A Litopenaeus vannamei Hemocyanin-Derived Antimicrobial Peptide (Peptide B11) Attenuates Cancer Cells’ Proliferation


 ,
,
Abstract
1. Introduction
2. Results
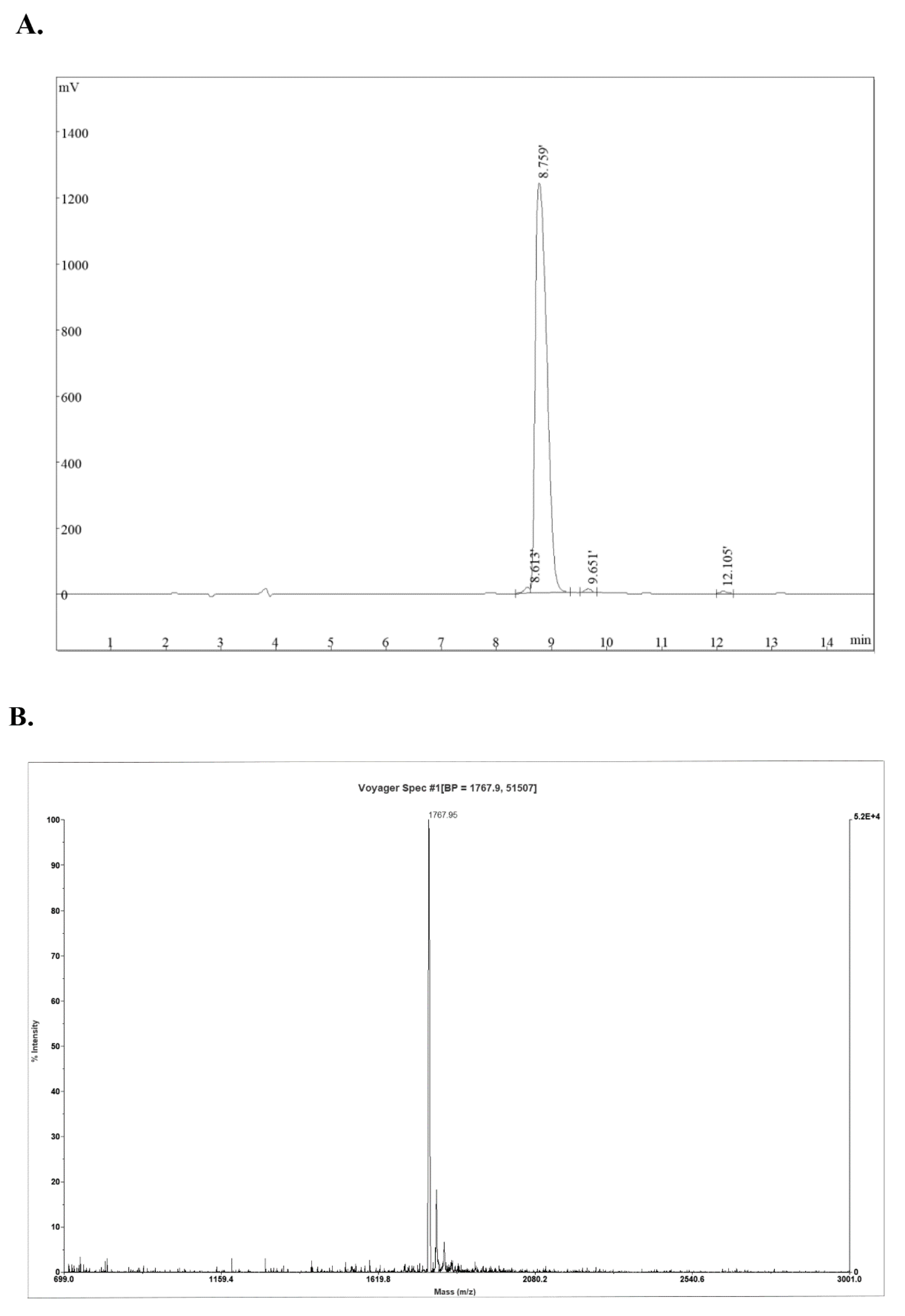
2.1. Synthesis and Characterization of Peptides
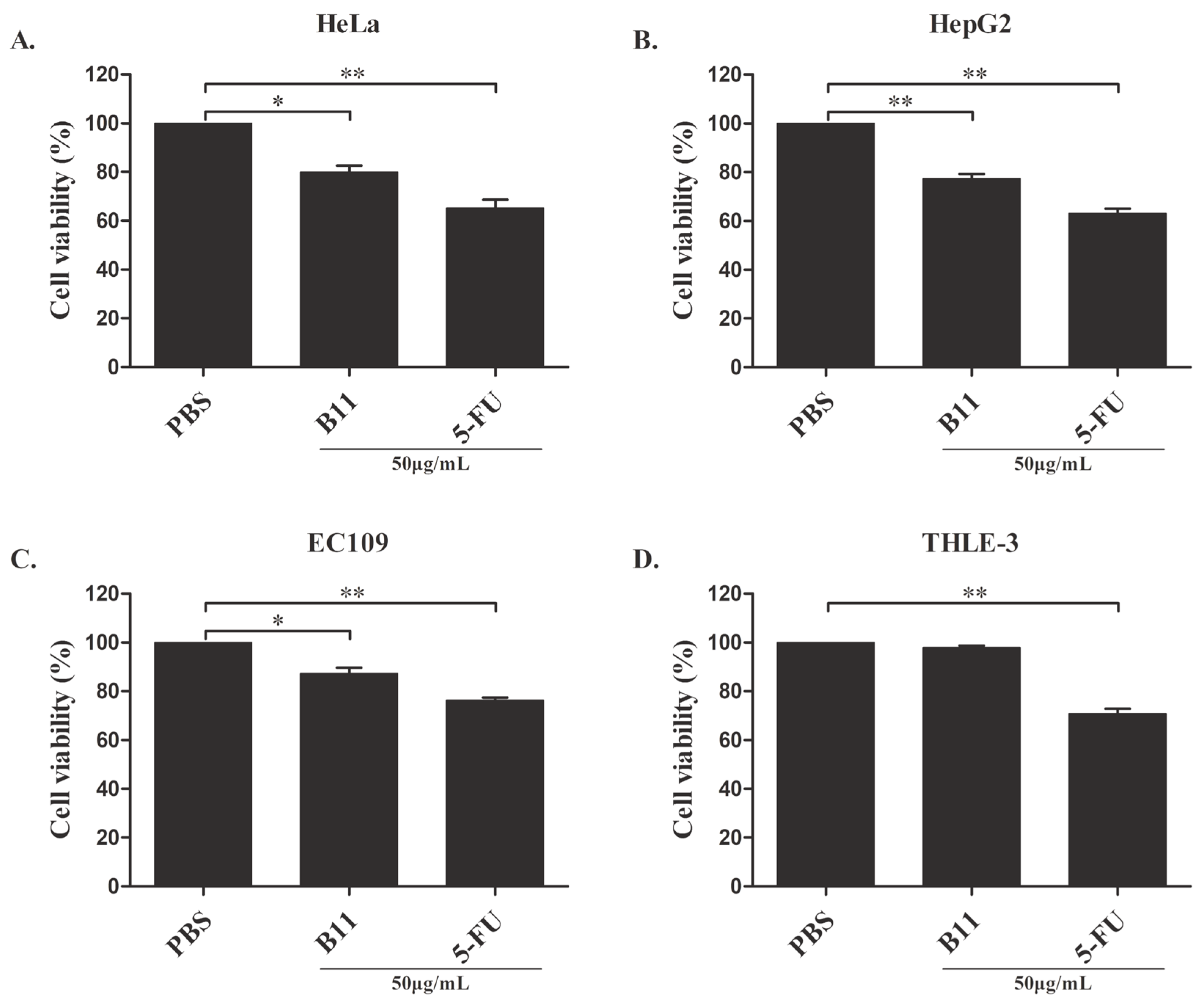
2.2. Effect of Peptide B11 on Cancer Cells’ Proliferation
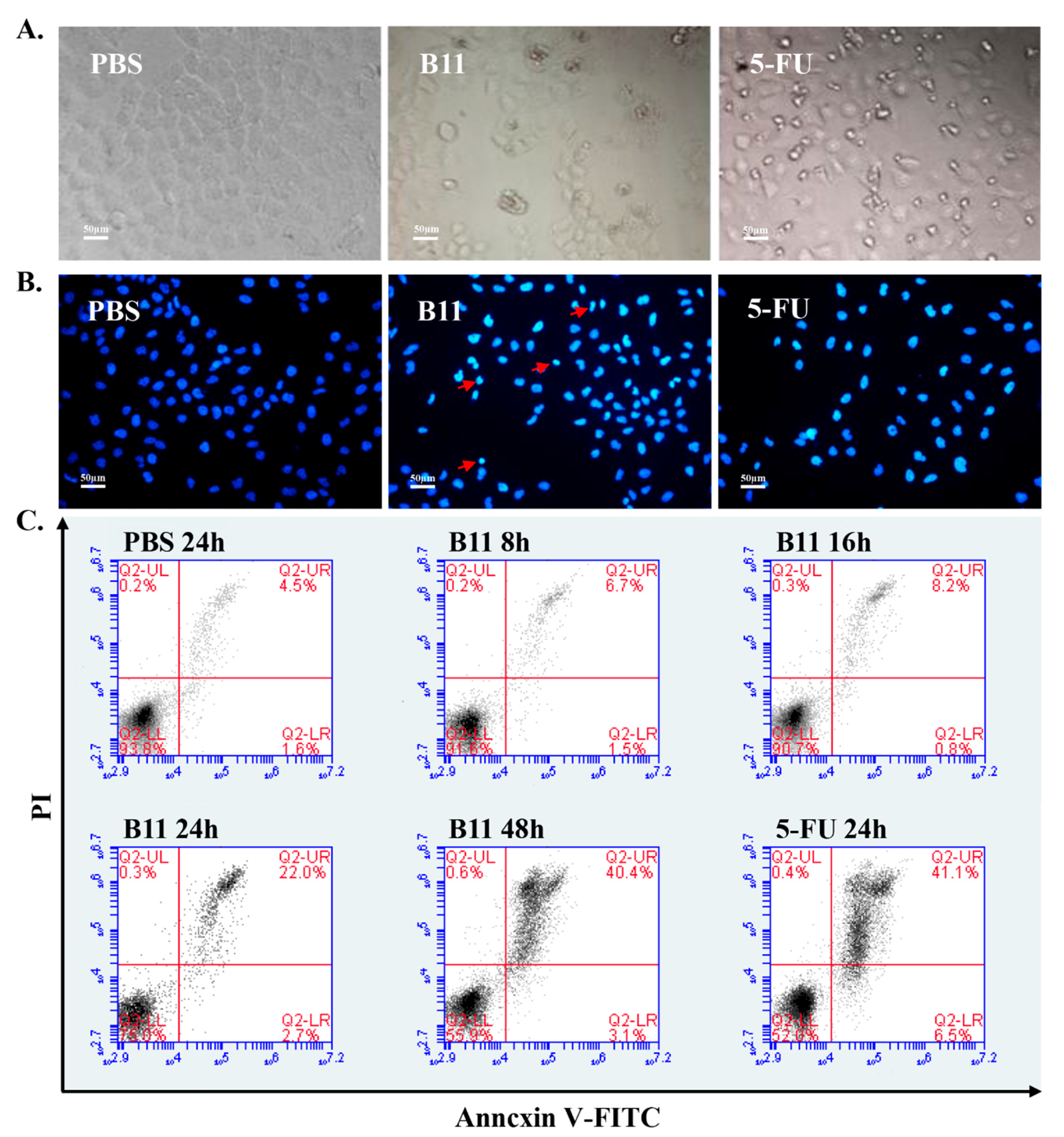
2.3. Peptide B11 Induces Apoptosis in HeLa Cells
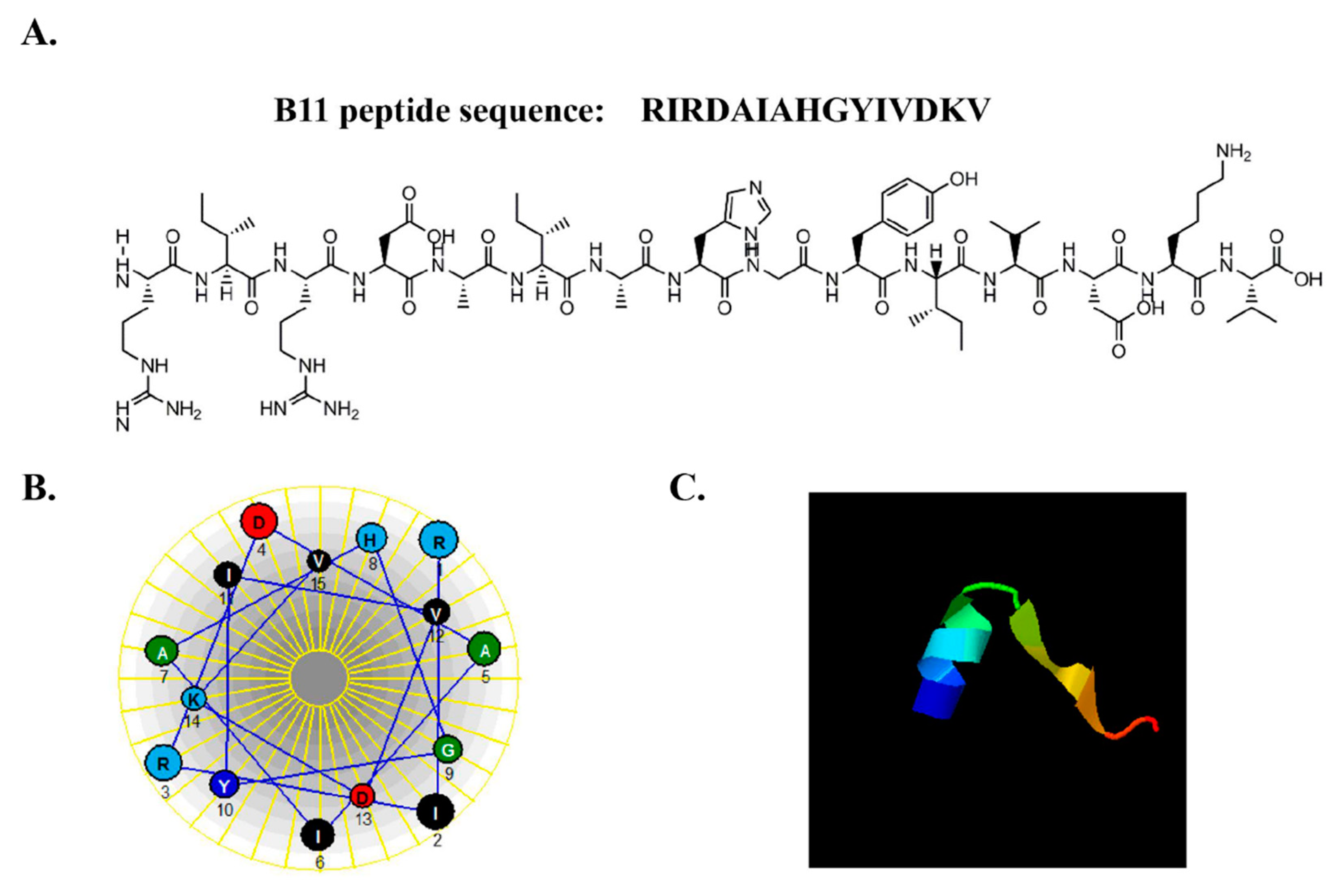
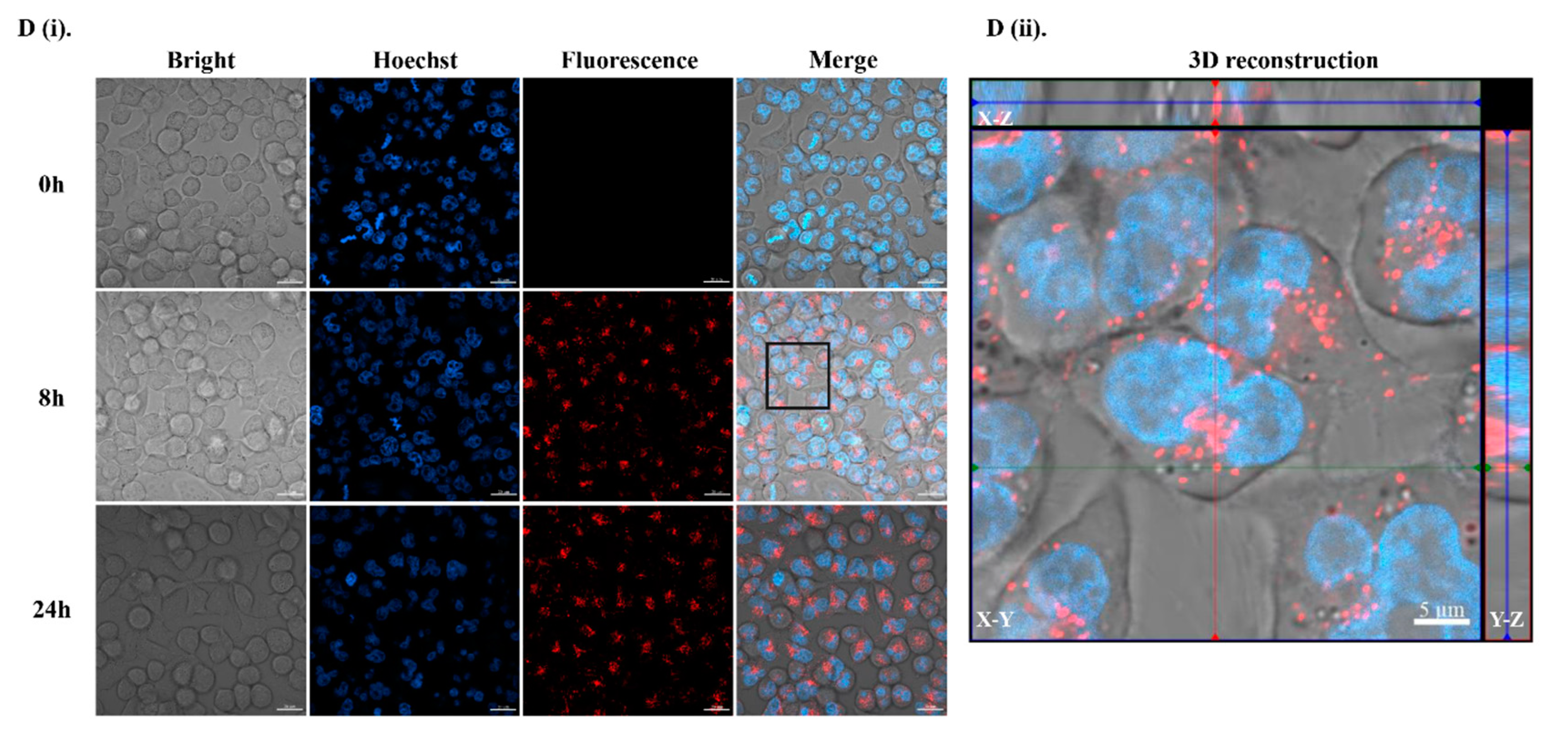
2.4. Physicochemical Properties of B11 and Cell Permeabilization Analysis
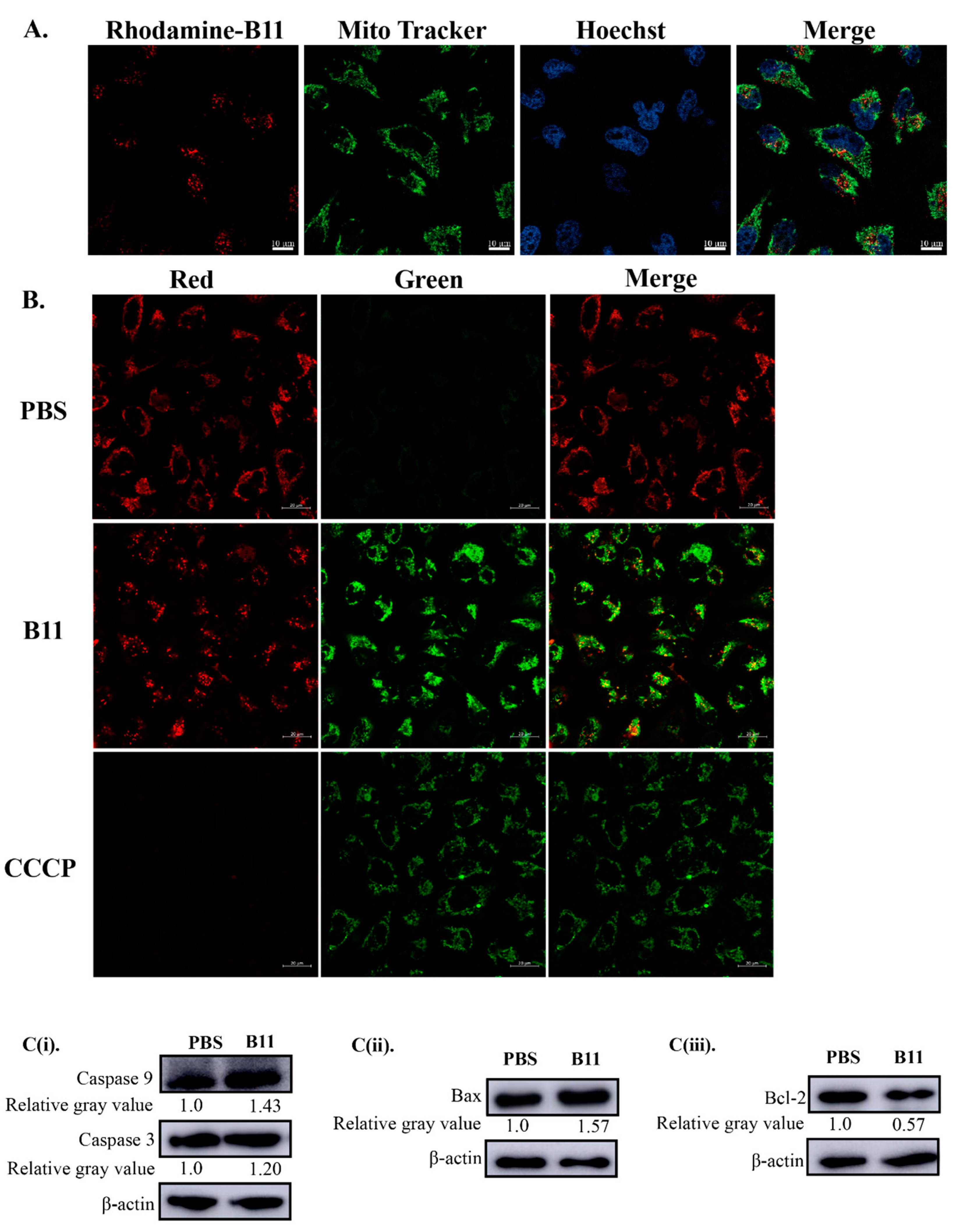
2.5. Peptide B11 Induces Mitochondrial-Dependent Apoptotic Cell Death in HeLa Cells through Lost of Mitochondrial Membrane Potential
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Cytological Effect of Peptide B11 on HeLa Cells
4.5. Annexin V-FITC/PI Apoptosis Detection Assay
4.6. Prediction of the Structural Characteristics and Features of Peptide B11
4.7. Cell Uptake Studies
4.8. Analysis of Subcellular Localization
4.9. JC-1 Dye Staining for Mitochondrial Membrane Potential Analysis
4.10. Western Blot Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Conflicts of Interest
References
- Jaenicke, E.; Föll, R.; Decker, H. Spider hemocyanin binds ecdysone and 20-OH-ecdysone. J. Biol. Chem. 1999, 274, 34267–34271. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.J.; Pirow, R. The physiological significance of respiratory proteins in invertebrates. Zoology 1997, 100, 298–306. [Google Scholar]
- Coates, C.J.; Decker, H. Immunological properties of oxygen-transport proteins: Hemoglobin, hemocyanin and hemerythrin. Cell Mol. Life Sci. 2016, 74, 1–25. [Google Scholar] [CrossRef]
