1. Introduction
Influenza is a heavy burden of global public health because of its strong infectivity and high morbidity. The most common symptoms of influenza include cough, fever, headache, and weakness [
1]. In addition, gastroenteritis-like symptoms, such as nausea, vomiting, abdominal pain, and diarrhea, often accompany influenza in patients [
2]. Influenza virus infection is believed to cause changes in intestinal flora, which might be responsible for the changes in excrement characteristics [
3]. What is worse, the abuse of antibiotics often exacerbates changes in the gut microbiome. Therefore, the alteration of intestinal flora induced by antibiotic interference may be an important and modifiable factor that contributes to the immune response in influenza.
Accumulating evidence suggests that commensal bacteria have a crucial impact on the host’s immune system [
4]. On one hand, intestinal flora and the host immune system jointly defend the invasion of pathogenic microorganisms. The healthy intestinal microenvironment contributes to the maintenance of the immunological status of the host. On the other hand, the deviations in microbial community composition increase the risk of pathogen infection, which can cause disease or influence its course and prognosis [
5]. Dysbacteriosis of the gut microbiota could cause damage to the gastrointestinal mucosal barrier, dysfunction of antibody synthesis or secretion, and development of diseases in the respiratory system. For instance, the intestinal microbiota is related to chronic intestinal inflammation in various inflammatory bowel diseases (IBDs) [
6]. What is more, studies in recent years also testified that the intestine microbiota affected the host’s immune response to extragastrointestinal tract infections [
7]. The intestinal microbiota of the host affects the replication and transmission of a diverse array of viral pathogens, such as influenza virus, dengue virus, and human immunodeficiency virus [
8]. For example, Ichinohe et al. revealed that alteration of intestinal flora resulted in defective cluster of differentiation 4 (CD4) T-cell, CD8 T-cell, and B-cell immunity in influenza virus infection [
9]. Abt et al. demonstrated that intestinal dysbacteriosis altered the expression of antiviral defense genes and interfered in the responsive pathways, which led to the failure to control viral infection and an increase in host morbidity and mortality [
10]. In addition, our previous study also found that dysbacteriosis downregulated the Toll-like receptor 7 (TLR7) signaling pathway, which affected the immunity of the respiratory mucosa against influenza A virus infection [
11]. Therefore, the alteration of the intestinal flora might interfere with the host immune function.
However, due to the complexity and diversity of commensal–host interactions, more study is needed to increase the knowledge of the joint modulating mechanism of immune homeostasis and intestinal microbiota. Interdomain molecular communication plays an important role in host and gut microorganism interactions, of which microRNAs (miRNAs) are considered to be a key mode [
12]. MicroRNAs have many regulatory roles in various cellular processes and they are implicated in various diseases. Researchers claimed that the microbiota regulated dendritic cell (DC) expression of interleukins 12 and 23p40 (IL-12/IL-23p40) through the inhibition of miR-10a, which could be important in the maintenance of host immune homeostasis [
13]. Therefore, the relationship between intestinal flora and miRNA expression should be discussed. The mechanism of the intestinal microbiota that affects the natural immunoreaction in influenza infection is interesting; however, the roles of the intestinal flora and microRNA communication in the influenza immune response are yet to be clarified. Therefore, in this study, mice were treated with antibiotics before viral infection to establish an animal model of dysbacteriosis, to investigate the mechanism of intestinal flora and miRNAs in the immune response to influenza.
2. Materials and Methods
2.1. Animals
Forty-eight SPF (Specefic pathogen Free, SPF) BALB/c mice, half males and half females, weighing 16–18 g, were purchased from Guangdong Medical Laboratory Animal Center (Guangdong, China). The mice were kept in the SPF laboratory animal room in a controlled environment (22 ± 1 °C and 55% relative humidity) with free access to food and water during the experiment. The experiment was performed under the supervision and assessment of the Laboratory Animal Ethics Committee of Jinan University. Every experiment operation abided by the statute on the Administration of Laboratory Animals approved by the China Council in 1988.
2.2. Grouping and Treatment
After sample size calculating, the mice were randomly divided into four groups: control group (C), virus infection model group (V), dysbacteriosis model group (D), and dysbacteriosis and virus infection group (D + V). Neomycin (0.30 g/kg, dissolved in normal saline (NS) administered via gavage for 14 days) was used to establish an intestinal dysbacteriosis mouse model in the D and D + V groups. The C and V groups received NS intragastrically as a comparison. The influenza A virus (A/FM1/1/47) was provided by the Department of Immunology and Microbiology of Jinan University (Guangzhou, China). After full anesthetization by inhalation of diethyl ether, 20% median lethal dose (LD
50) FM1 virus intranasal infection for four days was used to establish the virus infection model in the V group and D + V group from the 15th day of the experiment. Intranasal treatment with NS was also performed in the C and D groups to avoid the impact of the operation. All the mice were sacrificed on the 19th day after abstaining from food except water for 12 h (
Figure 1).
