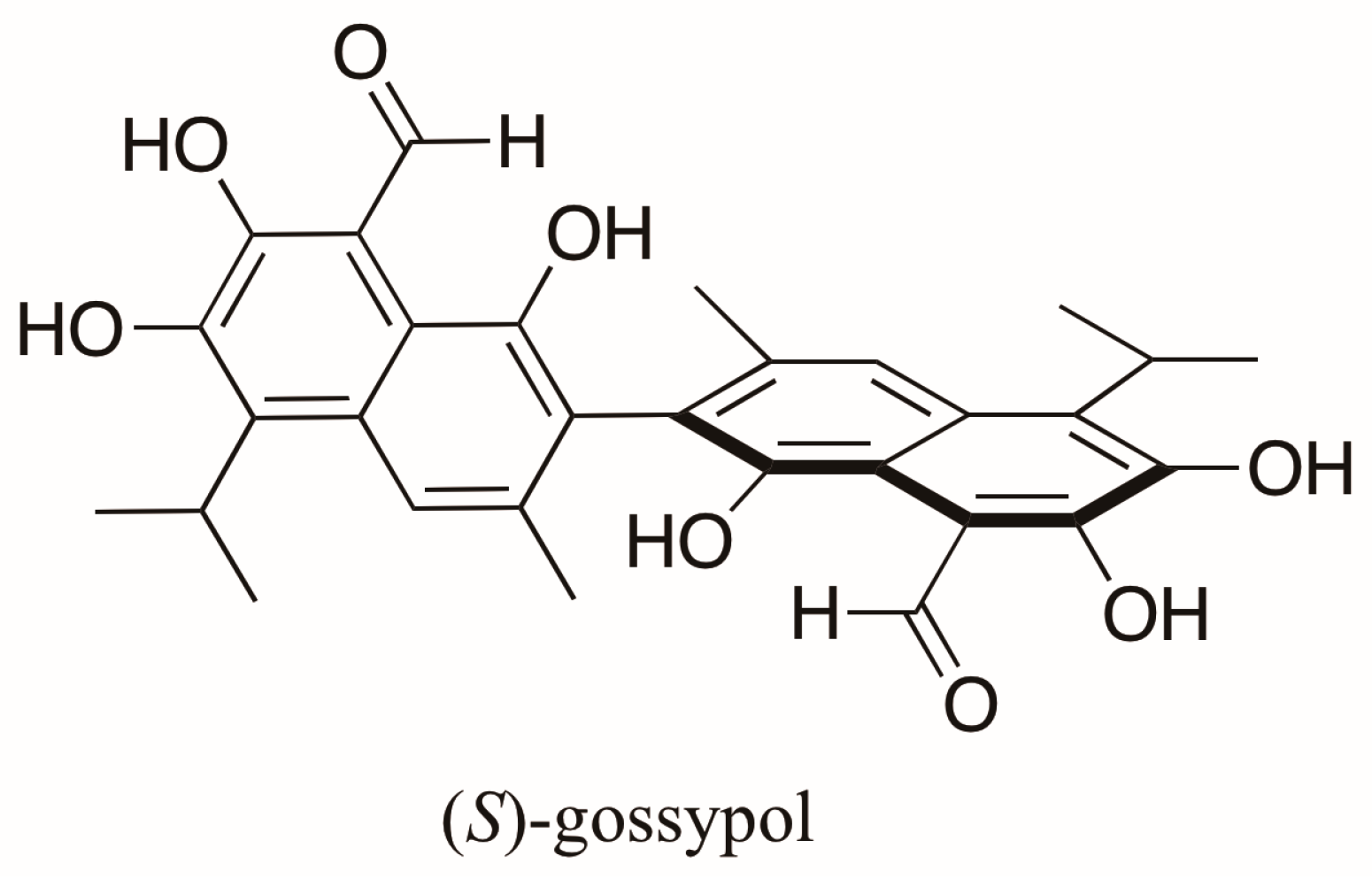
- Freedman, T.B.; Cao, X.; Oliveira, R.V.; Cass, Q.B.; Nafie, L.A. Determination of the absolute configuration and solution conformation of gossypol by vibrational circular dichroism. Chirality 2003, 15, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Keshmiri-Neghab, H.; Goliaei, B. Therapeutic potential of gossypol: An overview. Pharm. Biol. 2014, 52, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.E.A.; Matlin, S.A.; Knox, P. Cytotoxicity of enantiomers of gossypol. Br. J. Cancer 1986, 54, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Kenar, J.A. Reaction chemistry of gossypol and its derivatives. J. Am. Oil Chem. Soc. 2006, 83, 269–302. [Google Scholar] [CrossRef]
- Cass, Q.B.; Tiritan, E.; Matlint, S.A.; Freire, E.C. Gossypol enantiomer ratios in cotton seeds. Phytochemistry 1991, 30, 2655–2657. [Google Scholar] [CrossRef]
- Cass, Q.B.; Bassi, A.L.; Matlin, S.A. First direct resolution of gossypol enantiomers on a chiral high-performance liquid chromatography phase. Chirality 1999, 11, 46–49. [Google Scholar] [CrossRef]
- Cass, Q.B.; Oliveira, R.V.; De Pietro, A.C. Determination of gossypol enantiomer ratio in cotton plants by chiral higher-performance liquid chromatography. J. Agric. Food Chem. 2004, 52, 5822–5827. [Google Scholar] [CrossRef] [PubMed]
- Mota, J.S.; Leite, A.C.; Batista, J.M., Jr.; López, S.N.; Ambrósio, D.L.; Passerini, G.D.; Kato, M.J.; Bolzani, V.S.; Cicarelli, R.M.B.; Furlan, M. In vitro trypanocidal activity of phenolic derivatives from Peperomia obtusifolia. Planta Medica 2009, 75, 620–623. [Google Scholar] [CrossRef] [PubMed]
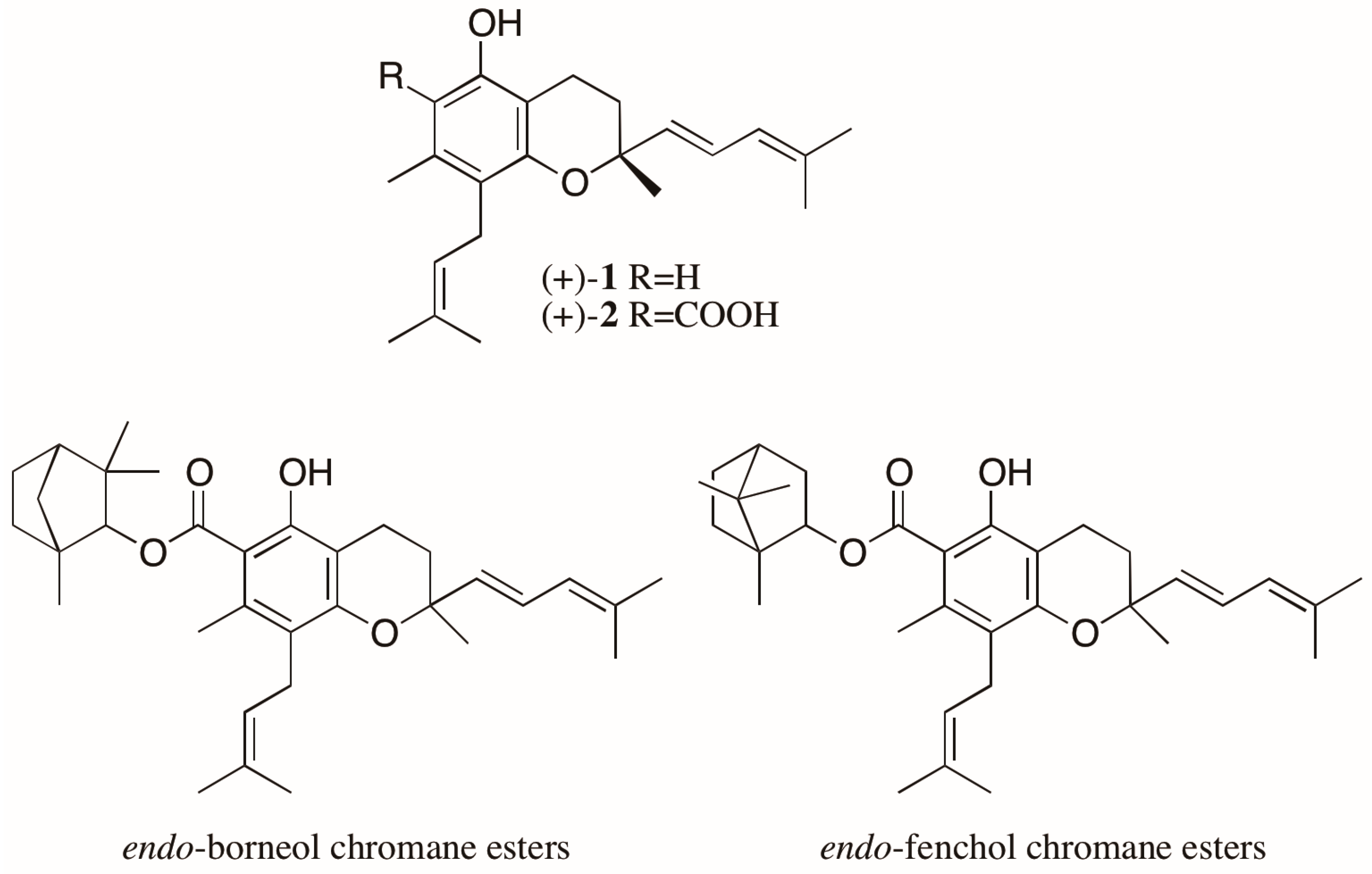
- Batista, J.M., Jr.; Batista, A.N.L.; Mota, J.S.; Cass, Q.B.; Kato, M.J.; Bolzani, V.S.; Freedman, T.B.; López, S.N.; Furlan, M.; Nafie, L.A. Structure elucidation and absolute stereochemistry of isomeric monoterpene chromane esters. J. Org. Chem. 2011, 76, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Batista, J.M., Jr.; Batista, A.N.L.; Kato, M.J.; Bolzani, V.S.; López, S.N.; Nafie, L.A.; Furlan, M. Further monoterpene chromane esters from Peperomia obtusifolia: VCD determination of the absolute configuration of a new diastereomeric mixture. Tetrahedron Lett. 2012, 53, 6051–6054. [Google Scholar] [CrossRef]
- Batista, J.M., Jr.; Batista, A.N.L.; Rinaldo, D.; Vilegas, W.; Ambrósio, D.L.; Cicarelli, R.M.B.; Bolzani, V.S.; Kato, M.J.; Nafie, L.A.; López, S.N.; et al. Absolute configuration and selective trypanocidal activity of gaudichaudianic acid enantiomers. J. Nat. Prod. 2011, 74, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.-J.; Zhang, H.-J.; Li, Q.; Li, Z.-H.; Zhang, Z.-H.; Wu, L.-Q.; Li, L.; Qin, H.-L. Six scalemic mixtures of 6-monosubstituted dihydrobenzophenanthridine alkaloids from Chelidonium majus and optically active structures of enantiomers. Phytochemistry 2017, 144, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Tshitenge, D.T.; Feineis, D.; Awale, S.; Bringmann, G. Gardenifolins A–H, scalemic neolignans from Gardenia ternifolia: Chiral resolution, configurational assignment, and cytotoxic activities against the HeLa cancer cell line. J. Nat. Prod. 2017, 80, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Allenmark, S.G. Chiroptical methods in the stereochemical analysis of natural products. Nat. Prod. Rep. 2000, 17, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, C.; Bang, S.H.; Ma, J.Y.; Kim, S.; Koh, Y.