- Coates, C.J.; Nairn, J. Diverse immune functions of hemocyanins. Dev. Comp. Immunol. 2014, 45, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Decker, H.; Rimke, T. Tarantula hemocyanin shows phenoloxidase activity. J. Biol. Chem. 1998, 273, 25889–25892. [Google Scholar] [CrossRef]
- Nesterova, N.V.; Zagorodnya, S.D.; Moshtanska, V.; Dolashka, P.; Baranova, G.V.; Golovan, A.V.; Kurova, A.O. Antiviral activity of hemocyanin isolated from marine snail Rapana venosa. Antivir. Res. 2011, 90, A38. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Xu, A.; Chen, J.; Lin, B.; Peng, X. Affinity proteomic Approach for identification of an IgA-like protein in Litopenaeus vannamei and study on its Agglutination characterization. J. Proteome. Res. 2006, 5, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Zhang, Y.; Jiang, R.; Zhong, M.; Hu, Z.; Du, H.; Lun, J.; Chen, J.; Li, Y. Identification and agglutination properties of hemocyanin from the mud crab (Scylla serrata). Fish Shellfish Immunol. 2011, 30, 354–360. [Google Scholar] [CrossRef]
- Zheng, L.; Zhao, X.; Zhang, P.; Chen, C.; Liu, S.; Huang, R.; Zhong, M.; Wei, C.; Zhang, Y. Hemocyanin from shrimp Litopenaeus vannamei has antiproliferative effect against HeLa cell in vitro. PLoS ONE 2016, 11, e0151801. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Peng, X. Identification of a type of human IgG-like protein in shrimp Penaeus vannamei by mass spectrometry. J. Exp. Mar. Biol. Ecol. 2004, 301, 39–54. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, F.; Hu, Z.; Zhao, X.; Min, S.; Du, Z.; Zhao, S.; Ye, X.; Li, Y. Hemocyanin from shrimp Litopenaeus vannamei shows hemolytic activity. Fish Shellfish Immunol. 2009, 27, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Du, Z.; Zhang, Y.; Du, H.; Guo, L.; Zhong, M.; Cao, J.; Wang, X. Proteomic identification of the related immune-enhancing proteins in shrimp Litopenaeus vannamei stimulated with vitamin C. and Chinese herbs. Fish Shellfish Immunol. 