2.3. 16S rRNA for Intestinal Flora
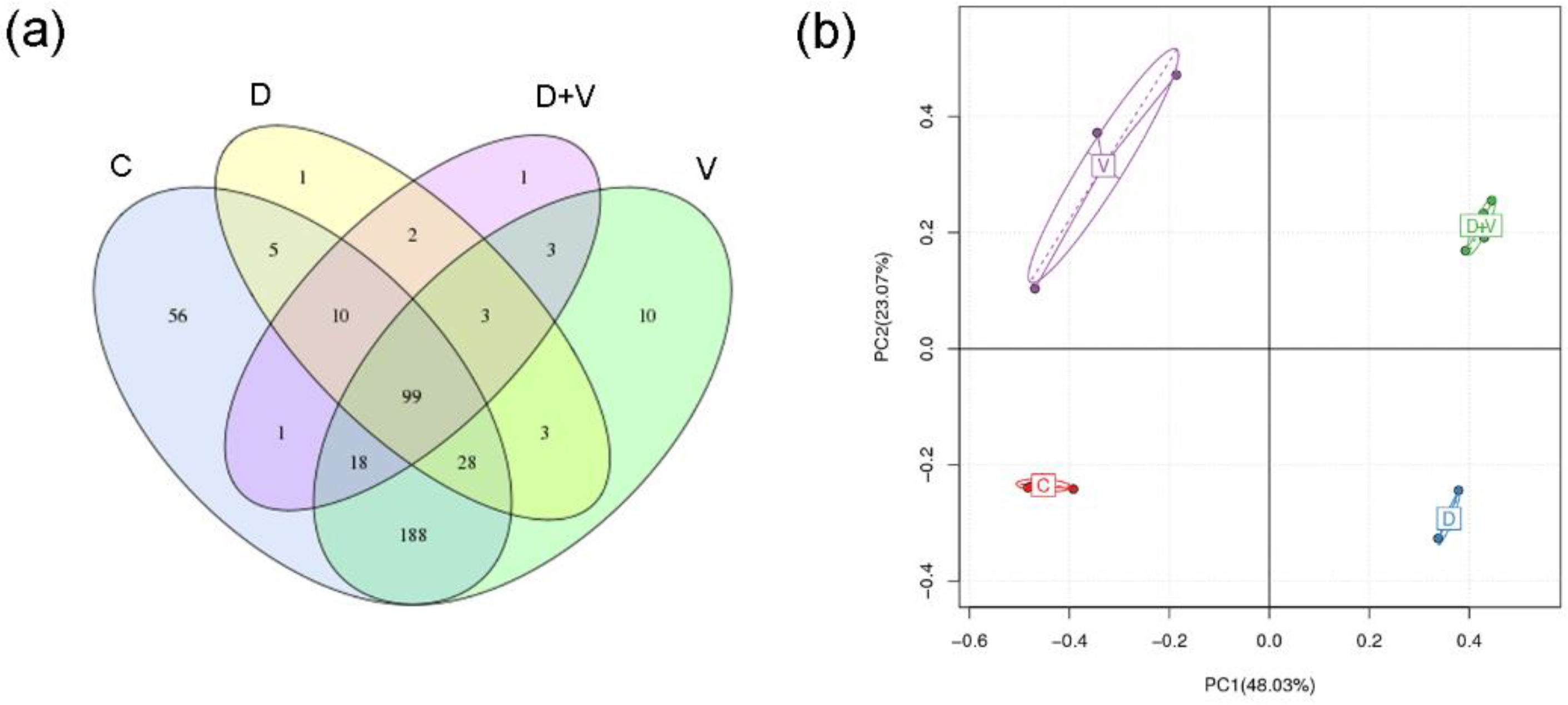
After the mice were sacrificed, all the stool samples were collected from the cecum into sterile centrifuge tubes. Bacterial genomic DNA was extracted using the QIAamp Fast DNA Stool Mini Kit (Cat: 51604, Qiagen, Germany) according to the manufacturer’s protocol. Then, 16S rRNA gene sequencing was carried out by BGI-Shenzhen, Shenzhen, China. A quality test was performed before the qualified DNA was used to construct a library. The 16S rDNA V4 region was amplified by polymerase chain reaction (PCR) and the primers flanking the hypervariable V4 region were 5′-AAATTTTTTTTCCCCCCCGG-3′ (forward primer) and 5′-GGGCCCCCTTTAAAAAAAAAC-3′ (reverse primer). The 2 × 250-bp paired-end reads were generated with the Illumina