Asymmetric Synthesis of Spirooxindoles via Nucleophilic Epoxidation Promoted by Bifunctional Organocatalysts

 , ,
, ,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
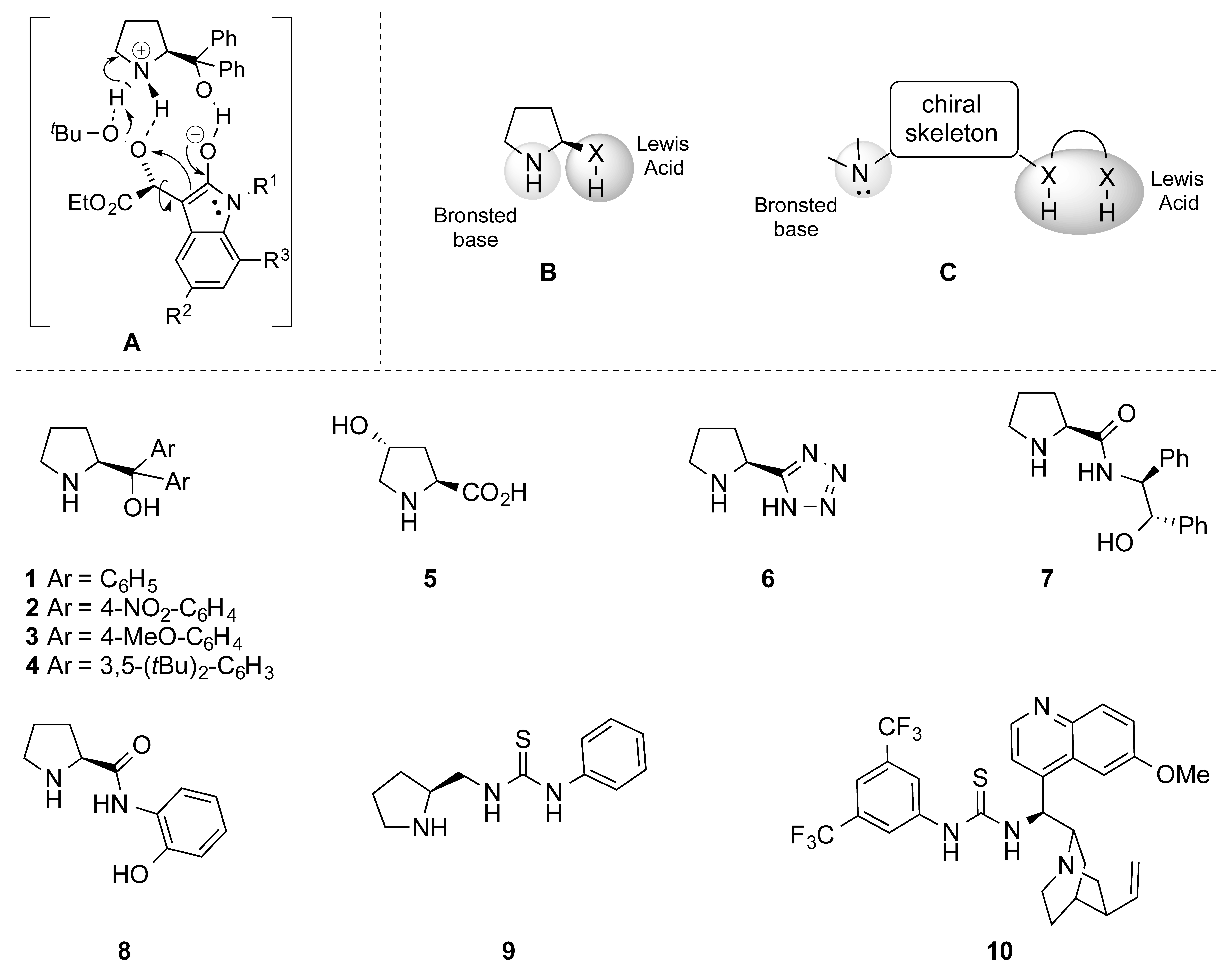
2.1. Evaluation of the Catalyst Modification
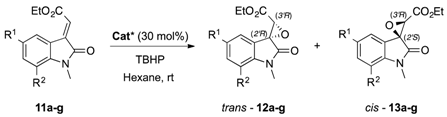
2.2. Evaluation of the Substrate Scope
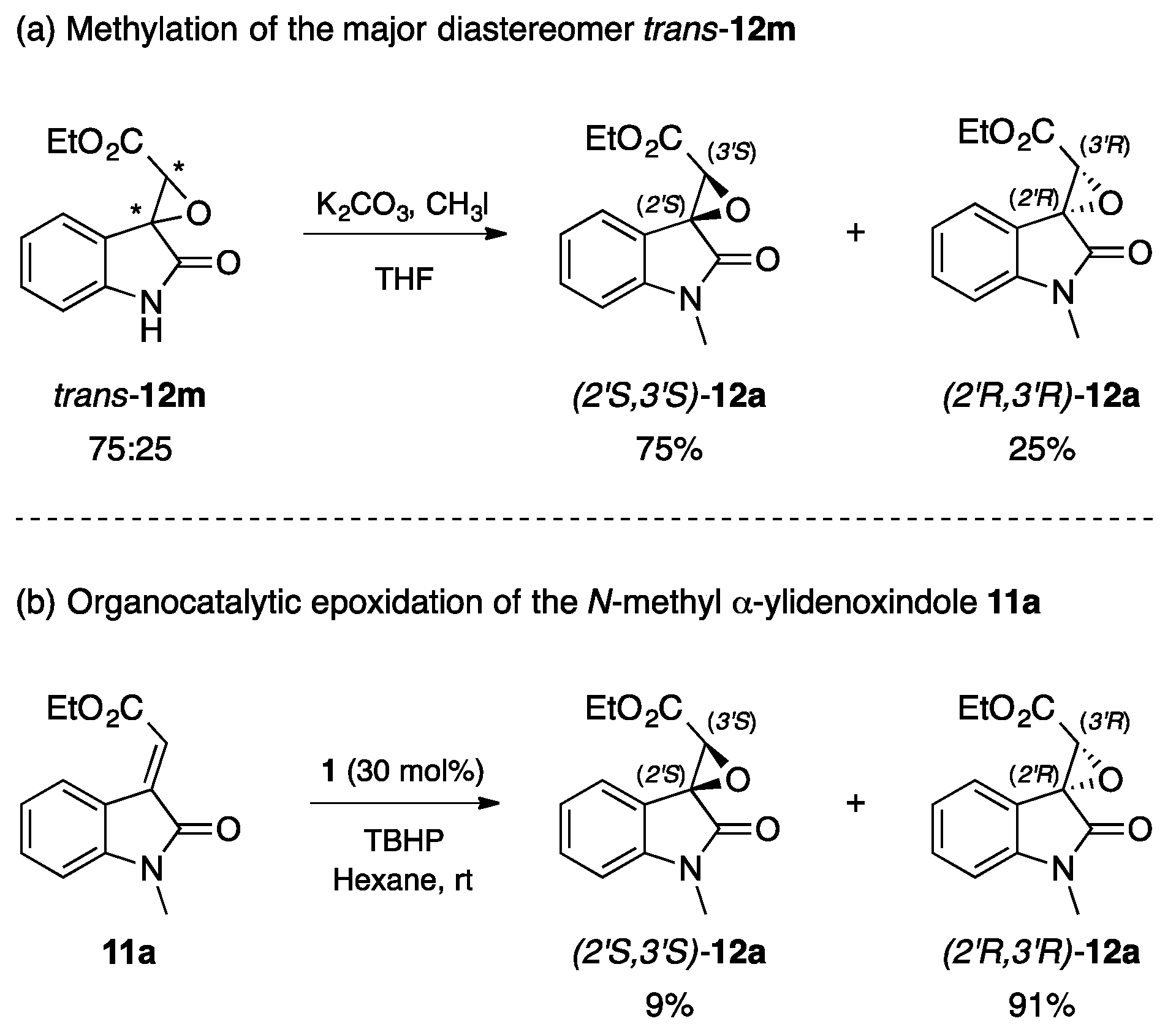
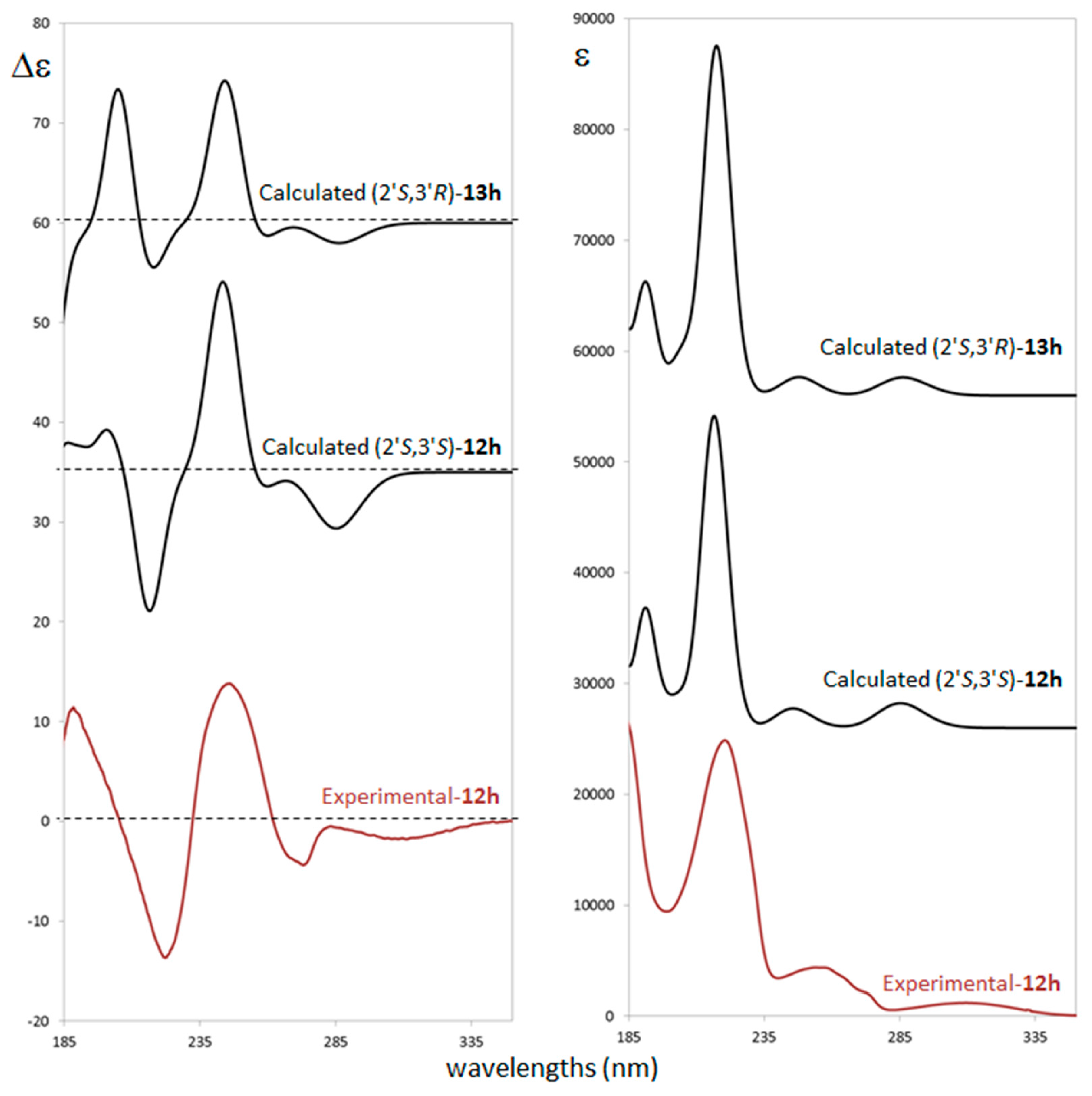
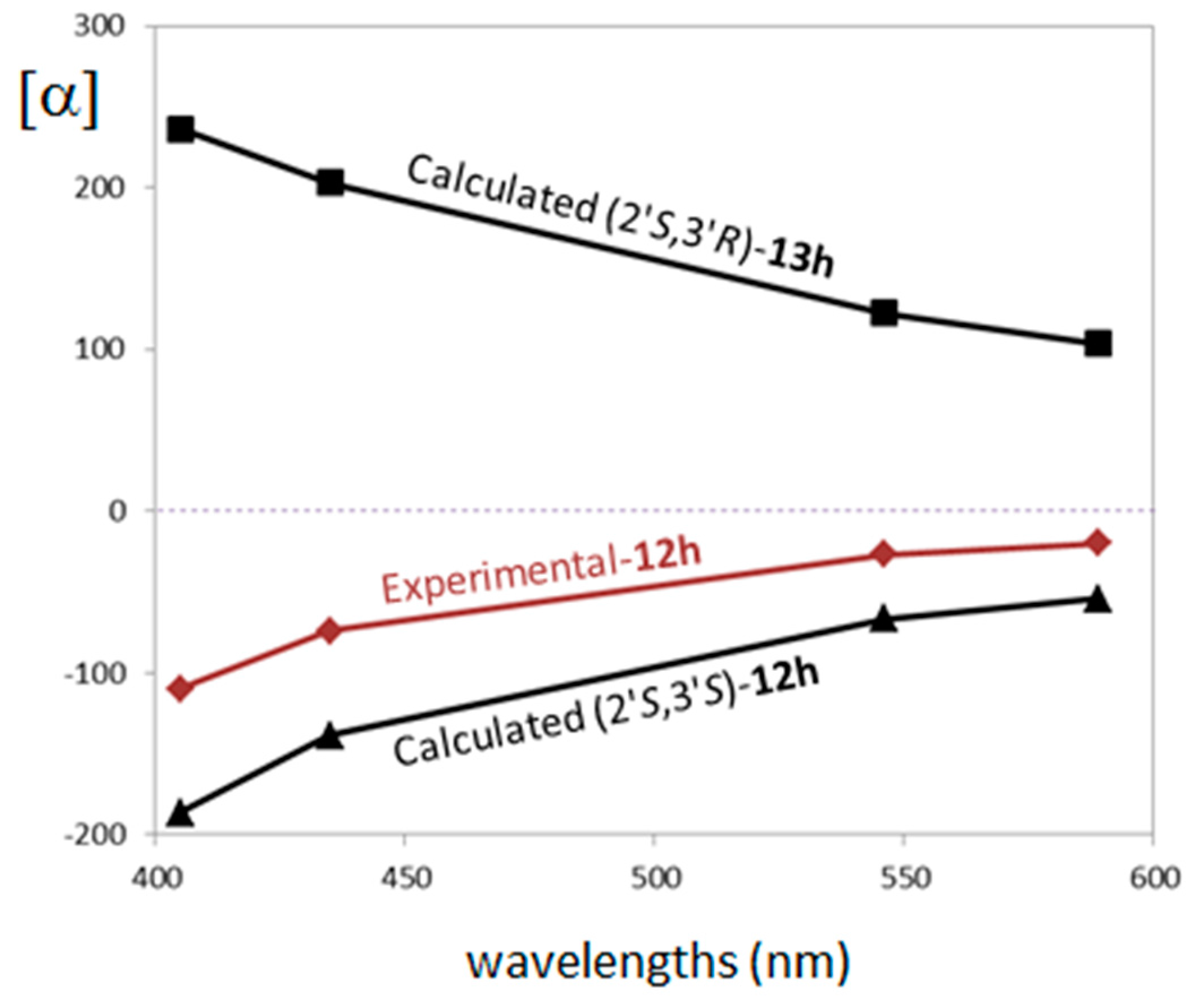
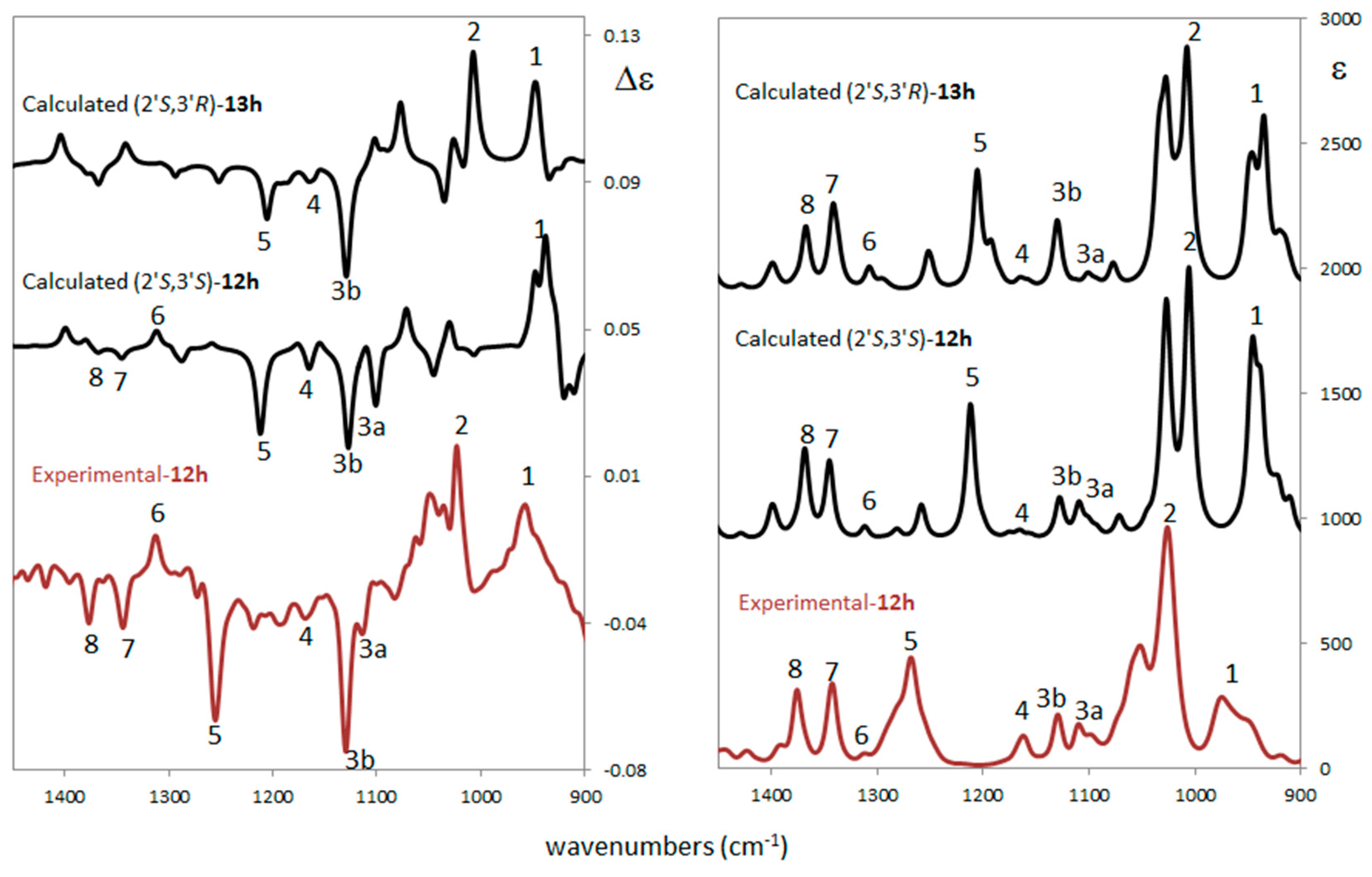
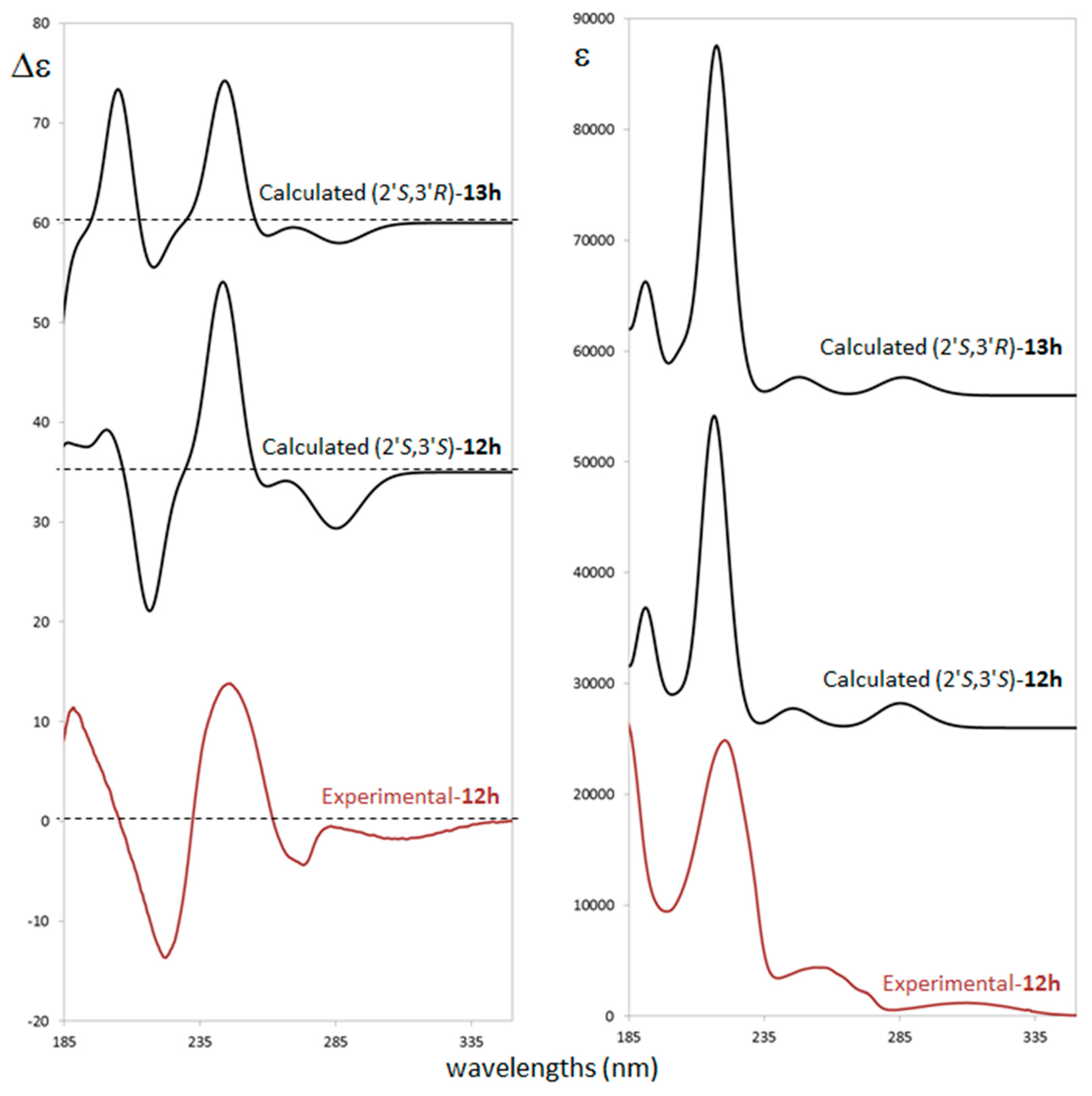
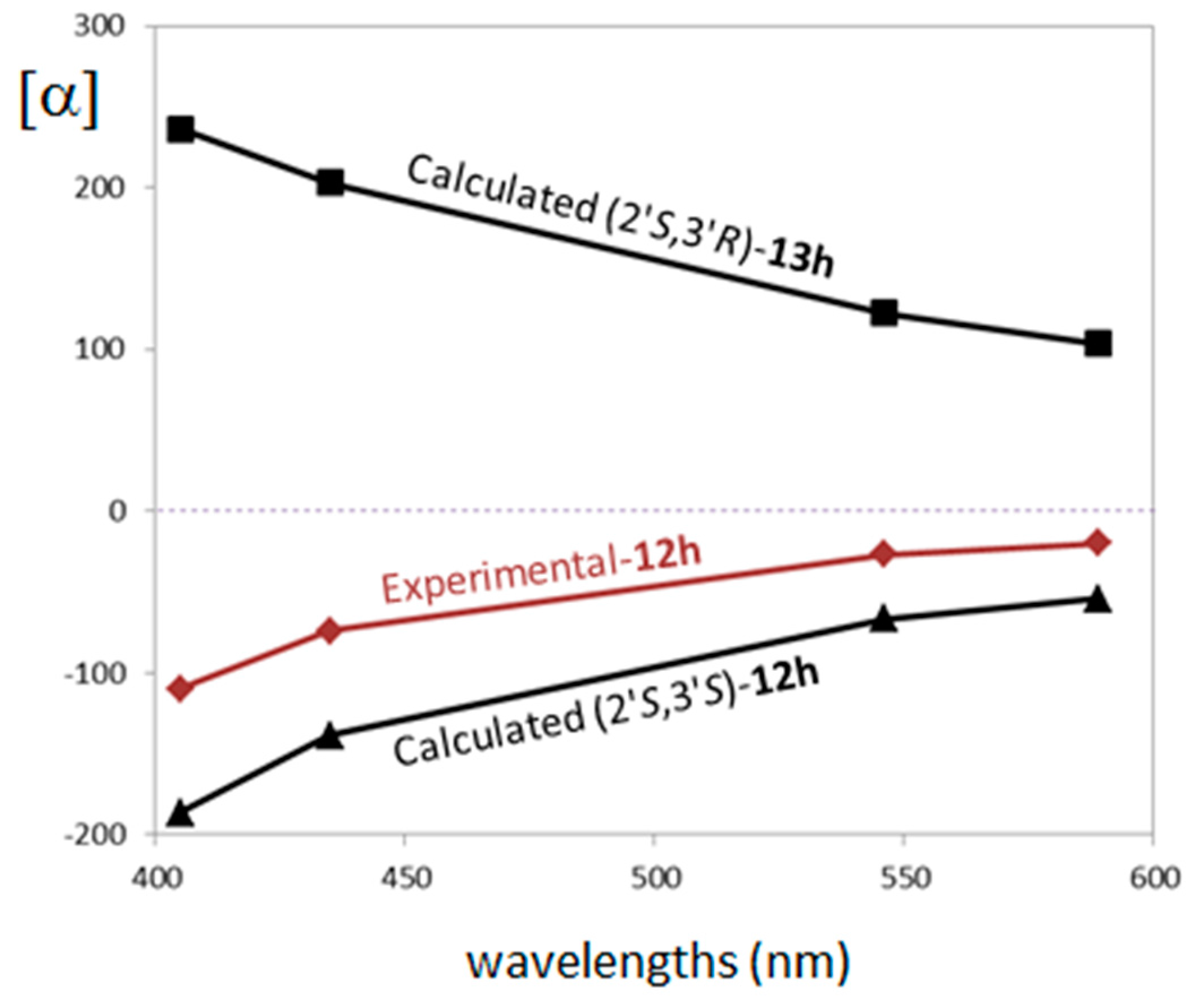
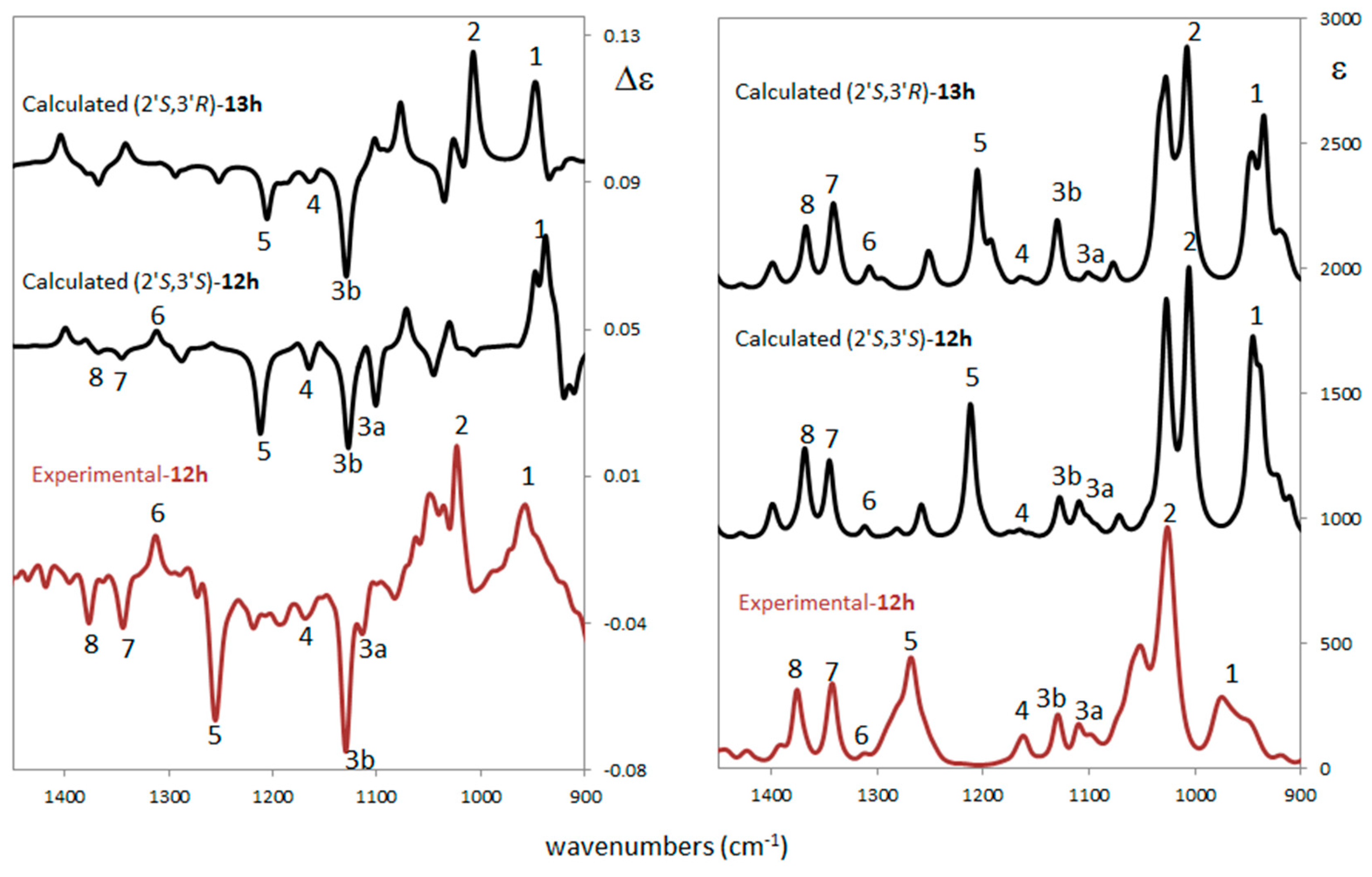
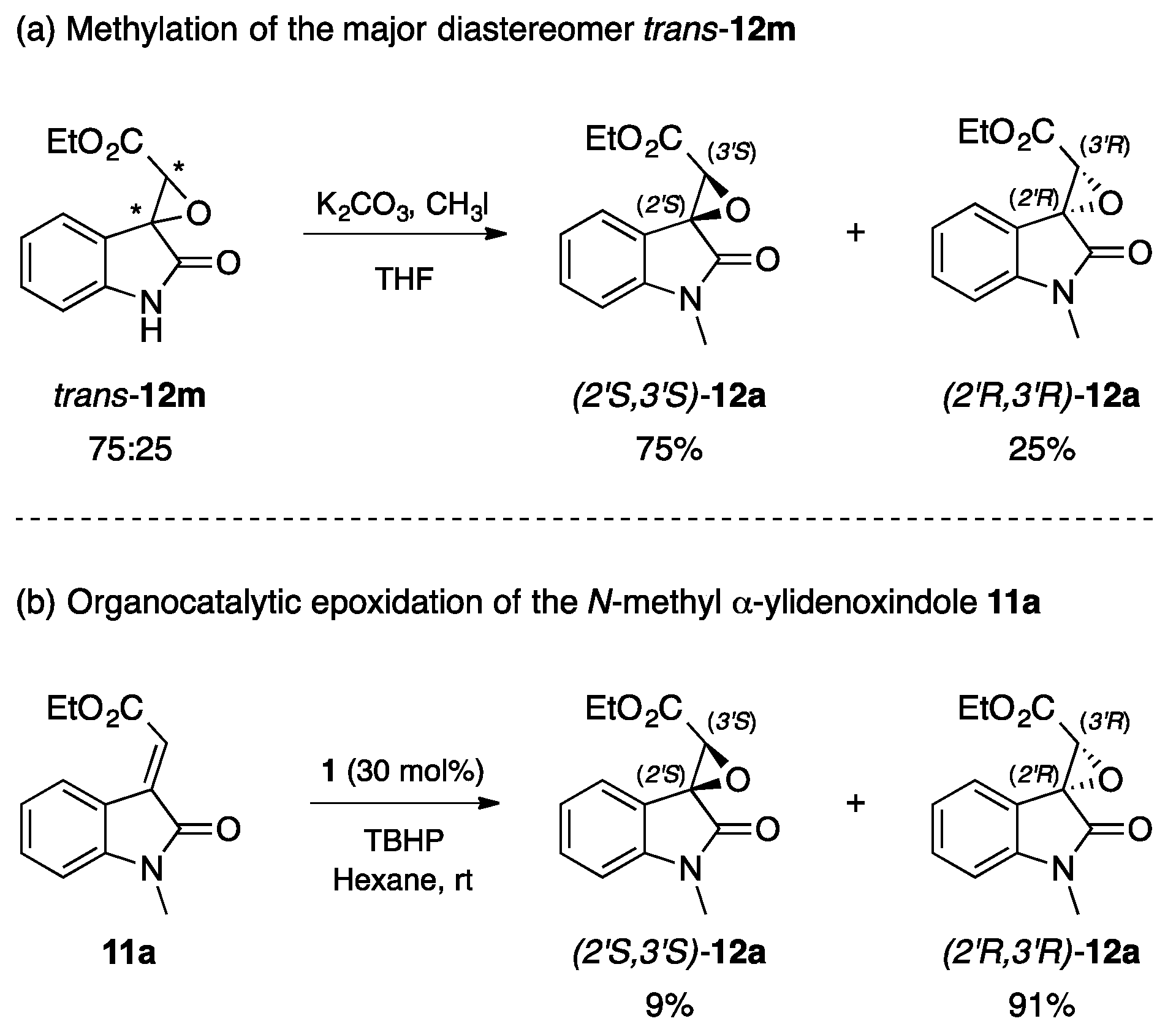
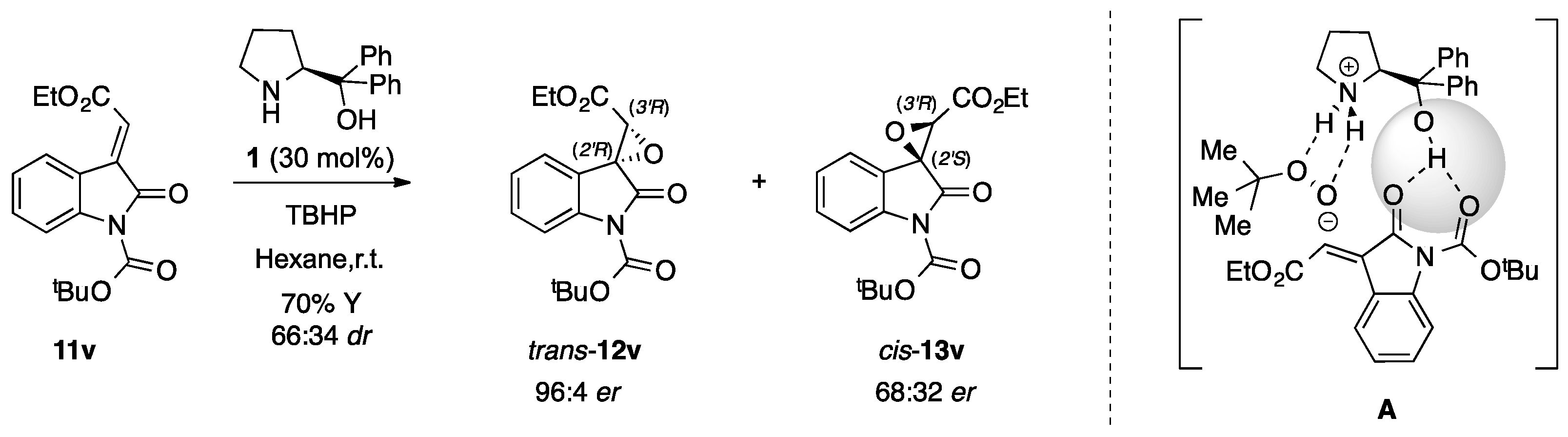
2.3. Stereochemistry Analysis
3. Materials and Methods
Synthesis and Characterization of Epoxides Trans-12a–v
Typical Experimental Procedure for the Synthesis of Epoxides
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seebach, D.; Weidmann, B. Modern Synthetic Methods; Otto Salle Verlag: Frankfurt, Germnay, 1983. [Google Scholar]
- De Figueiredo, R.M.; Mazziotta, A.; de Sant’Ana, D.P.; Palumbo, C.; Gasperi, T. Active methylene compounds in asymmetric organocatalytic synthesis of natural products and pharmaceutical scaffolds. Curr. Org. Chem. 2012, 16, 2231–2289. [Google Scholar] [CrossRef]
- Johnson, R.S.; Sharpless, K.B. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon Press: New York, NY, USA, 1991; Volume 7, p. 391. [Google Scholar]