2011, 31, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Petit, V.W.; Rolland, J.L.; Blond, A.; Cazevieille, C.; Djediat, C.; Peduzzi, J.; Goulard, C.; Bachère, E.; Dupont, J.; Destoumieuxgarzón, D. A hemocyanin-derived antimicrobial peptide from the penaeid shrimp adopts an alpha-helical structure that specifically permeabilizes fungal membranes. BBA-Gen Subjects 2016, 1860, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Destoumieuxgarzón, D.; Saulnier, D.; Garnier, J.; Jouffrey, C.; Bulet, P.; Bachère, E. Crustacean immunity. Antifungal peptides are generated from the C terminus of shrimp hemocyanin in response to microbial challenge. J. Biol. Chem. 2001, 276, 47070–47077. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, B.L.; Söderhäll, K. Processing of an antibacterial peptide from hemocyanin of the freshwater crayfish Pacifastacus leniusculus. J. Biol. Chem. 2003, 278, 7927–7933. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zhan, S.; Huang, H.; Zhong, M.; Chen, J.; You, C.; Wan, F.; Zhang, Y.L. Identification and characterization of an 18.4 kDa antimicrobial truncation from shrimp Litopenaeus vannamei hemocyanin upon Vibrio parahaemolyticus infection. Fish Shellfish Immunol. 2016, 56, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Bulet, P.; Stöcklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A.R.B. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.G.; Dobroff, A.S.; Cavarsan, C.F.; Paschoalin, T.; Nimrichter, L.; Mortara, R.A.; Santos, E.L.; Miranda, A.; Daffre, S.; Travassos, L.R. Effective topical treatment of subcutaneous murine B16F10-Nex2 melanoma by the antimicrobial peptide gomesin. Neoplasia. 