The Degradation Products of Ascorbic Acid Inhibit Amyloid Fibrillation of Insulin and Destabilize Preformed Fibrils


Abstract
1. Introduction
2. Results
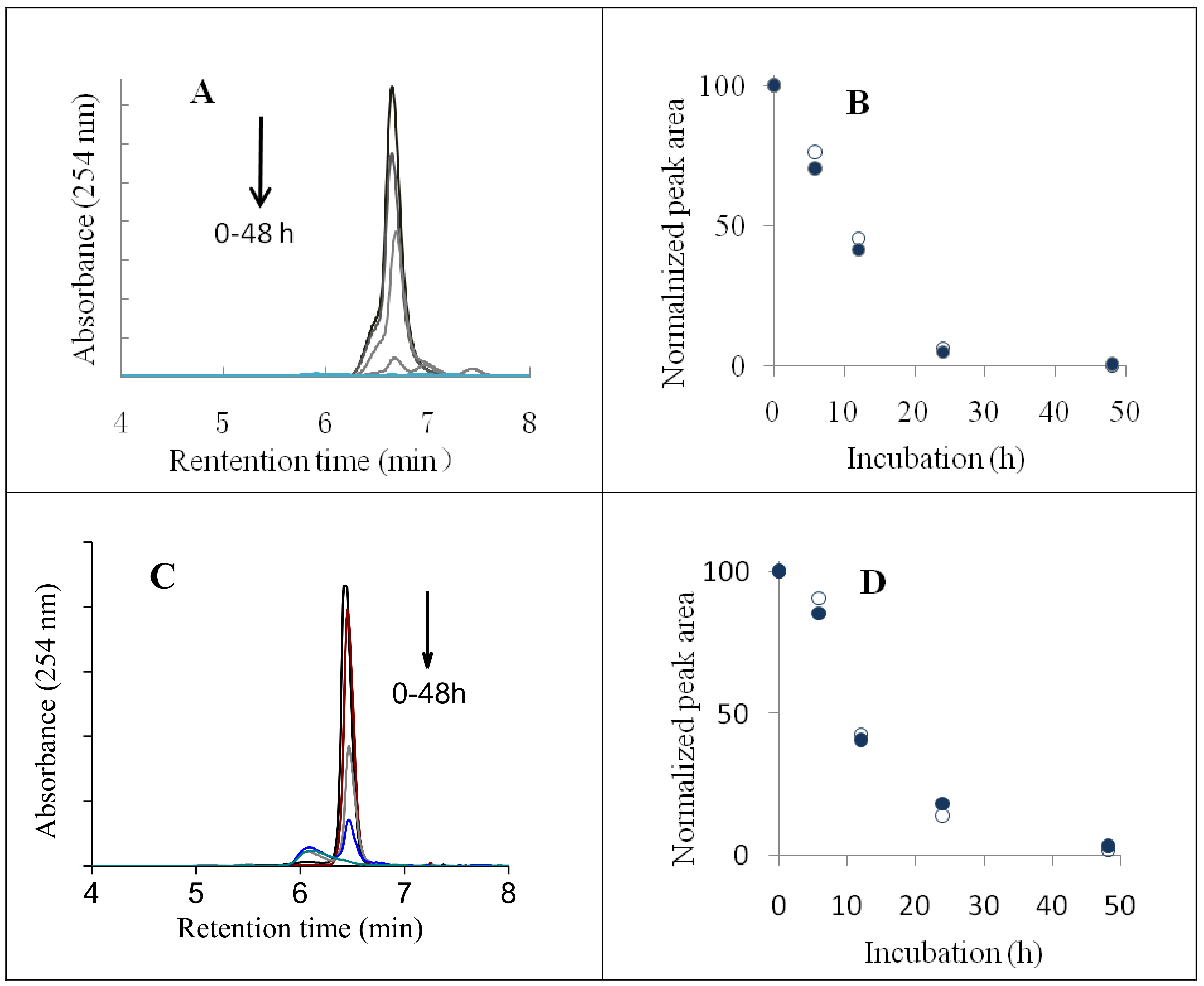
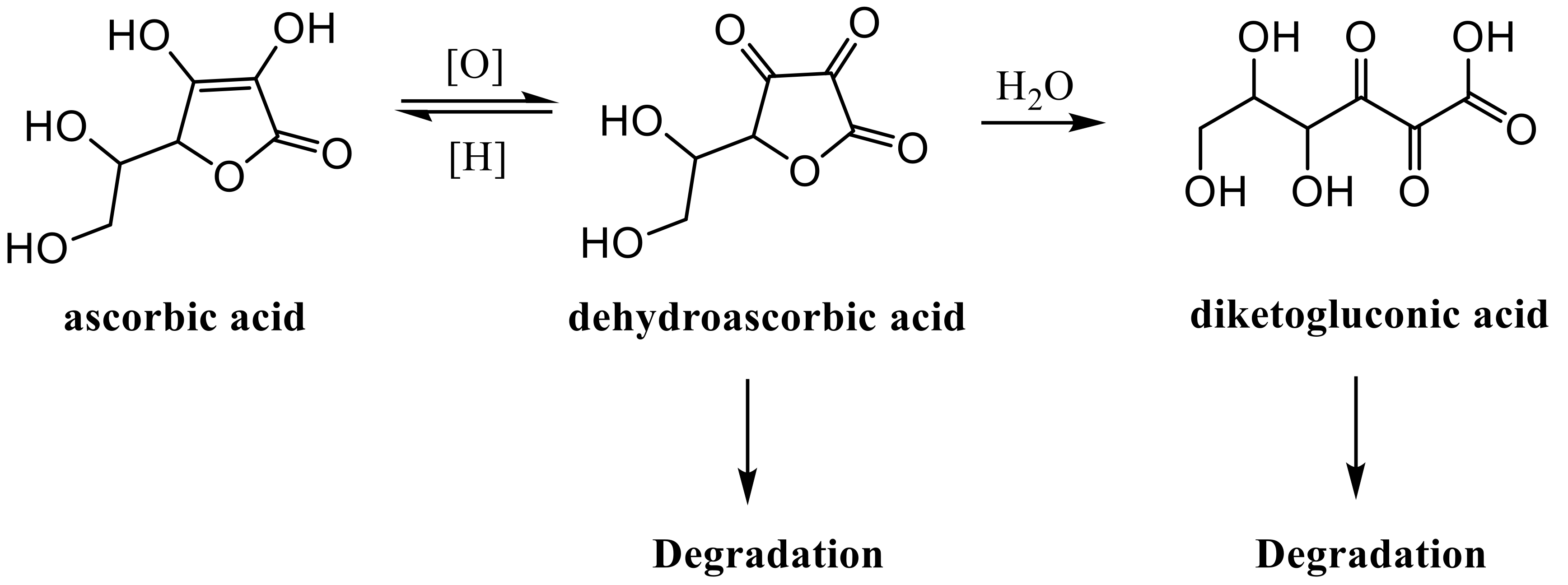
2.1. HPLC Analyses of AsA Degradation
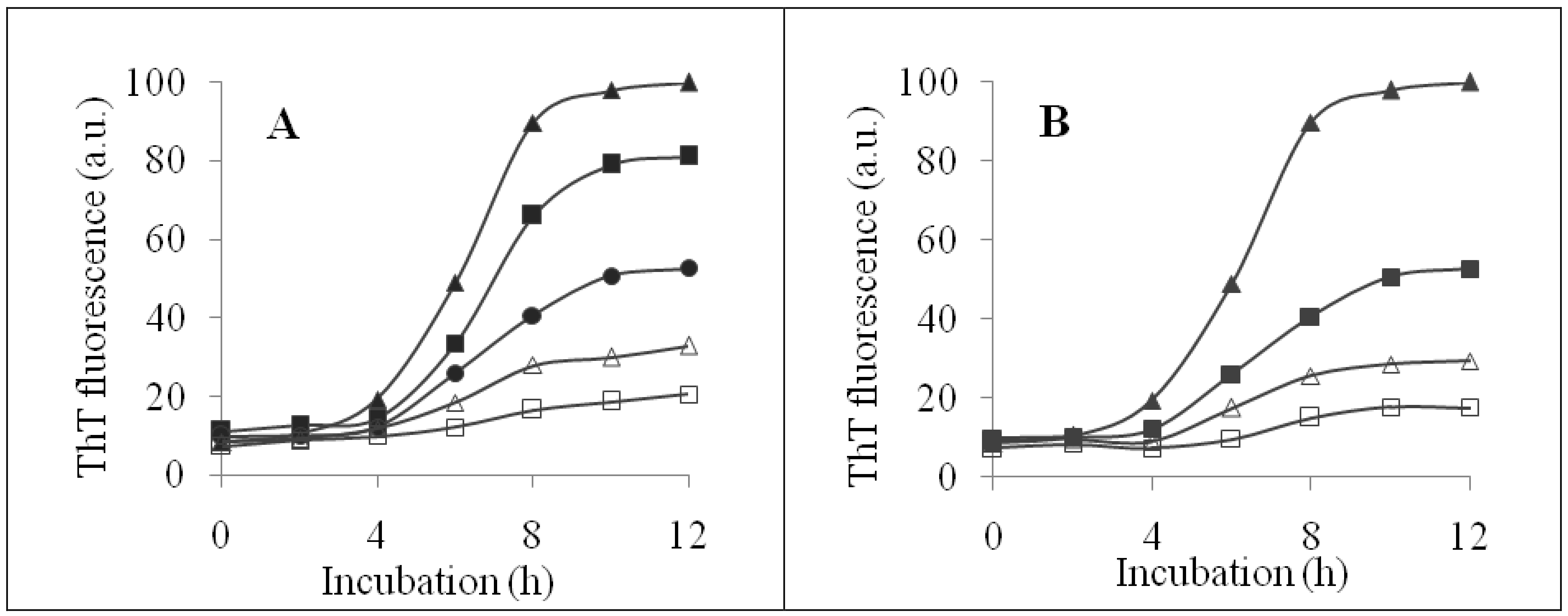
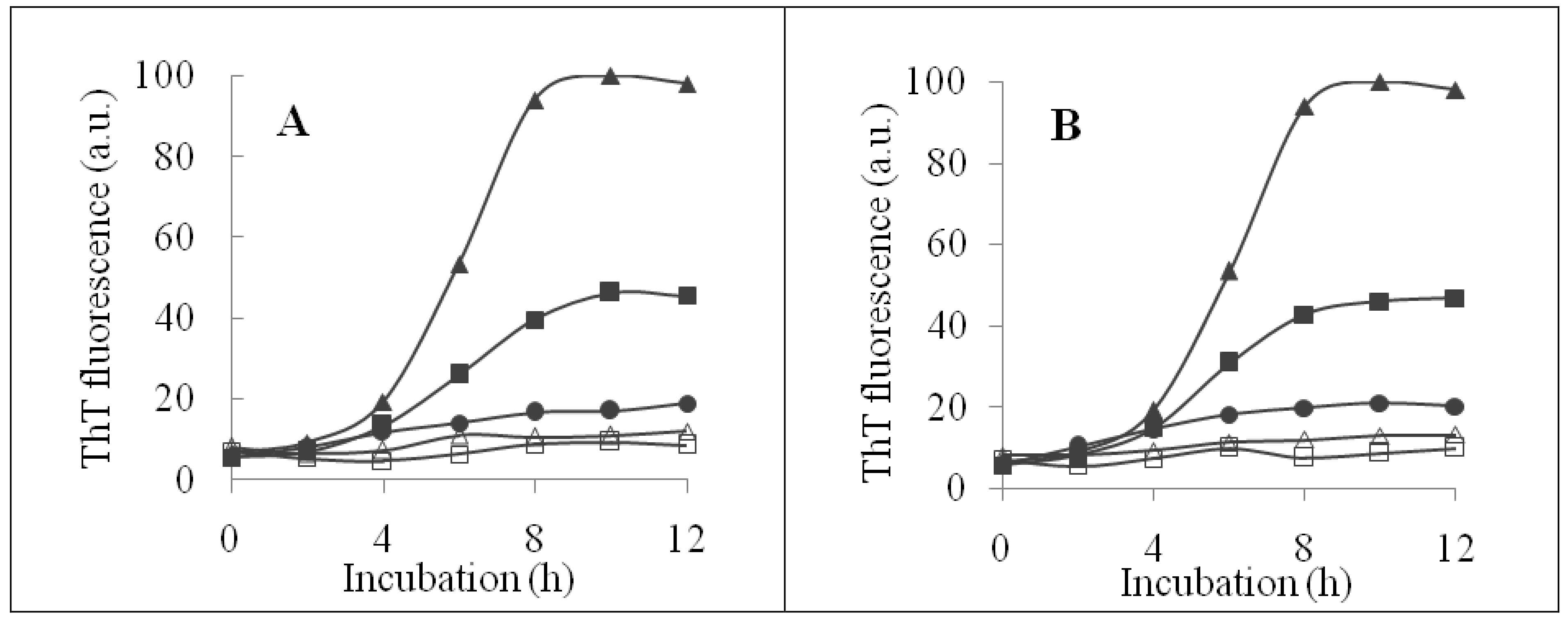
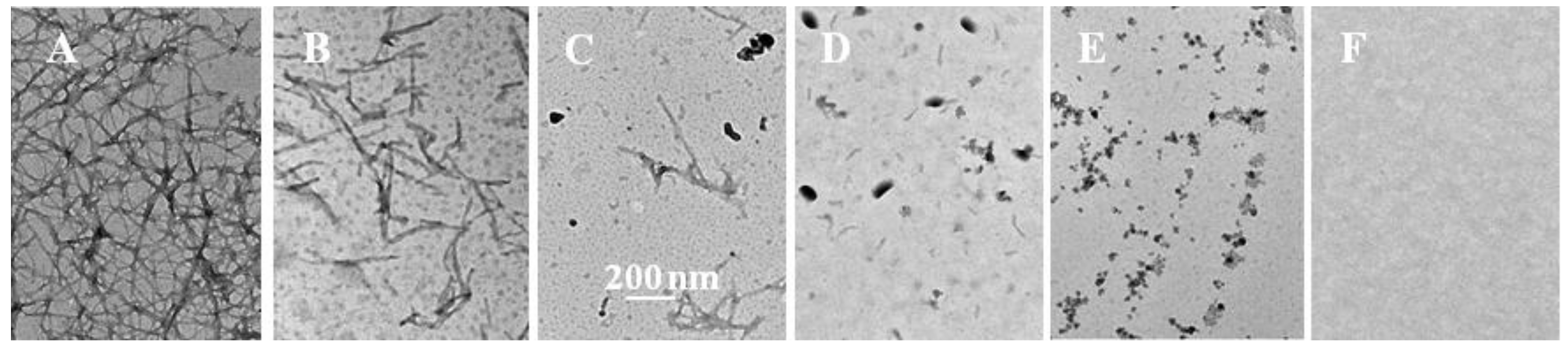
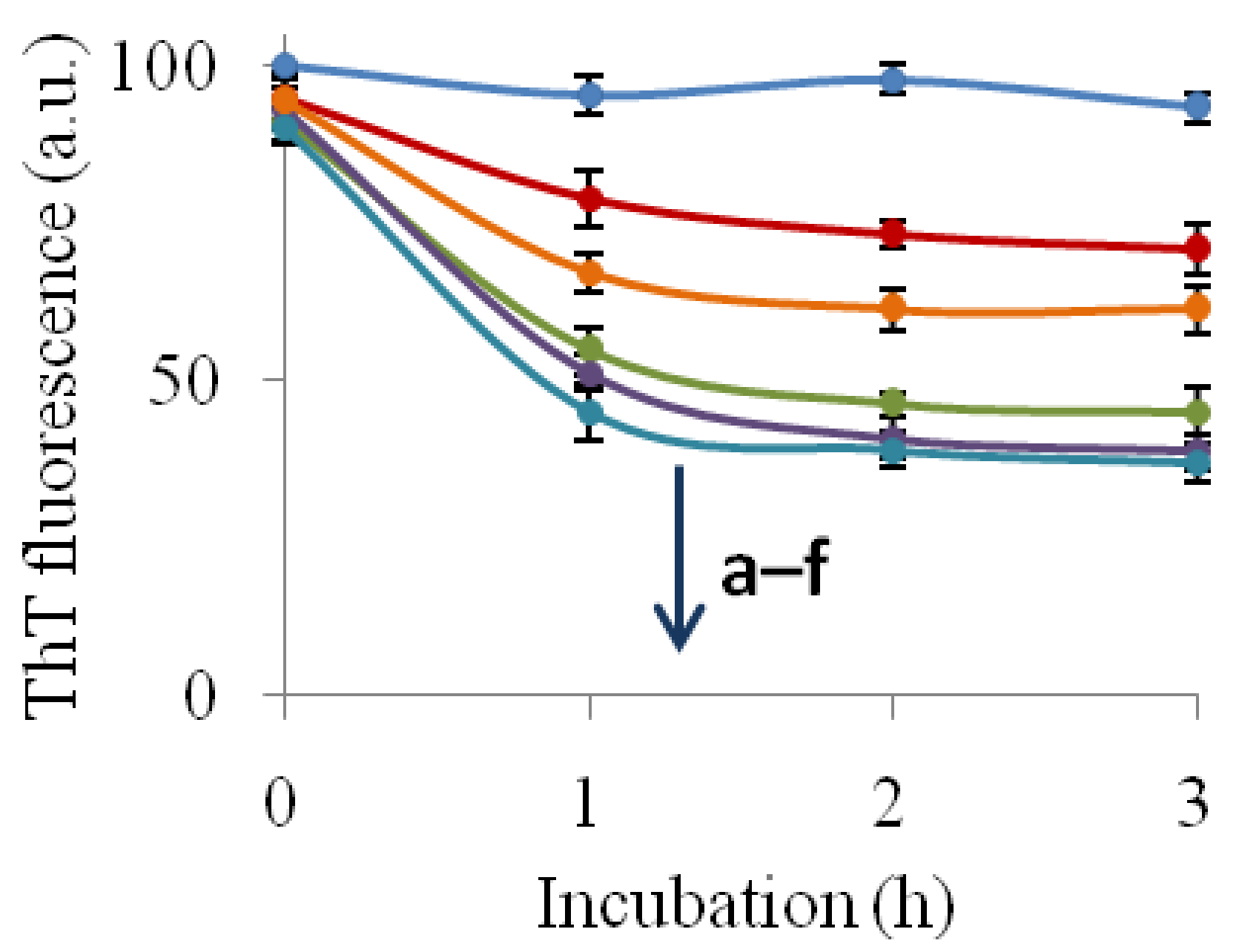
2.2. Inhibition of Insulin Fibrillation by AsA and Its Degradation Products
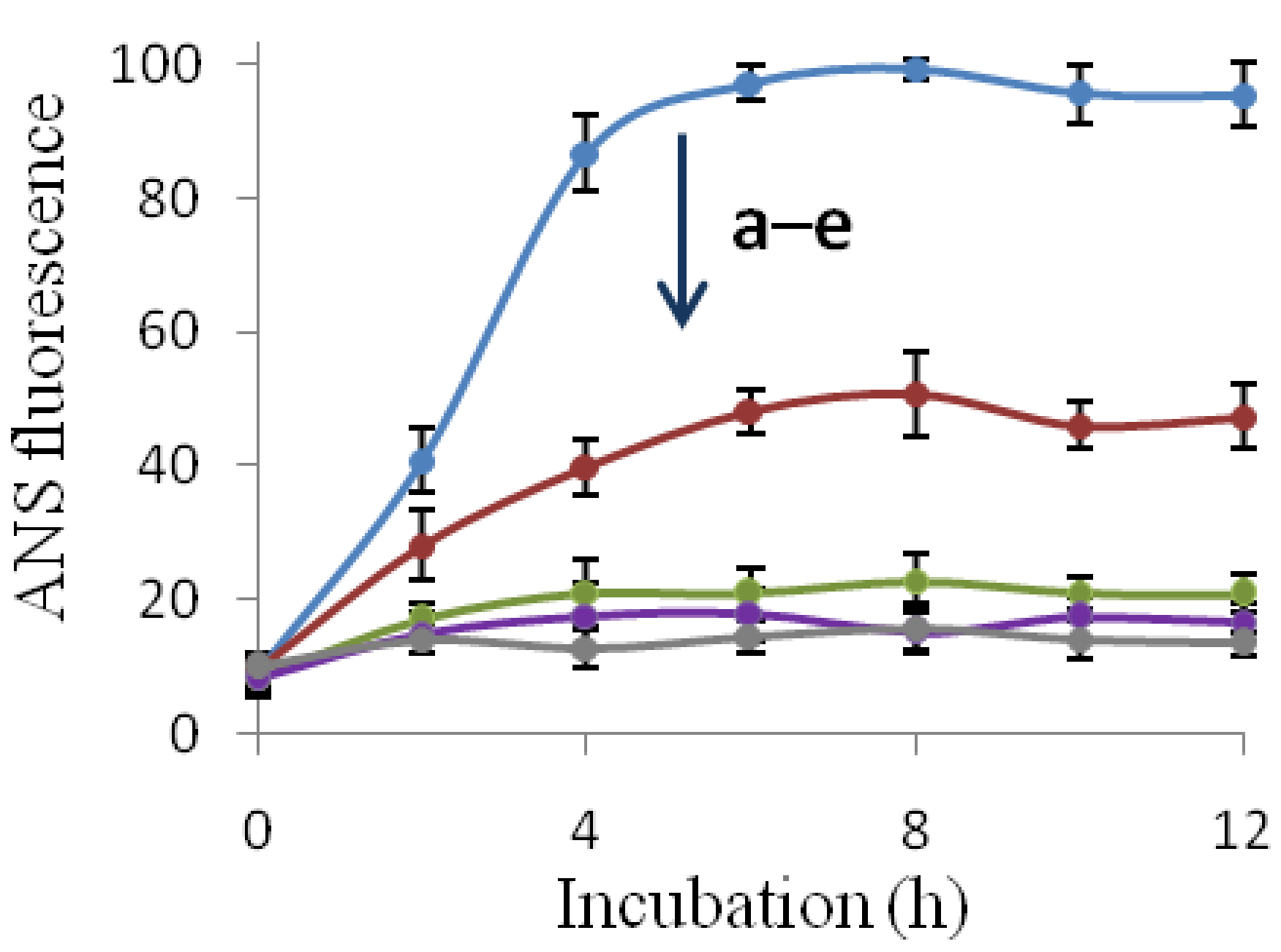
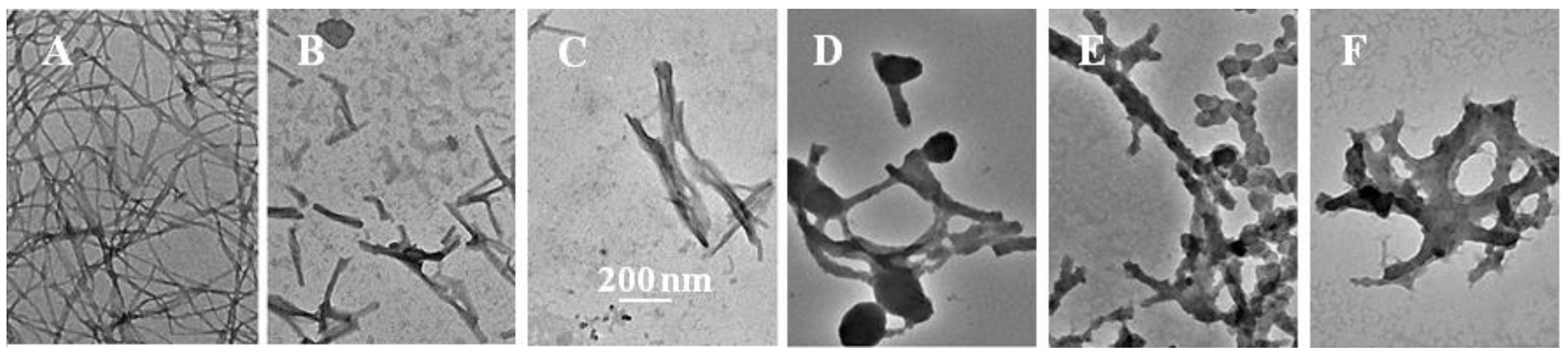
2.3. The Fibril-Disruptive Roles of AsA and Its Degradation Products
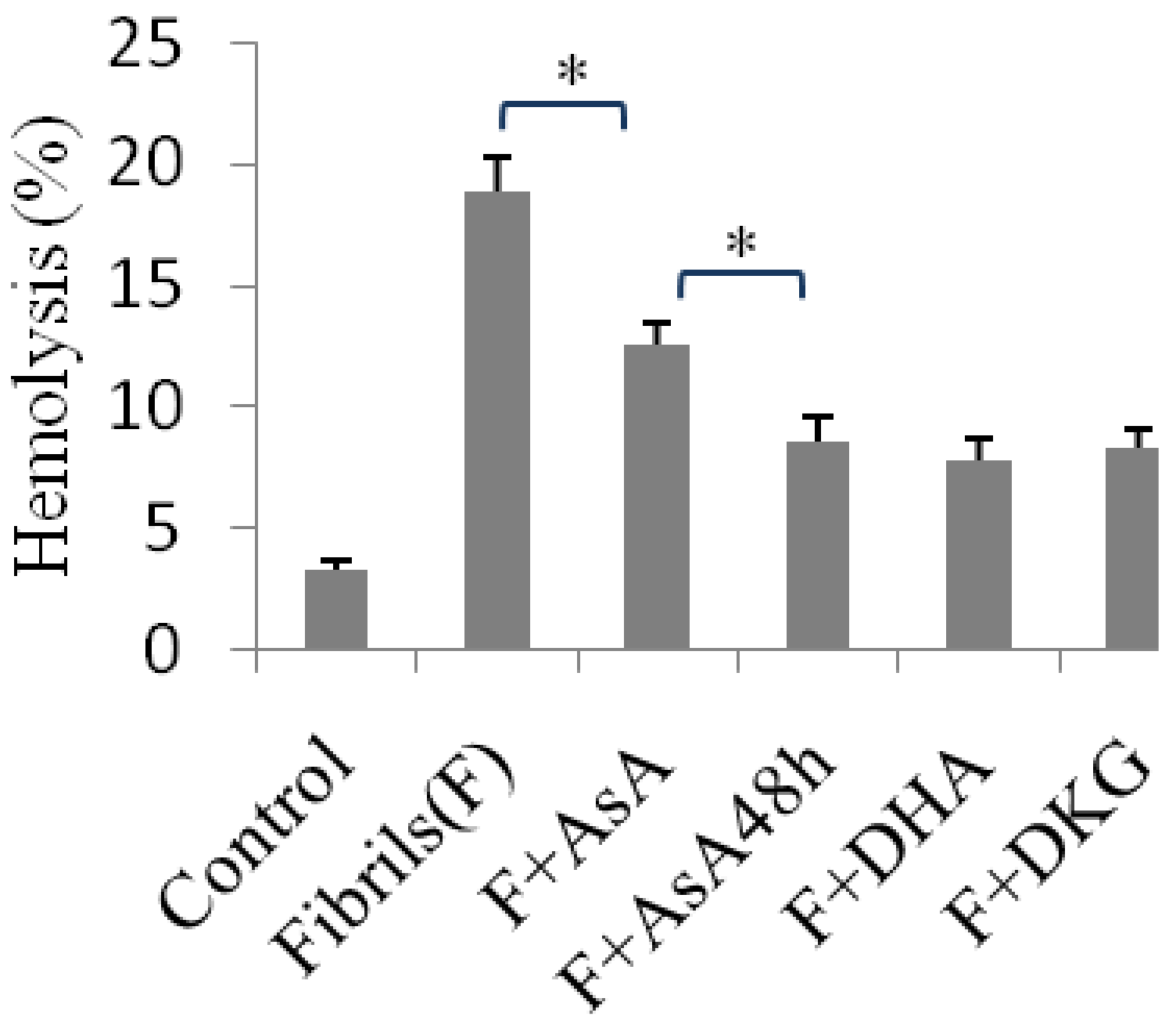
2.4. Hemolysis Induced Using Insulin Fibrils Pretreated with an Inhibitor
3. Materials and Methods
3.1. Chemicals
3.2. Stability of AsA
3.3. Preparation and Characterization of Insulin Fibrils
3.4. Fibril-Disruptive Assay
3.5. Hemolytic Assays
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stefani, M. Protein misfolding and aggregation: New examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 2004, 1739, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; He, J.; Ren, J.; Yan, X.Y.; Zeng, C.M. Cellular membrane disruption by amyloid fibrils involved intermolecular disulfide cross-linking. Biochemistry 2009, 48, 5794–5800. [Google Scholar] [CrossRef] [PubMed]