- Ojima, I. Catalytic Asymmetric Synthesis, 2nd ed.; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Ebner, C.; Carreira, E.M. Cyclopropanation strategi1es in recent total syntheses. Chem. Rev. 2017, 117, 11651–11679. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Stiller, J.; Naicker, T.; Jiang, H.; Jorgensen, K.A. Asymmetric organocatalytic epoxidations: Reactions, scope, mechanisms, and applications. Angew. Chem. Int. Ed. 2014, 53, 7406–7426. [Google Scholar] [CrossRef] [PubMed]
- Childers, M.I.; Longo, J.M.; Van Zee, N.J.; LaPointe, A.M.; Coates, G.W. Stereoselective epoxide polymerization and copolymerization. Chem. Rev. 2014, 114, 8129–8152. [Google Scholar] [CrossRef] [PubMed]
- Boucherif, A.; Yang, Q.Q.; Wang, Q.; Chen, J.R.; Lu, L.Q.; Xiao, W.J. Enantio- and diastereoselective synthesis of spiro-epoxyoxindoles. J. Org. Chem. 2014, 79, 3924–3929. [Google Scholar] [CrossRef] [PubMed]
- Hajra, S.; Maity, S.; Roy, S. Regioselective friedel-crafts reaction of electron-rich benzenoid arenes and spiroepoxyoxindole at the spiro-centre: Efficient synthesis of benzofuroindolines and 2h-spiro benzofuran -3,3′-oxindoles. Adv. Synth. Catal. 2016, 358, 2300–2306. [Google Scholar] [CrossRef]
- Kuang, Y.L.; Lu, Y.; Tang, Y.; Liu, X.H.; Lin, L.L.; Feng, X.M. Asymmetric synthesis of spiro-epoxyoxindoles by the catalytic darzens reaction of isatins with phenacyl bromides. Org. Lett. 2014, 16, 4244–4247. [Google Scholar] [CrossRef] [PubMed]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Brennan, M. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis 2009, 2009, 3003–3025. [Google Scholar] [CrossRef]