2008, 10, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Chien, Y.L.; Pan, C.Y.; Lin, T.L.; Chen, J.Y.; Chiu, S.J.; Hui, C.F. Epinecidin-1, an antimicrobial peptide from fish (Epinephelus coioides) which has an antitumor effect like lytic peptides in human fibrosarcoma cells. Peptides 2009, 30, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Hilchie, A.L.; Doucette, C.D.; Pinto, D.M.; Patrzykat, A.; Douglas, S.; Hoskin, D.W. Pleurocidin-family cationic antimicrobial peptides are cytolytic for breast carcinoma cells and prevent growth of tumor xenografts. Breast Cancer Res. 2011, 13, R102. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Huang, H.; Wang, F.; Aweya, J.J.; Zheng, Z.; Zhang, Y. Prediction and characterization of a novel hemocyanin-derived antimicrobial peptide from shrimp Litopenaeus vannamei. Amino Acids 2018, 50, 995–1005. [Google Scholar] [CrossRef]
- Coin, I.; Beyermann, M.; Bienert, M. Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. [Google Scholar] [CrossRef]
- Stark, M.; Liu, L.P.; Deber, C.M. Cationic hydrophobic peptides with antimicrobial activity. Antimicrob. Agents Chemother. 2002, 46, 3585–3590. [Google Scholar] [CrossRef]
- Koczulla, A.R.; Bals, R. Antimicrobial peptides. Drugs 2003, 63, 389–406. [Google Scholar] [CrossRef]
- Li, C.; Zhu, J.; Wang, Y.; Chen, Y.; Song, L.; Zheng, W.; Li, J.; Yu, R. Antibacterial activity of AI-Hemocidin 2, a novel N-terminal peptide of hemoglobin purified from Arca inflata. Mar. Drugs 2017, 15, 205. [Google Scholar] [CrossRef]