- Shewmaker, F.; McGlinchey, R.P.; Wickner, R.B. Structural insights into functional and pathological amyloid. J. Biol. Chem. 2011, 286, 16533–16540. [Google Scholar] [CrossRef] [PubMed]
- Martino, P.D. Bap: A new type of functional amyloid. Trends Microbiol. 2016, 24, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Millett, I.S.; Doniach, S.; Uversky, V.N.; Fink, A.L. Partially folded intermediates in insulin fibrillation. Biochemistry 2003, 42, 11404–11416. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.X.; Weiss, M.A. Mechanism of insulin fibrillation: The structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J. Biol. Chem. 2004, 279, 21449–21460. [Google Scholar] [CrossRef] [PubMed]
- Noormägi, A.; Valmsen, K.; Tõugu, V.; Palumaa, P. Insulin fibrillization at acidic and physiological pH values is controlled by different molecular mechanisms. Protein J. 2015, 34, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Sievers, S.A.; Sawaya, M.R.; Wall, J.S.; Eisenberg, D. Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. USA 2009, 106, 18990–18995. [Google Scholar] [CrossRef] [PubMed]
- Shikama, Y.; Kitazawa, J.; Yagihashi, N.; Uehara, O.; Murata, Y.; Yajima, N.; Wada, R.; Yagihashi, S. Localized amyloidosis at the site of repeated insulin injection in a diabetic patient. Intern. Med. 2010, 49, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Clos, A.L.; Midoro-Hiriuti, T.; Goldblum, R.M.; Jackson, G.R.; Kayed, R. Inhaled insulin forms toxic pulmonary amyloid aggregates. Endocrinology 2010, 151, 4717–4724. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, O.M.; Grishin, S.Y.; Glyakina, A.V.; Sadgyan, A.S.; Ushakova, N.I.; Galzitskaya, O.V. Analysis of insulin analogs and the strategy of their further development. Biochemistry (Moscow) 2018, 83, 146. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.S.; Liu, K.N.; Han, T.C. Amyloid fibrillation and cytotoxicity of insulin are inhibited by the amphiphilic surfactants. Biochim. Biophys. Acta 2010, 1802, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Kachooei, E.; Moosavi-Movahedi, A.A.; Khodagholi, F.; Mozaffarian, F.; Sadeghi, P.; Hadi-Alijanvand, H.; Ghasemi, A.; Saboury, A.A.; Farhadi, M.; Sheibani, N. Inhibition study on insulin fibrillation and cytotoxicity by paclitaxel. J. Biochem. 2014, 155, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Uversky, V.N.; Hong, D.; Fink, A.L. Early events in the fibrillation of monomeric insulin. J. Biol. Chem. 2005, 280, 42669–42675. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free. Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.; Lee, J.