-S.; Shim, S.H. Isochromans and related constituents from the endophytic fungus Annulohypoxylon truncatum of Zizania caduciflora and their anti-inflammatory effects. J. Nat. Prod. 2017, 80, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-L.; Shen, C.-C.; Shen, Y.-C.; Chiou, W.-F.; Chen, C.-C. Anti-inflammatory and antiosteoporosis flavonoids from the rhizomes of Helminthostachys zeylanica. J. Nat. Prod. 2017, 80, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Fois, B.; Bianco, G.; Sonar, V.P.; Distinto, S.; Maccioni, E.; Meleddu, R.; Melis, C.; Marras, L.; Pompei, R.; Floris, C.; et al. Phenylpropenoids from Bupleurum fruticosum as anti-human rhinovirus species A selective capsid binders. J. Nat. Prod. 2017, 80, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wu, Y.; Dalal, S.; Merino, E.F.; Liu, Q.-F.; Xu, C.-H.; Yuan, T.; Ding, J.; Kingston, D.G.I.; Cassera, M.B.; et al. Nanomolar antimalarial agents against chloroquine-resistant Plasmodium falciparum from medicinal plants and their structure-activity relationships. J. Nat. Prod. 2017, 80, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-L.; Hao, Z.-Y.; Liu, Y.-F.; Wang, Y.; Shi, G.-R.; Jiang, Z.-B.; Chen, R.-Y.; Cao, Z.-Y.; Yu, D.-Q. Polycycloiridals with a cyclopentane ring from Iris tectorum. J. Nat. Prod. 2017, 80, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Joyce, L.A.; Nawrat, C.C.; Sherer, E.C.; Biba, M.; Brunskill, A.; Martin, G.E.; Cohen, R.D.; Davies, I.W. Beyond optical rotation: What’s left is not always right in total synthesis. Chem. Sci. 2018, 9, 415–424. [Google Scholar] [CrossRef]
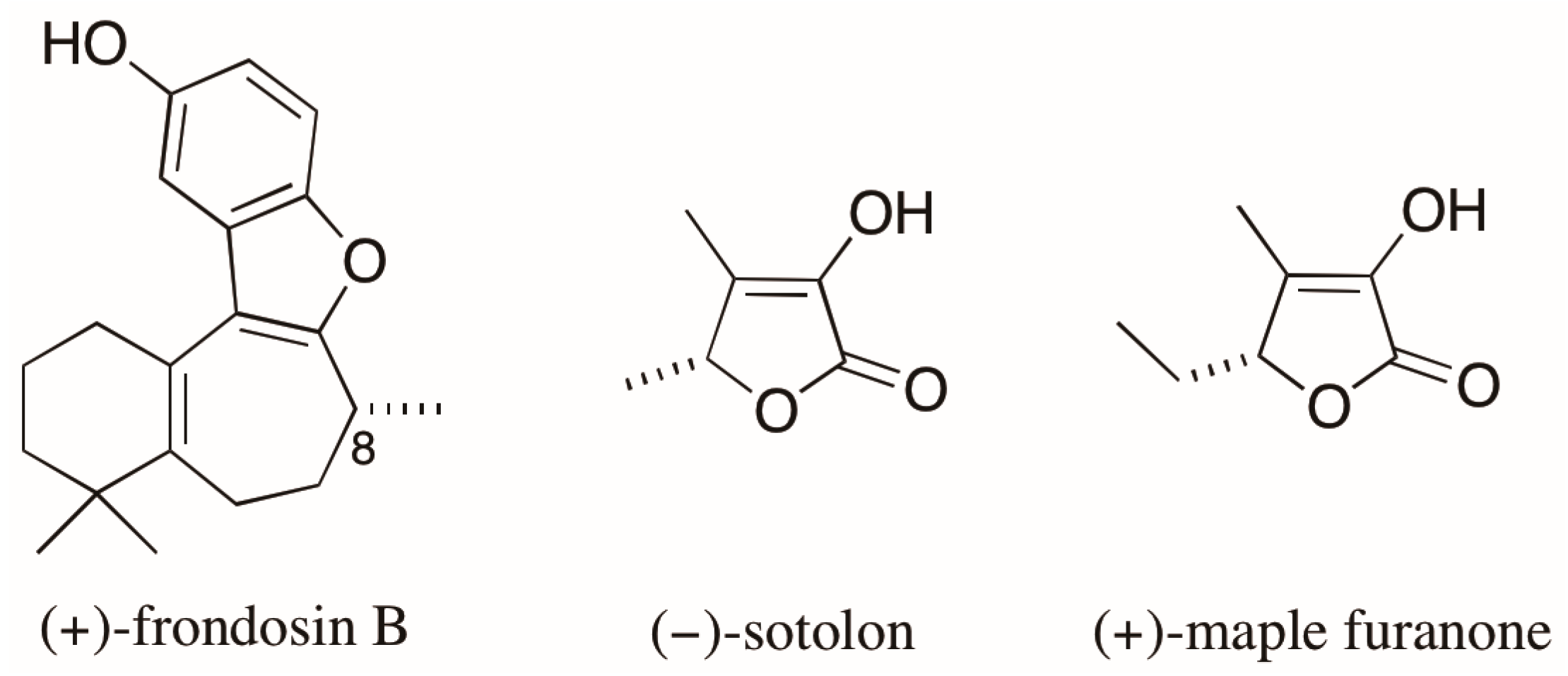
- Nakahashi, A.; Yaguchi, Y.; Miura, N.; Emura, M.; Monde, K. A vibrational circular dichroism approach to the determination of the absolute configurations of flavorous 5-substituted-2(5H)-furanones. J. Nat. Prod. 2011, 74, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Schurig, V. Terms for the quantitation of a mixture of stereoisomers. Top. Curr. Chem. 2013, 340, 21–40. [Google Scholar] [PubMed]
- Ribeiro, A.R.; Maia, A.S.; Cass, Q.B.; Tiritan, M.E. Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: an overview. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 968, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Scriba, G.K.E. Chiral recognition in separation science—An update. J. Chromatogr. A 2016, 1467, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Tiritan, M.E.; Pinto, M. Small molecules as chromatographic tools for HPLC enantiomeric resolution: Pirkle-type chiral stationary phases evolution. Chromatographia 2013, 76, 871–897. [Google Scholar] [CrossRef]
- Lourenco, T.D.; Cassiano, N.M.; Cass, Q.B. Chiral stationary phases for high-performance liquid chromatography. Quim. Nova 2010, 33, 2155–2164. [Google Scholar]