- Singh, G.S.; Desta, Z.Y. Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Wang, R. Recent advances in asymmetric organocatalytic construction of 3,3′-spirocyclic oxindoles. Adv. Synth. Catal. 2013, 355, 1023–1052. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Wang, Q.; Cornwall, R.G.; Shi, Y. Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev. 2014, 114, 8199–8256. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Yan, C.G. Facile synthesis of functionalized spiro[indoline-3,2′-oxiran]-2-ones by darzens reaction. Beilstein J. Org. Chem. 2013, 9, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Chouhan, M.; Pal, A.; Sharma, R.; Nair, V.A. Quinine as an organocatalytic dual activator for the diastereoselective synthesis of spiro-epoxyoxindoles. Tetrahedron Lett. 2013, 54, 7119–7123. [Google Scholar] [CrossRef]
- Wang, L.H.; Su, Y.B.; Xu, X.M.; Zhang, W. A comparison of the photosensitized rearrangement and the lewis-acid-catalyzed rearrangement of spirooxindole epoxides. Eur. J. Org. Chem. 2012, 6606–6611. [Google Scholar] [CrossRef]
- Wang, L.; Li, Z.; Lu, L.; Zhang, W. Synthesis of spiro[furan-3,3′-indolin]-2′-ones by pet-catalyzed [3+2] reactions of spiro[indoline-3,2′-oxiran]-2-ones with electron-rich olefins. Tetrahedron 2012, 68, 1483–1491. [Google Scholar] [CrossRef]
- Chouhan, M.; Senwar, K.R.; Sharma, R.; Grover, V.; Nair, V.A. Regiospecific epoxide opening: A facile approach for the synthesis of 3-hydroxy-3-aminomethylindolin-2-one derivatives. Green Chem. 2011, 13, 2553–2560. [Google Scholar] [CrossRef]