MiSeq platform (San Diego, CA, USA). To obtain clean reads, the raw data were filtered to eliminate the adapter pollution N base, poly-bases, and low-quality sequencing reads. The high-quality paired-end reads were combined with tags based on overlaps, and then the tags were clustered into operational taxonomic units (OTUs) at 97% sequence similarity by USEARCH (v7.0.1090,
http://www.drive5.com/usearch/). OTU representative sequences were taxonomically classified using the Ribosomal Database Project (RDP) Classifier v.2.2 trained on the Greengenes database (v201305,
http://greengenes.secondgenome.com/downloads). Finally, the alpha diversity and the different species screening were analyzed based on OTU and taxonomic ranks. Software R (v3.1.1,
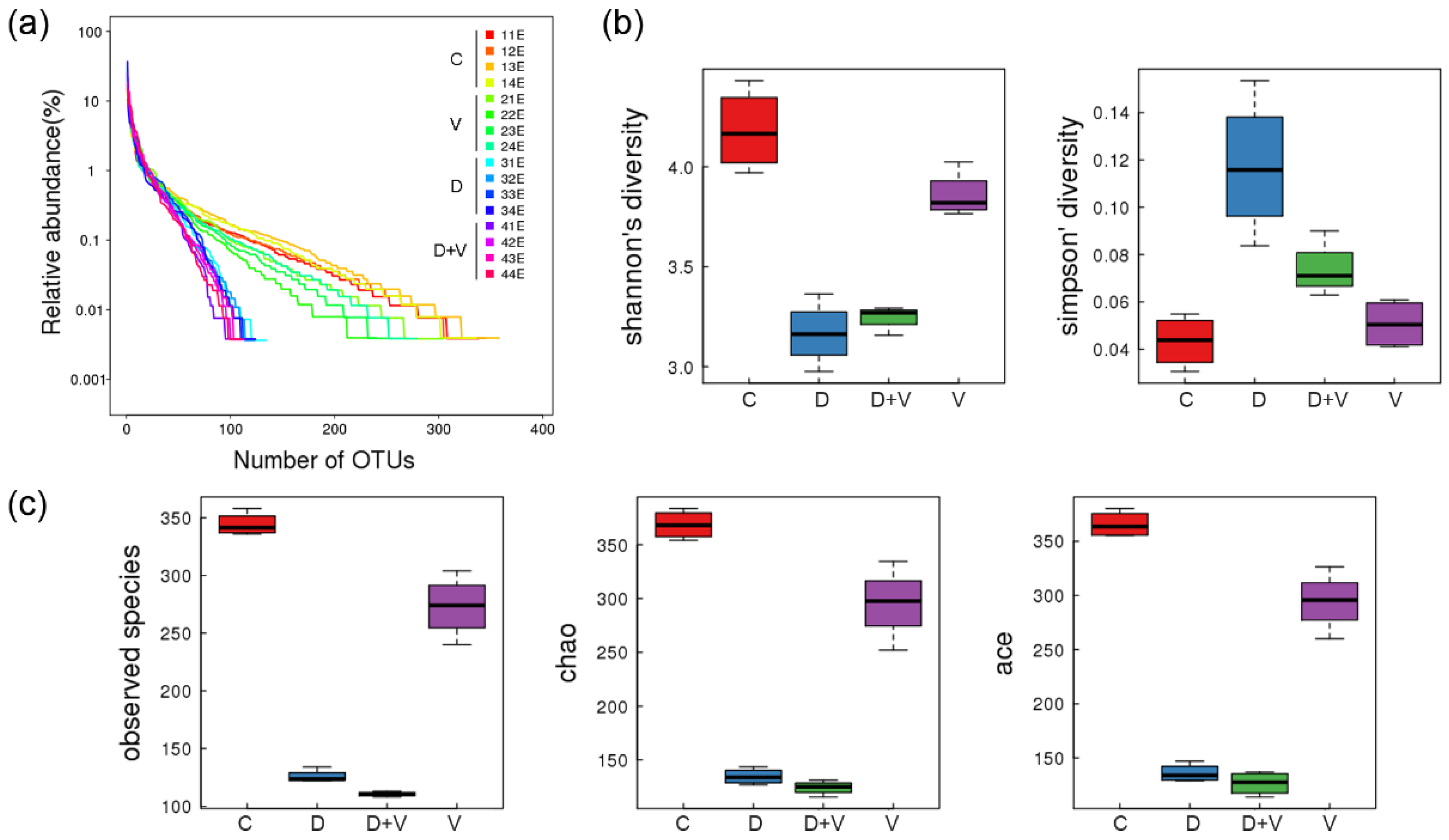