- Ward, T.J.; Ward, K.D. Chiral separations: A review of current topics and trends. Anal. Chem. 2012, 84, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Berthod, A. Chiral recognition mechanisms. Anal. Chem. 2006, 78, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Berthod, A. Chiral recognition mechanisms with macrocyclic glycopeptide selectors. Chirality 2009, 21, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Cavazzini, A.; Pasti, L.; Massi, A.; Marchetti, N.; Dondi, F. Recent applications in chiral high performance liquid chromatography: A review. Anal. Chim. Acta 2011, 706, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Wainer, I.W. Proposal for the classification of high-performance liquid-chromatographic chiral stationary phases—How to choose the right column. Trends Anal. Chem. 1987, 6, 125–134. [Google Scholar] [CrossRef]
- Wainer, I.W. HPLC chiral stationary phases for the stereochemical resolution of enantiomeric compounds. The current state of the art. Clin. Pharmacol. 1993, 18, 139–182. [Google Scholar]
- Lough, W.J. Classification of LC chiral stationary phases: Wainer Types I-V revisited. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 968, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, J.C.; Lourenco, T.C.; Silva, L.M.; Venancio, T.; Cass, Q.B. High resolution magic angle spinning NMR as a tool for unveiling the molecular enantiorecognition of omeprazole by amylose-based chiral phase. Analyst 2014, 139, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, J.C.; Paixao, M.W.; Lourenco, T.C.; Cass, Q.B.; Venancio, T. A high-resolution magic angle spinning NMR study of the enantiodiscrimination of 3,4-methylenedioxymethamphetamine (MDMA) by an immobilized polysaccharide-based chiral phase. PLoS ONE 2016, 11, e0162892. [Google Scholar] [CrossRef] [PubMed]
- Cass, Q.B.; Batigalhia, F. Enantiomeric resolution of a series of chiral sulfoxides by high-performance liquid chromatography on polysaccharide-based columns with multimodal elution. J. Chromatogr. A 2003, 987, 445–452. [Google Scholar] [CrossRef]
- Sheridan, R.P.; Piras, P.; Sherer, E.C.; Roussel, C.; Pirkle, W.H.; Welch, C.J. Mining chromatographic enantioseparation data using matched molecular pair analysis. Molecules 2016, 21, E1297. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.; Schafer, W.; Piras, P.; Zawatzky, K.; Sherer, E.C.; Roussel, C.; Welch, C.J. Toward structure-based predictive tools for the selection of chiral stationary phases for the chromatographic separation of enantiomers. J. Chromatogr. A 2016, 1467, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Montanari, C.A.; Cass, Q.B.; Tiritan, M.E.; Souza, A.L.S. A QSERR study on enantioselective separation of enantiomeric sulphoxides. Anal. Chim. Acta 2000, 419, 93–100. [Google Scholar] [CrossRef]
- Montanari, M.L.C.; Cass, Q.B.; Andricopulo, A.D.; Leitao, A.; Montanari, C.A. Identification of chiral selectors for improved enantioseparation based on molecular interaction fields. Anal. Chim. Acta 2005, 545, 33–45. [Google Scholar] [CrossRef]