- Hajra, S.; Roy, S.; Maity, S. Reversal of selectivity in c3-allylation and formal [3 + 2]-cycloaddition of spiro-epoxyoxindole: Unified synthesis of spiro-furanooxindole, (+/−)-n-methylcoerulescine, (+/−)-physovenine, and 3a-allylhexahydropyrrolo[2,3-b]indole. Org. Lett. 2017, 19, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.M.; Bao, G.J.; Li, Y.P.; Sun, W.S.; Li, J.; Hong, L.; Wang, R. Efficient catalytic kinetic resolution of spiro-epoxyoxindoles with concomitant asymmetric friedel-crafts alkylation of indoles. Angew. Chem. Int. Ed. 2017, 56, 5332–5335. [Google Scholar] [CrossRef] [PubMed]
- Schulz, V.; Davoust, M.; Lemarie, M.; Lohier, J.F.; Santos, J.S.D.; Metzner, P.; Briere, J.F. Straightforward stereoselective synthesis of spiro-epoxyoxindoles. Org. Lett. 2007, 9, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.L.; Han, J.W.; Wong, H.N.C. Asymmetric darzens reaction of isatins with diazoacetamides catalyzed by chiral binol-titanium complex. J. Org. Chem. 2017, 82, 12647–12654. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, C.; Mazzeo, G.; Mazziotta, A.; Gambacorta, A.; Loreto, M.A.; Migliorini, A.; Superchi, S.; Tofani, D.; Gasperi, T. Noncovalent organocatalysis: A powerful tool for the nucleophilic epoxidation of α-ylideneoxindoles. Org. Lett. 2011, 13, 6248–6251. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, T.; Loreto, M.A.; Migliorini, A.; Ventura, C. Synthesis of aziridine- and oxirane-2-phosphonates spiro-fused with oxindoles. Eur. J. Org. Chem. 2011, 385–391. [Google Scholar] [CrossRef]