- Wang, Y.H.; Yu, H.T.; Pu, X.P.; Du, G.H. Baicalein prevents 6-hydroxydopamine-induced mitochondrial dysfunction in SH-SY5Y cells via inhibition of mitochondrial oxidation and up-regulation of DJ-1 protein expression. Molecules 2013, 18, 14726–14738. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, E.; Koch, G.; Wagner, R.M.; Fekete, A.; Stengel, S.T.; Schneider, J.; Mielich-Süss, B.; Geibel, S.; Markert, S.M.; Stigloher, C.; et al. Membrane microdomain disassembly inhibits MRSA antibiotic resistance. Cell 2017, 171, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Semina, S.E.; Scherbakov, A.M.; Vnukova, A.A.; Bagrov, D.V.; Evtushenko, E.G.; Safronova, V.M.; Golovina, D.A.; Lyubchenko, L.N.; Gudkova, M.V.; Krasil’nikov, M.A. Exosome-mediated transfer of cancer cell resistance to antiestrogen drugs. Molecules 2018, 23, 829. [Google Scholar] [CrossRef] [PubMed]
- Rubinchik, E.; Dugourd, D.; Algara, T.; Pasetka, C.; Friedland, H.D. Antimicrobial and antifungal activities of a novel cationic antimicrobial peptide, omiganan, in experimental skin colonisation models. Int. J. Antimicrob. Agents 2009, 34, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.Y.; Gellatly, S.L.; Hancock, R.E.W. Multifunctional cationic host defence peptides and their clinical applications. Cell Mol. Life Sci. 2011, 68, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wong, E.S.W.; Whitley, J.C.; Li, J.; Stringer, J.M.; Short, K.R.; Renfree, M.B.; Belov, K.; Cocks, B.G. Ancient antimicrobial peptides kill antibiotic-resistant pathogens: Australian mammals provide new options. PLoS ONE 2011, 6, e24030. [Google Scholar] [CrossRef] [PubMed]
- Standley, S.M.; Toft, D.J.; Cheng, H.; Soukasene, S.; Chen, J.; Raja, S.M.; Band, V.; Band, H.; Cryns, V.L.; Stupp, S.I. Induction of cancer cell death by self-assembling nanostructures incorporating a cytotoxic peptide. Cancer Res. 2010, 70, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, C.; Zhang, S.; Zhao, X.; Xu, H.; Zhao, X.; Lu, J.R. Designed antimicrobial and antitumor peptides with high selectivity. Biomacromolecules 2011, 12, 3839–3843. [Google Scholar] [CrossRef]