- Belaz, K.R.A.; Cass, Q.B.; Oliveira, R.V. Determination of albendazole metabolites by direct injection of bovine plasma and multidimensional achiral-chiral high performance liquid chromatography. Talanta 2008, 76, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Sousa, E.P.; Tiritan, M.E.; Oliveira, R.V.; Afonso, C.M.; Cass, Q.B.; Pinto, M.M. Enantiomeric resolution of kielcorin derivatives by HPLC on polysaccharide stationary phases using multimodal elution. Chirality 2004, 16, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; Zhang, T. Common approaches for efficient method development with immobilised polysaccharide-derived chiral stationary phases. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 875, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Tiritan, M.E.; Cass, Q.; Kairys, V.; Fernandes, M.X.; Pinto, M. Enantioseparation and chiral recognition mechanism of new chiral derivatives of xanthones on macrocyclic antibiotic stationary phases. J. Chromatogr. A 2012, 1241, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.A.; Ates, H.; Mangelings, D.; Heyden, Y.V. A separation strategy combining three HPLC modes and polysaccharide-based chiral stationary phases. J. Pharm. Biomed. Anal. 2013, 75, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.J. Microscale chiral HPLC in support of pharmaceutical process research. Chirality 2009, 21, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.C.; Wahab, M.F.; Armstrong, D.W.; Breitbach, Z.S. Advances in high-throughput and high-efficiency chiral liquid chromatographic separations. J. Chromatogr. A 2016, 1467, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Barhate, C.L.; Joyce, L.A.; Makarov, A.A.; Zawatzky, K.; Bernardoni, F.; Schafer, W.A.; Armstrong, D.W.; Welch, C.J.; Regalado, E.L. Ultrafast chiral separations for high throughput enantiopurity analysis. Chem. Commun. 2017, 53, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, M.; Kagan, H.B. Recent advances in the measurement of enantiomeric excesses. Adv. Synth. Catal. 2002, 344, 453–463. [Google Scholar] [CrossRef]
- Gao, R.Q.; Fan, J.; Tan, Q.; Guo, D.; Chen, T.; He, R.J.; Li, D.; Zhang, H.; Zhang, W.G. Reliable HPLC separation, vibrational circular dichroism spectra, and absolute configurations of isoborneol enantiomers. Chirality 2017, 29, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Cretin, B.N.; Dubourdieu, D.; Marchal, A. Development of a quantitation method to assay both lyoniresinol enantiomers in wines, spirits, and oak wood by liquid chromatography-high resolution mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kawashima, M.; Hatada, K. Chromatographic resolution. 7. Useful chiral packing materials for high-performance liquid chromatographic resolution of enantiomers: Phenylcarbamates of polysaccharides coated on silica gel. J. Am. Chem. Soc. 1984, 106, 5357–5359. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kawashima, M.; Yamamoto, K.; Hatada, K. Chromatographic resolution. 6. Useful chiral packing materials for high-performance liquid chromatographic resolution. Cellulose triacetate and tribenzoate coated on macroporous silica gel. Chem. Lett. 1984, 13, 739–742. [Google Scholar] [CrossRef]