- Dandia, A.; Singh, R.; Bhaskaran, S. Ultrasound promoted greener synthesis of spiro indole-3,5′-1,3 oxathiolanes in water. Ultrason. Sonochem. 2010, 17, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.N.; Xu, Q.; Wei, Y.; Shi, M. Exploration of a new zwitterion: Phosphine-catalyzed 2+1+2 cycloaddition reaction. Adv. Synth. Catal. 2017, 359, 1663–1671. [Google Scholar] [CrossRef]
- Cao, Y.M.; Jiang, X.X.; Liu, L.P.; Shen, F.F.; Zhang, F.T.; Wang, R. Enantioselective michael/cyclization reaction sequence: Scaffold-inspired synthesis of spirooxindoles with multiple stereocenters. Angew. Chem. Int. Ed. 2011, 50, 9124–9127. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Kuhen, K.L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Wu, T.Y.H.; He, Y. Design, synthesis and biological evaluations of novel oxindoles as hiv-1 non-nucleoside reverse transcriptase inhibitors. Part I. Bioorg. Med. Chem. Lett. 2006, 16, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Nafie, L.A. Vibrational Optical Activity-Principles and Application; Wiley: New Yrok, NY, USA, 2011. [Google Scholar]
- Polavarapu, P.L. Chiroptical Spectroscopy Fundamentals and Applications; CRC Publisher: Boca Raton, FL, USA, 2017. [Google Scholar]
- Stephens, P.J. The theory of vibrational circular dichroism. J. Phys. Chem. 1985, 89, 748–750. [Google Scholar] [CrossRef]
- Mazzeo, G.; Longhi, G.; Abbate, S.; Buonerba, F.; Ruzziconi, R. Chiroptical signatures of planar and central chirality in 2 paracyclo 2 (5,8)-quinolinophane derivatives. Eur. J. Org. Chem. 2014, 7353–7363. [Google Scholar] [CrossRef]