- Pushpanathan, M.; Gunasekaran, P.; Rajendhran, J. Antimicrobial peptides: Versatile biological properties. Int. J. Pept. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.D.S.; Silvapereira, I.; Kyaw, C.M. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.C.L.; Lima, L.A.; Miranda, V.J.; Dias, S.C.; Franco, O.L. Current scenario of peptide-based drugs: The key roles of cationic antitumor and antiviral peptides. Front. Microbiol. 2013, 4, 321. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. BBA-Biomembranes 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Guzmánrodríguez, J.J.; Ochoazarzosa, A.; Lópezgómez, R.; Lópezmeza, J.E. Plant antimicrobial peptides as potential anticancer agents. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Li, C.; Wang, F.; Aweya, J.J.; Yao, D.; Zheng, Z.; Huang, H.; Li, S.; Zhang, Y. Trypsin of Litopenaeus vannamei is required for the generation of hemocyanin-derived peptides. Dev. Comp. Immunol. 2018, 79, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Mader, J.S.; Hoskin, D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Investig. Drugs 2006, 15, 933–946. [Google Scholar] [CrossRef]
- Wang, H.; Ma, J.L.; Yang, Y.G.; Song, Y.; Wu, J.; Qin, Y.Y.; Zhao, X.L.; Wang, J.; Zou, L.L.; Wu, J.F. Efficient therapeutic delivery by a novel cell-permeant peptide derived from KDM4A protein for antitumor and antifibrosis. Oncotarget 2016, 7, 49075–49090. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2015, 16, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yuan, H.; Guo, C.; Lu, Y.; Deng, S.; Yang, Y.; Wei, Q.; Wen, L.; He, Z. Zearalenone induces apoptosis and necrosis in porcine granulosa cells via a caspase-3- and caspase-9-dependent mitochondrial signaling pathway. J. Cell Physiol. 