- Shen, J.; Ikai, T.; Okamoto, Y. Synthesis and application of immobilized polysaccharide-based chiral stationary phases for enantioseparation by high-performance liquid chromatography. J. Chromatogr. A 2014, 1363, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Cass, Q.B.; Degani, A.L.G.; Cassiano, N.M. Effects on enantioselectivity by the use of polysaccharide-based columns by multimodal elution. J. Liq. Chromatogr. Relat. Technol. 2003, 26, 2083–2101. [Google Scholar] [CrossRef]
- Zhang, T.; Franco, P. Analytical and preparative potential of immobilized polysaccharide-derived chiral stationary phases. Chiral separation techniques. In Chiral Separation Techniques: A Practical Approach, 3rd ed.; Subramanian, G., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 99–134. [Google Scholar]
- Zhang, T.; Franco, P.; Nguyen, D.; Hamasaki, R.; Miyamoto, S.; Ohnishi, A.; Murakami, T. Complementary enantiorecognition patterns and specific method optimization aspects nn immobilized polysaccharide-derived chiral stationary phases. J. Chromatogr. A 2012, 1269, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Chankvetadze, B. Recent developments on polysaccharide-based chiral stationary phases for liquid-phase separation of enantiomers. J. Chromatogr. A 2012, 1269, 26–51. [Google Scholar] [CrossRef] [PubMed]
- Bicchi, C.; Cagliero, C.; Liberto, E.; Sgorbini, B.; Martina, K.; Cravotto, G.; Rubiolo, P. New asymmetrical per-substituted cyclodextrins (2-O-methyl-3-O-ethyl- and 2-O-ethyl-3-O-methyl-6-O-t-butyldimethylsilyl-beta-derivatives) as chiral selectors for enantioselective gas chromatography in the flavour and fragrance field. J. Chromatogr. A 2010, 1217, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, Y.; Liu, J.; Yao, G.; Li, D.; Sun, B.; Zhang, J.; Liu, Y.; Qi, C.; Xiang, M.; et al. (±)-Acortatarinowins A–F, norlignan, neolignan, and lignan enantiomers from Acorus tatarinowii. J. Nat. Prod. 2015, 78, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.W.; Tang, Y.; Chen, S.; Zhou, Y.; Bagwill, C.; Chen, J.-R. Macrocyclic antibiotics as a new class of chiral selectors for liquid chromatography. Anal. Chem. 1994, 66, 1473–1484. [Google Scholar] [CrossRef]
- Berthod, A.; Liu, Y.; Bagwill, C.; Armstrong, D.W. Facile LC enantioresolution of native amino acids and peptides using a teicoplanin chiral stationary phase. J. Chromatogr. A 1996, 731, 123–137. [Google Scholar] [CrossRef]
- Prompanya, C.; Fernandes, C.; Cravo, S.; Pinto, M.M.; Dethoup, T.; Silva, A.M.; Kijjoa, A. A new cyclic hexapeptide and a new isocoumarin derivative from the marine sponge-associated fungus Aspergillus similanensis KUFA 0013. Mar. Drugs 2015, 13, 1432–1450. [Google Scholar] [CrossRef] [PubMed]
- Pirkle, W.H.; Welch, C.J.; Lamm, B. Design, synthesis, and evaluation of an improved enantioselective naproxen selector. J. Org. Chem. 1992, 57, 3854–3860. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Welch, C.J. An improved chiral stationary phase for the chromatographic separation of underivatized naproxen enantiomers. J. Liq. Chromatogr. 1992, 15, 1947–1955. [Google Scholar] [CrossRef]