- Mazzeo, G.; Cimmino, A.; Masi, M.; Longhi, G.; Maddau, L.; Memo, M.; Evidente, A.; Abbate, S. Importance and difficulties in the use of chiroptical methods to assign the absolute configuration of natural products: The case of phytotoxic pyrones and furanones produced by diplodia corticola. J. Nat. Prod. 2017, 80, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Mazzeo, G.; Longhi, G.; Abbate, S.; Palomba, M.; Bagnoli, L.; Marini, F.; Santi, C.; Han, J.L.; Soloshonok, V.A.; Di Crescenzo, E.; et al. Solvent-free, uncatalyzed asymmetric “ene” reactions of n-tert-butylsulfinyl-3,3,3-trifluoroacetaldimines: A general approach to enantiomerically pure alpha-(trifluoromethyl) tryptamines. Org. Biomol. Chem. 2017, 15, 3930–3937. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Kuppens, T.; Langenaeker, W.; Tollenaere, J.P.; Bultinck, P. Determination of the stereochemistry of 3-hydroxymethyl-2,3-dihydro- 1,4 dioxino 2,3-b pyridine by vibrational circular dichroism and the effect of dft integration grids. J. Phys. Chem. A 2003, 107, 542–553. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds trans-12a–h, and 12m–u are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Sub | R1 | R2 | Cat * | Time (h) | Yield (%) c | dr d | trans-12 er e | cis-13 er e |
|---|---|---|---|---|---|---|---|---|---|
| 1 | a | H | H | 1 | 72 | 92 | 64:36 | 91:9 | 60:40 |
| 2 | a | H | H | 2 | 168 | 56 | 85:15 | 56:44 | 67:33 |
| 3 | a | H | H | 3 | 48 | 44 | 55:45 | 81:19 | 72:28 |
| 4 | a | H | H | 4 | 48 | 30 | 25:75 | 69:31 | 80:20 |