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.A.; Kang, J.Q.; Kennard, J.A.; Harrison, F.E. Low brain ascorbic acid increases susceptibility to seizures in mouse models of decreased brain ascorbic acid transport and Alzheimer’s disease. Epilepsy Res. 2015, 110, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, aging and Alzheimer’s disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; May, J.M. Ascorbic acid protects SH-SY5Y neuroblastoma cells from apoptosis and death induced by β-amyloid. Brain Res. 2006, 1097, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Hasanbašić, S.; Jahić, A.; Berbić, S.; Žnidarič, M.T.; Žerovnik, E. Inhibition of protein aggregation by several antioxidants. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Beg, A.Z.; Siddiqi, M.K.; Chaturvedi, S.K.; Rajpoot, R.K.; Ajmal, M.R.; Zaman, M.; Abdelhameed, A.S.; Khan, R.H. Ascorbic acid inhibits human insulin aggregation and protects against amyloid induced cytotoxicity. Arch. Biochem. Biophys. 2017, 621, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Parmar, K.; Patel, D.; Kumar, S.; Trivedi, M.; Das, M. Inhibition of amyloid fibril formation of lysozyme by ascorbic acid and a probable mechanism of action. Int. J. Biol. Macromol. 2018, 114, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Azzam, S.K.; Jang, H.; Choi, M.C.; Alsafar, H.; Lukman, S.; Lee, S. Inhibition of human amylin aggregation and cellular toxicity by lipoic acid and ascorbic acid. Mol. Pharm. 