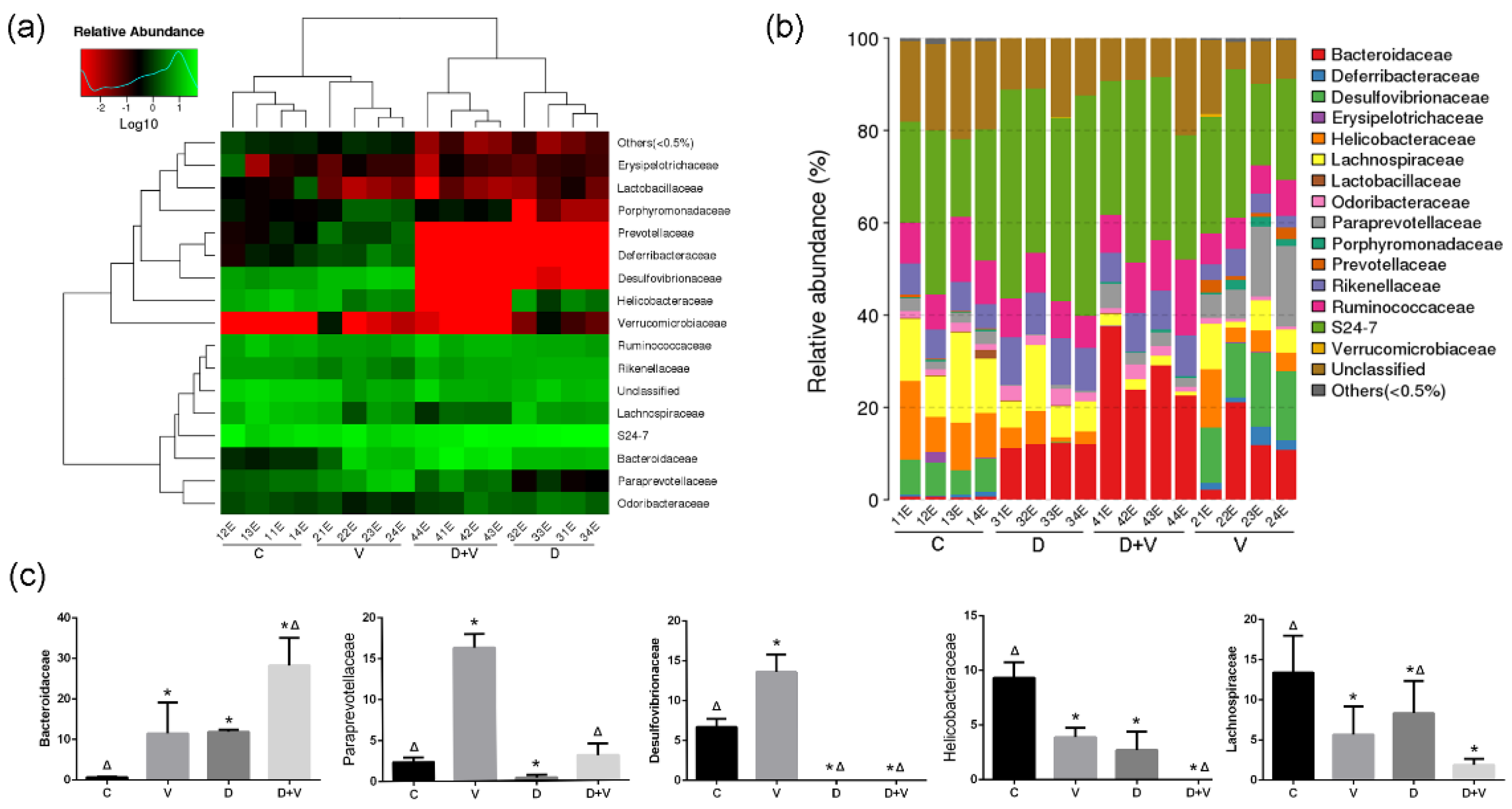
http://www.r-project.org/) was used to draw the Venn diagram, PCA (principal component analysis, PCA, package “ade4”), and the histogram of family taxonomic rank. The species heatmaps were generated using the package “gplots” of software R (v3.1.1). The distance algorithm was “Euclidean”, and the clustering method was “complete”.
2.4. 16S rRNA for Lung Flora
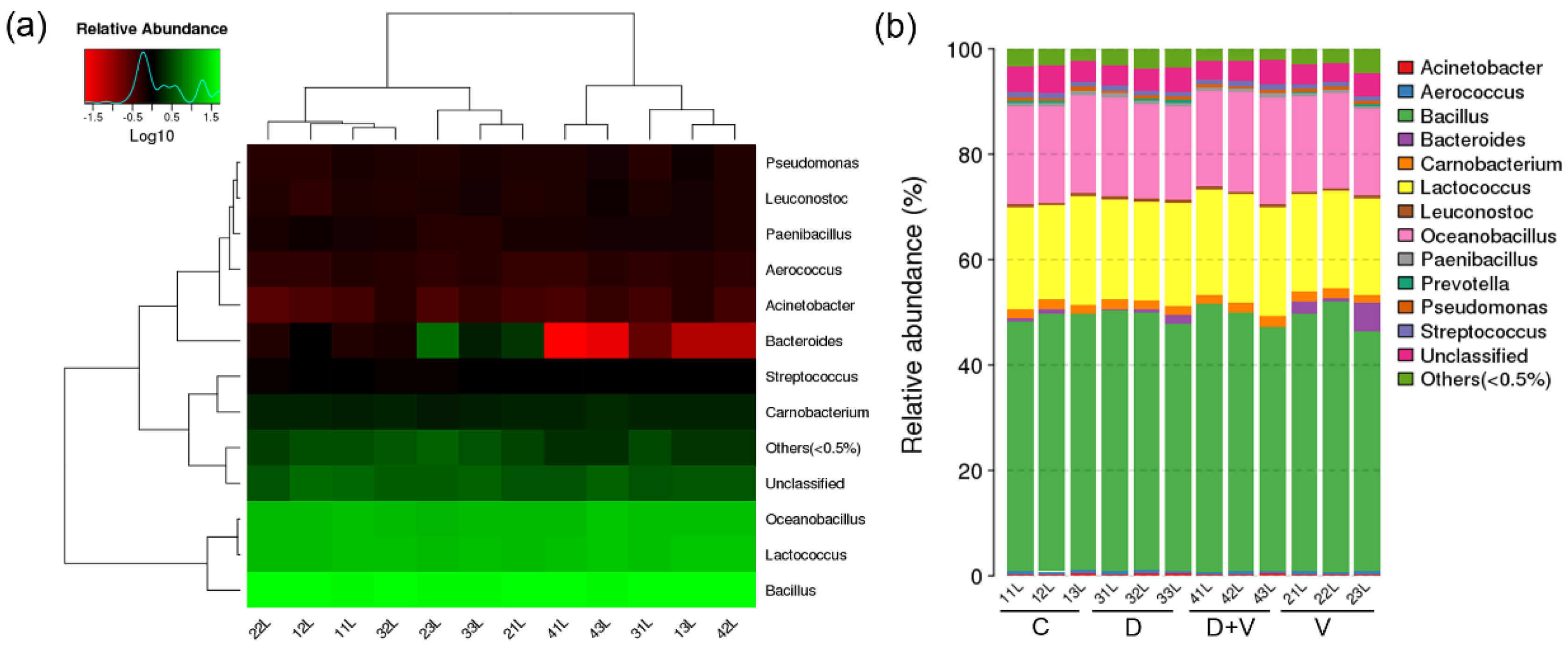
A portion of lung tissue was dissected after the mice were euthanized. Pulmonary bacterial genomic DNA extraction and 16S rRNA gene sequencing were carried out by BGI-Shenzhen, Shenzhen, China. The specific detection methods are as described in
Section 2.2. A heat map and a histogram of genus taxonomic rank were generated using software R (v3.1.1).