- Fernandes, C.; Palmeira, A.; Santos, A.; Tiritan, M.E.; Afonso, C.; Pinto, M.M. Enantioresolution of chiral derivatives of xanthones on (S,S)-Whelk-O1 and l-phenylglycine stationary phases and chiral recognition mechanism by docking approach for (S,S)-Whelk-O1. Chirality 2013, 25, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Badaloni, E.; Cabri, W.; Ciogli, A.; D’Acquarica, I.; Deias, R.; Gasparrini, F.; Giorgi, F.; Kotoni, D.; Villani, C. Extending the use of “inverted chirality columns approach” for enantiomeric excess determination in absence of reference samples: application to a water-soluble camptothecin derivative. J. Chromatogr. A 2010, 1217, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccanti, G.; Ismail, O.H.; D’Acquarica, I.; Villani, C.; Manzo, C.; Wilcox, M.; Cavazzini, A.; Gasparrini, F. Cannabis through the looking glass: chemo- and enantio-selective separation of phytocannabinoids by enantioselective ultra high performance supercritical fluid chromatography. Chem. Commun. 2017, 53, 12262–12265. [Google Scholar] [CrossRef] [PubMed]
- Si-Ahmed, K.; Tazerouti, F.; Badjah-Hadj-Ahmed, A.Y.; Aturki, Z.; D’Orazio, G.; Rocco, A.; Fanali, S. Analysis of hesperetin enantiomers in human urine after ingestion of blood orange juice by using nano-liquid chromatography. J. Pharm. Biomed. Anal. 2010, 51, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S. A strategy for developing HPLC methods for chiral drugs. LCGC N. Am. 2007, 25, 1112–1128. [Google Scholar]
- Scatena, G.S.; Cassiano, N.M.; Netto, C.D.; Costa, P.R.R.; Cass, Q.B.; Batista, J.M. Preparative chiral separation and absolute configuration of the synthetic pterocarpanquinone LQB-118. Chirality 2017, 29, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.; Kang, S.O.; Anslyn, E.V. Rapid determination of enantiomeric excess: A focus on optical approaches. Chem. Soc. Rev. 2012, 41, 448–479. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.M.; Edmonds, R.B.; Rauh, R.D.; Carrabba, M.M. Analytical determination of enantiomeric purity using Raman optical activity. Anal. Chem. 1994, 66, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Brittain, H.G. Applications of chiroptical spectroscopy in the characterization of compounds having pharmaceutical importance. In Circular Dichroism: Principles and Applications; Berova, N., Nakanishi, K., Woody, R.W., Eds.; John Wiley and Sons: New York, NY, USA, 2000; pp. 819–844. [Google Scholar]
- Polavarapu, P.L. Optical rotation: recent advances in determining the absolute configuration. Chirality 2002, 14, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Shah, R.D.; Dukor, R.K.; Cao, X.; Freedman, T.B.; Nafie, L.A. Determination of enantiomeric excess in samples of chiral molecules using fourier transform vibrational circular dichroism spectroscopy: Simulation of real-time reaction monitoring. Anal. Chem. 2005, 76, 6956–6966. [Google Scholar] [CrossRef] [PubMed]