2018, 15, 2098–2106. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, I.; Lee, S.W.; Lee, H.; Lee, G.; Kim, S.; Lee, S.W.; Yoon, D.S. Quantifying l-ascorbic acid-driven inhibitory effect on amyloid fibrillation. Macromol. Res. 2016, 24, 868–873. [Google Scholar] [CrossRef]
- Yuan, J.P.; Chen, F. Degradation of ascorbic acid in aqueous solution. J. Agric. Food Chem. 1998, 46, 5078–5082. [Google Scholar] [CrossRef]
- Szultka, M.; Buszewska-Forajta, M.; Kaliszan, R.; Buszewski, B. Determination of ascorbic acid and its degradation products by high-performance liquid chromatography-triple quadrupole mass spectrometry. Electrophoresis 2014, 35, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Koshiishi, I.; Mamura, Y.; Liu, J.; Imanari, T. Degradation of dehydroascorbate to 2,3-diketogulonate in blood circulation. Biochim. Biophys. Acta 1998, 1425, 209–214. [Google Scholar] [CrossRef]
- Wang, J.B.; Wang, Y.M.; Zeng, C.M. Quercetin inhibits amyloid fibrillation of bovine insulin and destabilizes preformed fibrils. Biochem. Biophys. Res. Commun. 2011, 415, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Cunningham, L.; Rose, R.C. Spontaneous decay of oxidized ascorbic acid (dehydro-L-ascorbic acid) evaluated by high-pressure liquid chromatography. Clin. Chem. 1990, 36, 1807–1809. [Google Scholar] [PubMed]