2.5. Lung Index and Virus Quantification
The animals were weighed before being sacrificed. The lung tissue of mice was removed and washed with normal saline (NS). After soaking up the water on the surface with a filter, the lung tissue was weighed with an electronic balance. The following formula was used to calculate the lung index: lung index = (lung weight/body weight) × 100%.
Approximately 20 mg of lung tissue was separated from the lung, and the total RNA was extracted with 1 mL of Beyozol reagent (Beyotime Biotechnology, Haimen, China). Nucleic acid concentrations were quantified with a Thermo Nanodrop ND-1000 spectrophotometer (Rockland, DE, USA). The purity of RNA samples was assessed by measuring the optical density ratio at 260 and 280 nm (OD260/OD280). The RNA of each sample was diluted to the same concentration and was reverse-transcribed to complementary DNA (cDNA) using the Reverse Transcription Kit (Tiangen, Biotech Co. Ltd, Beijing, China) according to the manufacturer’s protocol. The viral RNA expression was examined using the real master mix (Tiangen Biotech Co. Ltd, Beijing, China). PCR was performed, and the amplification products were analyzed on a Bio-Rad Mini-Opticon detection system (#CPB3120EDU, Hercules, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was taken as an internal parameter and the viral primer sequences were designed according to the hemagglutinin (HA) gene of influenza A virus. The forward primer sequence of the virus was 5′-GACCAATCCTGTCACCTCTGAC-3′, and the reverse primer sequence was 5′-GGGCATTTGGACAAACGTCTACG-3′. The primers were synthesized by Generay Biotech Co., Ltd, Shanghai, China. The protocol was as follows: 1 min at 95 °C, followed by 40 cycles of 15 s at 94 °C, 30 s at 60 °C, and 30 s at 72 °C. After RT-qPCR, the 2−ΔΔCT method was used for analysis of the relative virus amplification. Each sample was run in triplicate and averaged to ensure accuracy.
2.6. Microarray Analysis, RT-PCR Validation, and Prediction of Differentially Expressed microRNA Targets
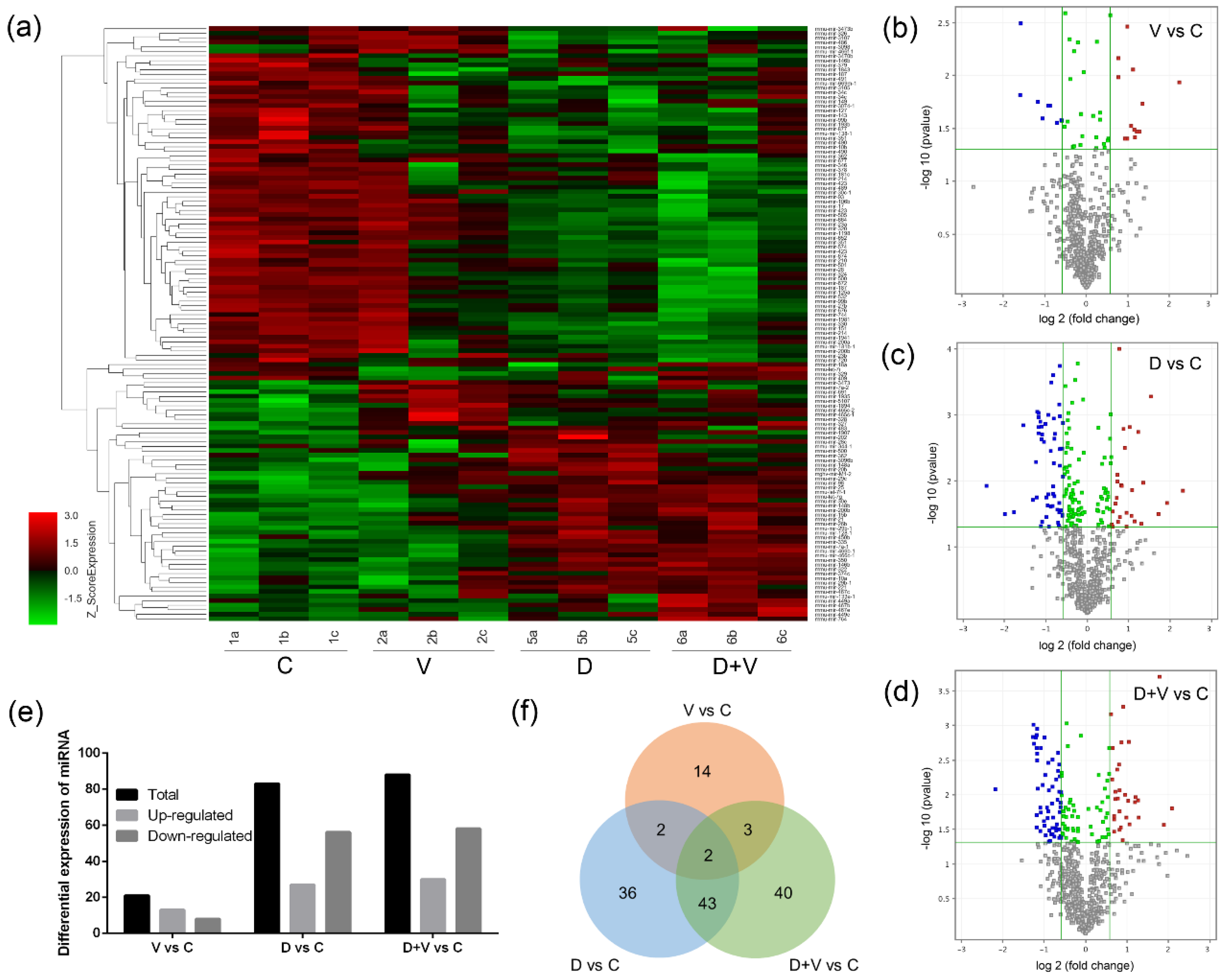