- Kott, L.; Petrovic, J.; Phelps, D.; Roginski, R.; Schubert, J. Determination of a low-level percent enantiomer of a compound with no ultraviolet chromophore using vibrational circular dichroism (VCD): Enantiomeric purity by VCD of a compound with three chiral centers. Appl. Spectrosc. 2014, 68, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Tranter, G.E.; Le Pevelen, D.D. Chiroptical spectroscopy and the validation of crystal structure stereochemical assignments. Tetrahedron Asymmetry 2017, 28, 1192–1198. [Google Scholar] [CrossRef]
- Bijvoet, J.M.; Peerdeman, A.F.; Van Bommel, A.J. Determination of the absolute configuration of optically active compounds by means of X-rays. Nature 1951, 168, 271–272. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. The use of X-ray crystallography to determine absolute configuration. Chirality 2008, 20, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Harada, N. Determination of absolute configurations by X-ray crystallography and 1H NMR anisotropy. Chirality 2008, 20, 691–723. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S. Determination of absolute configuration using X-ray diffraction. Tetrahedron Asymmetry 2017, 28, 1304–1313. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Chisholm, C.D. Assignment of absolute configuration using chiral reagents and NMR spectroscopy. Chirality 2011, 23, 190–214. [Google Scholar] [CrossRef] [PubMed]
- Sadlej, J.; Dobrowolski, J.C.; Rode, J.E. VCD Spectroscopy as a novel probe for chirality transfer in molecular interactions. Chem. Soc. Rev. 2010, 39, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Polavarapu, P.L. Renaissance in chiroptical spectroscopic methods for molecular structure determination. Chem. Rec. 2007, 7, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.D. Ab initio calculation of molecular chiroptical properties. Theor. Chem. Acc. 2006, 115, 227–245. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Nafie, L.A.; McConnel, O.; Minick, D.; Kellenbach, E.; He, Y.; Wang, B.; Dukor, R.K.; Bartberger, M.D.; Pappa, H.N. Vibrational circular dichroism as a new technology for determining the absolute configuration, conformation, and enantiomeric purity of chiral pharmaceutical ingredients. Pharmacopeial Forum 2013, 39, 311–452. [Google Scholar]
- Joseph-Nathan, P.; Gordillo-Román, B. Vibrational Circular Dichroism Absolute Configuration Determination of Natural Products in Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Falk, H., Kobayashi, J., Eds.; Springer: Basel, Switzerland, 2015; Volume 100, pp. 311–451. [Google Scholar]
- Taniguchi, T.; Monde, K. Exciton chirality method in vibrational circular dichroism. J. Am. Chem. Soc. 2012, 134, 3695–3698. [Google Scholar] [CrossRef] [PubMed]
- Passareli, F.; Batista, A.N.L.; Cavalheiro, A.J.; Herrebout, W.A.; Batista, J.M., Jr. Vibrational spectroscopy as a direct stereochemical probe for polyhydroxylated molecules. Phys. Chem. Chem. Phys. 2016, 18, 30903–30906. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.A.; Nafie, L.A. Observation and calculation of vibrational circular birefringence: A new form of vibrational optical activity. Chirality 2009, 21, E277–E286. [Google Scholar] [CrossRef] [PubMed]