- Simpson, G.L.W.; Ortwerth, B.J. The non-oxidative degradation of ascorbic acid at physiological conditions. Biochim. Biophys. Acta 2000, 1501, 12–24. [Google Scholar] [CrossRef]
- Kong, L.X.; Zeng, C.M. Effects of seeding on lysozyme amyloid fibrillation in the presence of epigallocatechin and polyethylene glycol. Biochemistry (Moscow) 2017, 82, 156–167. [Google Scholar] [CrossRef] [PubMed]
- An, T.T.; Feng, S.; Zeng, C.M. Oxidized epigallocatechin gallate inhibited lysozyme fibrillation more strongly than the native form. Redox Biol. 2017, 11, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Kärkönen, A.; Dewhirst, R.A.; Mackay, C.L.; Fry, S.C. Metabolites of 2,3-diketogulonate delay peroxidase action and induce non-enzymic H2O2 generation: Potential roles in the plant cell wall. Arch. Biochem. Biophys. 2017, 620, 12–22. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xing, Y.F.; Huang, B.; Zhang, Y.Z.; Zeng, C.M. Tea catechins induced the conversion of preformed lysozyme amyloid fibrils to amorphous aggregates. J. Agric. Food Chem. 2009, 57, 11391–11396. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Yoshiike, Y.; Takashima, A.; Yamada, M.; Naiki, H. Nordihydroguaiaretic acid potently breaks down pre-formed Alzheimer’s beta-amyloid fibrils in vitro. J. Neurochem. 