RNA samples were isolated from the lungs of groups C, V, D, and D + V. Microarray analysis of RNA was performed using an Affymetrix Mouse Command Console Chip v4.0 (OE Biotech. Co., Ltd., Shanghai, China). Three biological replicates were processed from each group. The raw data from the miRNA-chip were normalized separately using Agilent GeneSpring GX v12.5 (Santa Clara, CA, USA). Both the on chip and on-gene median methods were used to normalize the gene expression data. Microarray data were then joined into one GeneSpring genome, and samples were assigned to one of two groups. Probes that had at least 100.0% of samples in any one out of two conditions that had flags in “p-value” were chosen for further analysis. Differentially expressed miRNA was then identified through fold change, as well as the p-value calculated using the t-test. The threshold set for up- and downregulated genes was a fold change ≥ 1.5 and a p-value ≤ 0.05. Hierarchical clustering was performed to show the distinguishable miRNA expression pattern among samples.
The miRNAs that changed in all four groups were screened, and the expression levels of all samples were verified by RT-qPCR. The quantification was performed with reverse transcription (RT) and PCR. Each RT reaction consisted of 1 μg of RNA, 4 μL of miScript HiSpec Buffer, 2 μL of Nucleics Mix, and 2 μL of miScript Reverse Transcriptase Mix (Qiagen, Duesseldorf, Germany), for a total volume of 20 μL. Reactions were performed in a GeneAmp
® 9700 PCR System (Applied Biosystems, Waltham, MA, USA) for 60 min at 37 °C, followed by heat inactivation of RT for 5 min at 95 °C. The conditions for RT-qPCR were incubation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s, and 60 °C for 30 s. The miRNA-specific primer sequences were designed based on the miRNA sequences obtained from the miRBase database (Release 20.0,
http://www.mirbase.org/) and synthesized by Generay Biotech (Shanghai, China). Expression analysis was performed in triplicate for each sample. The expression levels of miRNAs were normalized to 18S rRNA and were calculated using the 2
−ΔΔCt method. Furthermore, target genes of differentially expressed miRNAs were predicted using the intersection of the TargetScan (
http://www.targetscan.org/vert_71/) and microRNA.org databases (
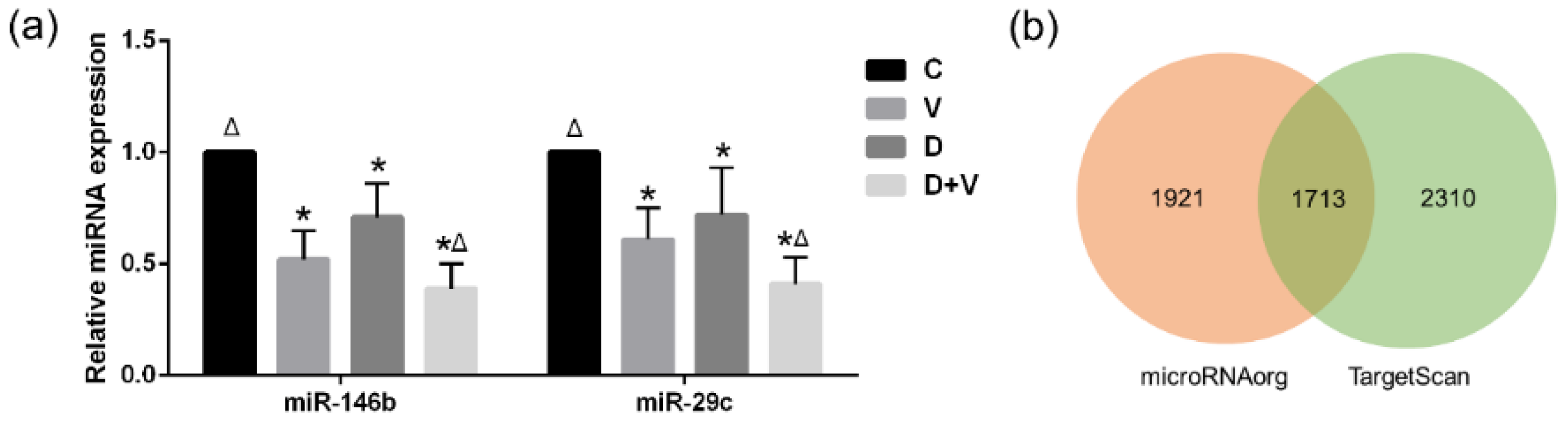
http://www.microrna.org/microrna/microrna/home.do). Target gene prediction was performed using the microRNA.org (version 2012.06.10, default parameters, conservation > 0.25, mirSVR score > −0.1) and microRNA.org (version 2016.06, context score > 50%) databases.