- Merten, C. Vibrational optical activity as probe for intermolecular interactions. Phys. Chem. Chem. Phys. 2017, 19, 18803–18812. [Google Scholar] [CrossRef] [PubMed]
- Batista, J.M., Jr.; Batista, A.N.L.; Rinaldo, D.; Vilegas, W.; Cass, Q.B.; Bolzani, V.S.; Kato, M.J.; López, S.N.; Furlan, M.; Nafie, L.A. Absolute configuration reassignment of two chromanes from Peperomia obtusifolia (Piperaceae) using VCD and DFT calculations. Tetrahedron Asymmetry 2010, 21, 2402–2407. [Google Scholar] [CrossRef]
- Ccana-Ccapatinta, G.V.; Sampaio, B.L.; Santos, F.M., Jr.; Batista, J.M., Jr.; Costa, F.B. Absolute configuration assignment of caffeic acid ester derivatives from Tithonia diversifolia by vibrational circular dichroism: The pitfalls of deuteration. Tetrahedron Asymmetry 2017, 28, 1823–1828. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Pan, J.-J. The Determination of the absolute configurations of chiral molecules using vibrational circular dichroism (VCD) spectroscopy. Chirality 2008, 20, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhu, C.; Reiling, S.; Vaz, R. A novel computational method for comparing vibrational circular dichroism spectra. Spectrochim. Acta Part A 2010, 76, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Debie, E.; De Gussem, E.; Dukor, R.K.; Herrebout, W.; Nafie, L.A.; Bultinck, P. A confidence level algorithm for the determination of absolute configuration using vibrational circular dichroism or Raman optical activity. ChemPhysChem 2011, 12, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Covington, C.L.; Polavarapu, P.L. Similarity in dissymmetry factor spectra: A quantitative measure of comparison between experimental and predicted vibrational circular dichroism. J. Phys. Chem. A 2013, 117, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; (the “Gold Book”); McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997; ISBN 0-9678550-9-8. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
N. L. Batista, A.; M. dos Santos, F.; Batista, J.M.; Cass, Q.B. Enantiomeric Mixtures in Natural Product Chemistry: Separation and Absolute Configuration Assignment. Molecules 2018, 23, 492. https://doi.org/10.3390/molecules23020492
N. L. Batista A, M. dos Santos F, Batista JM, Cass QB. Enantiomeric Mixtures in Natural Product Chemistry: Separation and Absolute Configuration Assignment. Molecules. 2018; 23(2):492. https://doi.org/10.3390/molecules23020492
Chicago/Turabian StyleN. L. Batista, Andrea, Fernando M. dos Santos, João M. Batista, and Quezia B. Cass. 2018. "Enantiomeric Mixtures in Natural Product Chemistry: Separation and Absolute Configuration Assignment" Molecules 23, no. 2: 492. https://doi.org/10.3390/molecules23020492
APA StyleN. L. Batista, A., M. dos Santos, F., Batista, J. M., & Cass, Q. B. (2018). Enantiomeric Mixtures in Natural Product Chemistry: Separation and Absolute Configuration Assignment. Molecules, 23(2), 492. https://doi.org/10.3390/molecules23020492