| 5 | a | H | H | 5 | 144 | 36 | 33:67 | 43:57 | 47:53 |
| 6 | a | H | H | 6 | 240 | 4 | 79:21 | 92:8 | 39:61 |
| 7 | a | H | H | 7 | 72 | 20 | 60:40 | 49:51 | 56:44 |
| 8 | a | H | H | 8 | 72 | 33 | 78:22 | 53:47 | 51:49 |
| 9 | a | H | H | 9 | 72 | 10 | 65:35 | 67:33 | 71:29 |
| 10 | a | H | H | 10 | 144 | 36 | 40:60 | 90:10 | 58:42 |
| 11 | b f | I | H | 1 | 24 | 97 | 33:67 | 80:20 | 66:34 |
| 12 | c g | iPr | H | 1 | 96 | 90 | 42:58 | 89:11 | 64:36 |
| 13 | d g | Cl | Cl | 1 | 33 | 79 | 37:63 | 93:7 | 77:23 |
| 14 | e h | OCH3 | H | 1 | 48 | 98 | 51:49 | 90:10 | 62:38 |
| 15 | e h | NO2 | H | 1 | 48 | 72 | 27:73 | 85:15 | 62:38 |
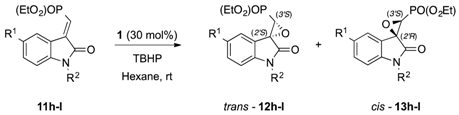
| 16 | g g |  | 1 | 24 | 98 | 51:49 | 91:9 | 55:45 | |
| Sub | R1 | R2 | Yield (%) b | dr c | trans-12 er d | cis-13 er d | |
|---|---|---|---|---|---|---|---|
| 1 | 11h e | H | CH3 | 80 | 56:44 | 80:20 | 61:39 |
| 2 | 11i e | H | Ph | 95 | 60:40 | 79:21 | 62:38 |
| 3 | 11j e | H | Bn | 80 | 56:44 | 74:26 | 55:45 |
| 4 | 11k e | H | 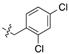 | 83 | 65:35 | 72:28 | 55:45 |
| 5 | 11l f | Cl | CH3 | 65 | 40:60 | 77:23 | 57:43 |
| Sub | R1 | R2 | Time (h) | Yield (%) b | dr c | trans-12 er d | cis-13 er d | |
|---|---|---|---|---|---|---|---|---|
| 1 | 11me | H | H | 144 | 96 | 65:35 | 75:25 | 61:39 |
| 2 | 11ne | F | H | 43 | 97 | 34:66 | 93:7 | 61:39 |
| 3 | 11oe | Cl | H | 140 | 83 | 40:60 | 73:27 | 59:41 |
| 4 | 11pe | I | H | 170 | 91 | 78:22 | 68:32 | 59:41 |
| 5 | 11qe | OCF3 | H | 96 | 76 | 38:62 | 87:13 | 66:34 |