2.7. Statistical Analysis
Data analysis was performed with SPSS for Windows (version 19, IBM Software, New York, NY, USA). The significance of differences between groups was evaluated using one-way ANOVA, and the differences were considered statistically significant at p < 0.05. All values are represented as the means ± SD.
4. Discussion
In recent years, the relationship between the homeostatic maintenance of human health and the intestinal microbiota attracted increasing attention [
14]. Perhaps the symbiotic relationship we share with the intestinal flora has indeed coevolved, as the coexistence of the host with intestinal flora represents an active and mutually beneficial relationship [
15]. The healthy intestinal flora is suggested to be composed of a well-balanced community of three highly evolved groups of bacteria, which were termed symbionts, commensals, and pathobionts [
9]. However, dysbiosis of intestinal flora may result in the loss of host–microbiota symbiosis, which often leads to a shift from symbiont to pathobiont. It means that an entire microbial community under the influence of specific conditions can tip the balance from homeostasis to destructive inflammation [
16].
In this study, neomycin was used to establish the model of intestinal flora imbalance. Although the absorption of neomycin through the intact gastrointestinal mucosa is poor, it can significantly change the intestinal flora [
17]. After neomycin treatment, abundance and species diversity of intestinal flora declined significantly. However, when virus infection was combined with dysbacteriosis, species diversity did not show a further decline, and even raised a little. When the species richness is the same, the greater the species evenness, and the greater the community diversity will be. As the species richness of intestinal flora shows a downward trend, the abnormality indicated that group D + V had a greater species evenness that group D. The higher species evenness indicated that the number of individual species in the community was relatively close. Combined with the species taxonomic results, it might be related to an outgrowth of potential pathogenic bacteria (pathobionts) and a decrease in the number of beneficial bacteria.
As a characteristic change in flora, the bacterial count of Bacteroidaceae was elevated in both group V and group D, and the greatest elevation was shown in group D + V. Bacteroidaceae contains some commensal bacteria with pathogenic potential, such as
Bacteroides fragilis.
B. Fragilis was found be able to promote regulatory T cell (Treg) function, and the phenomenon was associated with the capacity of this bacterium to limit T helper cell 17 (Th17) responses, which could suppress the immune response [
18]. In addition, recent results suggested commensal microbes could induce constitutive and physiological inflammation through pattern-recognition receptors (PRRs) [
19]. Nod2 is a receptor of nucleotide-binding oligomerization domain-like receptors (NLRs), which regulate the receptor-interacting protein 2 (RIP2)-dependent nuclear factor kappa B (NF-κB) pathway, and its deficiency was reported to lead to dysbiosis characterized by the outgrowth of Bacteroidetes [
20]. Thus, the increase of Bacteroidaceae might be related to the weakened antiviral ability the under alteration of intestinal flora. Furthermore, antibiotic interference and virus infection significantly decreased the relative abundance of Desulfovibrionaceae, Helicobacteraceae, and Lachnospiraceae in the murine colon microbiota. Although neomycin treatment decreased the opportunistic pathogens (Desulfovibrionaceae), it destroyed the normal colony structure and species abundance. Therefore, the alteration of intestinal flora induced by antibiotics and the influenza virus not only reduced species abundance, but also destroyed the normal symbiotic relationship of the intestinal flora, including the reduction of beneficial bacteria (Lachnospiraceae) and an increase of opportunistic pathogen (Bacteroidaceae).
Soner Yildiz found that influenza A virus (IAV) infection resulted in a reduction of commensal community richness, and also implicated that probiotics treatment could potentially balance the IAV-induced dysbiosis [
21]. The changes in intestinal flora could be related to the abnormity of immune function and imbalance of immunologic homeostasis. In this study, higher viral load and lung index in the D + V group compared with group V were found. This phenomenon suggested that pulmonary virus infection could affect the intestinal microbiome; in turn, the imbalance of the intestinal flora could affect antiviral immunity in the lung and increase its susceptibility to influenza.