2012, 227, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Follis, A.V.; Kriwacki, R.W.; Green, D.R. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014, 39, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Wang, X.; Chen, C.; Zhou, G.; Ye, J.; Yin, R.; Feng, D.; Zhang, S.; Wang, X.; Zhao, X.; Zhang, Z. Sepia ink oligopeptide induces apoptosis of lung cancer cells via mitochondrial pathway. Cell Physiol. Biochem. 2018, 45, 2095–2106. [Google Scholar] [CrossRef]
- Ow, Y.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Fisher, J.C.; Dillon, C.P.; Kriwacki, R.W.; Kuwana, T.; Green, D.R. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 20327–20332. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P. Bcl-2 Inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef]
- Huertas, N.J.; Monroy, Z.J.R.; Medina, R.F.; Castañeda, J.E.G. Antimicrobial activity of truncated and polyvalent peptides derived from the FKCRRQWQWRMKKGLA sequence against Escherichia coli ATCC 25922 and Staphylococcus aureus ATCC 25923. Molecules 2017, 22, 987. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.R.; Chua, K.N.; Sim, W.J.; Ng, H.C.; Bi, C.; Ho, J.; Nga, M.E.; Pang, Y.H.; Ong, W.R.; Soo, R.A. MEK inhibition overcomes cisplatin resistance conferred by SOS/MAPK pathway activation in squamous cell carcinoma. Mol. Cancer Ther. 2015, 14, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.J.; Zhang, Y.Y.; Fan, Y.Y.; Jin, S.J.; Yan, J.; Zheng, X.W.; Chen, J.; Yao, X.; Zhou, L.M. β, β-Dimethylacrylshikonin exerts antitumor activity via Notch-1 signaling pathway in vitro and in vivo. Biochem. Pharmacol. 2012, 84, 507–512. [Google Scholar]