| 6 | 11re | H | F | 66 | 93 | 23:77 | 92:8 | 70:30 |
| 7 | 11se | H | Cl | 48 | 99 | 36:64 | 88:12 | 62:38 |
| 8 | 11te | H | Br | 86 | 87 | 56:44 | 92:8 | 65:35 |
| 9 | 11ue | Cl | Cl | 78 | 42 | 35:65 | 83:17 | 86:14 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miceli, M.; Mazziotta, A.; Palumbo, C.; Roma, E.; Tosi, E.; Longhi, G.; Abbate, S.; Lupattelli, P.; Mazzeo, G.; Gasperi, T. Asymmetric Synthesis of Spirooxindoles via Nucleophilic Epoxidation Promoted by Bifunctional Organocatalysts. Molecules 2018, 23, 438. https://doi.org/10.3390/molecules23020438
Miceli M, Mazziotta A, Palumbo C, Roma E, Tosi E, Longhi G, Abbate S, Lupattelli P, Mazzeo G, Gasperi T. Asymmetric Synthesis of Spirooxindoles via Nucleophilic Epoxidation Promoted by Bifunctional Organocatalysts. Molecules. 2018; 23(2):438. https://doi.org/10.3390/molecules23020438
Chicago/Turabian StyleMiceli, Martina, Andrea Mazziotta, Chiara Palumbo, Elia Roma, Eleonora Tosi, Giovanna Longhi, Sergio Abbate, Paolo Lupattelli, Giuseppe Mazzeo, and Tecla Gasperi. 2018. "Asymmetric Synthesis of Spirooxindoles via Nucleophilic Epoxidation Promoted by Bifunctional Organocatalysts" Molecules 23, no. 2: 438. https://doi.org/10.3390/molecules23020438
APA StyleMiceli, M., Mazziotta, A., Palumbo, C., Roma, E., Tosi, E., Longhi, G., Abbate, S., Lupattelli, P., Mazzeo, G., & Gasperi, T. (2018). Asymmetric Synthesis of Spirooxindoles via Nucleophilic Epoxidation Promoted by Bifunctional Organocatalysts. Molecules, 23(2), 438. https://doi.org/10.3390/molecules23020438