MicroRNAs are a class of highly conserved noncoding single-stranded RNA molecules approximately 18–25 nucleotides in length, which play roles in the regulation of gene and protein expression at the post-transcriptional level [
22]. As miRNAs participate in the modulation of both the innate and adaptive immune response, their abnormal expression may lead to disorder of the immune system [
23]. Studies showed that miRNA plays important roles in the regulation of T-cell differentiation, the TLR7 signaling pathway, and the expression of inflammatory cytokines [
24]. Thus, the decrease in host antivirus immune function might be related to the change in miRNAs. Based on the complex and precise physiological response of the human body, the relationship between intestinal flora and miRNA may be bidirectional. Williams et al. [
25] raised three possible interactions between the host and gut microbiome involving miRNAs, which include miRNA regulation of host gene expression, influence of host miRNA expression by the gut microbiota, and influence of the gut microbiota through the release of miRNAs by the host. We sought to understand the role miRNA played in the interactions between intestinal flora and influenza infections.
Generally, it is believed that local microflora is more closely related to vicinal miRNA. However, the similar composition and structure of pulmonary flora based on 16S rRNA gene sequencing proved that neomycin had little effect on pulmonary flora. Since the oral administration of neomycin had almost no effect on the lung, it proved that it was not the local pulmonary bacterial flora changes that affected the expression of miRNAs. The miRNA microarray results showed that pulmonary miRNA expression was significantly changed after alteration of the intestinal flora. Therefore, it was the maladjusted intestinal flora that impacted the expression of pulmonary miRNAs. Combined with the important role of small RNAs in immunity, we speculate that the disorder of intestinal flora could affect the pulmonary immune response through miRNAs. After comparison and screening the differentially expressed miRNAs among the four groups, two common miRNAs were found: miR-146b and miR-29c. They are both related to immune response and might be suitable as therapeutic targets.
The miR-146 family includes miR-146a and miR-146b. Analysis of miR-146 gene expression unveiled its important role in response to a variety of microbial components and proinflammatory cytokines. Specifically, miR-146a was found to be an NF-κB-dependent gene, which could downregulate the protein levels of interleukin-1 receptor-associated kinase 1 (IRAK1) and tumor necrosis factor receptor-associated factor 6 (TRAF6) in Toll-like receptor signaling [
26]. Moreover, Park et al. reported that miR-146a and miR-146b modulated DC apoptosis and cytokine production, which was inversely correlated with TRAF6 and IRAK1 expression [
27]. In addition, our previous work revealed the negative effect of dysbacteriosis on the activation of the TLR7 signaling pathway and the release of type I interferon [
11]. In this study, we found that miR-146a expression was reduced during viral infection, and it showed a more significant decreasing trend when viral infection was combined with dysbacteriosis. Therefore, under dysbacteriosis in influenza virus infection, miR-146a might work in connection with the suppression of TLR signaling pathway activation, which downregulates the host immune recognition mechanism-induced antiviral function.
The miR-29 family could affect helper T-cell differentiation and regulate type I interferon secretion to exert an antivirus role [
28]. Ma et al. found that miR-29 expression was substantially downregulated in activated interferon gamma (IFN-γ)-producing natural killer (NK) cells and T-cell subsets [
29]. Through the regulation of IFN-γ mRNA, miR-29 participated in the regulation of NK cell function and the Th1 cell response mechanism. As miR-29a and miR-29b were downregulated in IFN-γ secretion-type T cells, the increases in IFN-γ and Th1 inhibited the expression of miR-29. This study showed that, after viral infection, the expression level of miR-29c exhibited a decreasing trend. Combined with the previous studies, the mechanism might be associated with an increase in IFN-γ and the activation of NF-κB. Dysbacteriosis also led to a decrease in miR-29c. As observed in previous studies, the NF-κB expression in the dysbacteriosis group was higher than normal [
11], and the associated inflammatory response might be responsible for the decrease in miR-29c. However, this trend was not significant. In addition, dysbacteriosis with viral infection also decreased the expression level of miR-29c. Thus, dysbacteriosis inhibited the expression of miR-29c during influenza virus infection, which could induce the enhancement of IFN-γ and NF-κB and result in a more serious inflammatory reaction.
Here, we only verified and analyzed miR-146b and miR-29c, which were found to have differential expression between group V vs. C, group D vs. C, and group D + V vs. C. Based on the discussion of their function, we believe that miR-146b and miR-29c might be good resources of resistance to influenza with intestinal flora alteration. However, the results of miRNA microarray analysis provided more information about the miRNAs and their target genes, which could help us find new approaches to fight influenza. For instance, miR-465c, miR-187, and miR-327 are also worth exploring under the effects of influenza and antibiotic abuse. As shown previously, miR-187 is a cancer-related microRNA, which was reported to play promoting or suppressive roles in different malignancies [
30,
31]. On the other hand, miR-327 was reported to indirectly suppress the TLR4 and TLR2 signaling pathways, and subsequently resulted in reduced myocardial infarct size and alleviated inflammation [
32]. The results suggest that miR-465c, miR-187, and miR-327 could also serve as a promising therapeutic targets for influenza with or without intestinal flora alteration.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