- Golks, A.; Brenner, D.; Schmitz, I.; Watzl, C.; Krueger, A.; Krammer, P.H.; Lavrik, I.N. The role of CAP3 in CD95 signaling: New insights into the mechanism of procaspase-8 activation. Cell Death Differ. 2006, 13, 489–498. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors as they are patented. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Parameter |
|---|---|
| Sequence | RIRDAIAHGYIVDKV |
| Molecular weight (Da) | 1767 |
| Total hydrophobic ratio | 46% |
| Charge at pH 7.0 | +1 |
| Hydrophobicity <H> | 0.333 |
| Hydrophobic moment <µH> | 0.119 |
| Average hydropathy value | 0.0466 |
| Molar extinction coefficient | 1490 |
| Isoelectric point | 8.862 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Aweya, J.J.; Zheng, L.; Wang, F.; Zheng, Z.; Zhong, M.; Lun, J.; Zhang, Y. A Litopenaeus vannamei Hemocyanin-Derived Antimicrobial Peptide (Peptide B11) Attenuates Cancer Cells’ Proliferation. Molecules 2018, 23, 3202. https://doi.org/10.3390/molecules23123202
Liu S, Aweya JJ, Zheng L, Wang F, Zheng Z, Zhong M, Lun J, Zhang Y. A Litopenaeus vannamei Hemocyanin-Derived Antimicrobial Peptide (Peptide B11) Attenuates Cancer Cells’ Proliferation. Molecules. 2018; 23(12):3202. https://doi.org/10.3390/molecules23123202
Chicago/Turabian StyleLiu, Shangjie, Jude Juventus Aweya, Liyuan Zheng, Fan Wang, Zhou Zheng, Mingqi Zhong, Jingsheng Lun, and Yueling Zhang. 2018. "A Litopenaeus vannamei Hemocyanin-Derived Antimicrobial Peptide (Peptide B11) Attenuates Cancer Cells’ Proliferation" Molecules 23, no. 12: 3202. https://doi.org/10.3390/molecules23123202
APA StyleLiu, S., Aweya, J. J., Zheng, L., Wang, F., Zheng, Z., Zhong, M., Lun, J., & Zhang, Y. (2018). A Litopenaeus vannamei Hemocyanin-Derived Antimicrobial Peptide (Peptide B11) Attenuates Cancer Cells’ Proliferation. Molecules, 23(12), 3202. https://doi.org/10.3390/molecules23123202