2002, 81, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; Van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Toyoshima, Y.; Kurata, T. Identification of 3,4-dihydroxy-2-oxo-butanal (l-threosone) as an intermediate compound in oxidative degradation of dehydro-l-ascorbic acid and 2,3-diketo-l-gulonic acid in a deuterium oxide phosphate buffer. Biosci. Biotechnol. Biochem. 2001, 65, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-β oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Omar, S.H. Biophenols pharmacology against the amyloidogenic activity in Alzheimer’s disease. Biomed. Pharmacother. 2017, 89, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibrildestabilizing effects for α-synuclein fibrils in vitro. J. Neurochem. 2006, 97, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Shoval, H.; Weiner, L.; Gazit, E.; Levy, M.; Pinchuk, I.; Lichtenberg, D. Polyphenol-induced dissociation of various amyloid fibrils results in a methionine-independent formation of ROS. Biochim. Biophys. Acta 2008, 1784, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, M.; Hasegawa, K.; Tsutsumi-Yasuhara, S.; Ohhashi, Y.; Ookoshi, T.; Ono, K.; Yamada, M.; Naiki, H. The anti-amyloidogenic effect is exerted against Alzheimer’s β-amyloid fibrils in vitro by preferential and reversible binding of flavonoids to the amyloid fibril structure. Biochemistry 2007, 46, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Song, X.H.; Zeng, C.M. Inhibition of amyloid fibrillation of lysozyme by phenolic compounds involves quinoprotein formation. FEBS Lett. 2012, 586, 3951–3955. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Zhang, Y.J.; Feng, S.; Zeng, C.M. Quinopeptide formation associated with the disruptive effect of epigallocatechin gallate on lysozyme fibrils. Int. J. Biol. Macromol. 2015, 78, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Schoneich, C. Methionine oxidation by reactive oxygen species: Reaction mechanisms and relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1703, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270–3274. [Google Scholar] [CrossRef] [PubMed]
- Shoval, H.; Lichtenberg, D.; Gazit, E. The molecular mechanisms of the anti-amyloid effects of phenols. Amyloid 2007, 14, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006, 67, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.J.; Lee, K.W. Naturally occurring phytochemicals for the prevention of Alzheimer’s disease. J. Neurochem. 2010, 112, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Velander, P.; Wu, L.; Henderson, F.; Zhang, S.; Bevan, D.R.; Xu, B. Natural product-based amyloid inhibitors. Biochem. Pharmacol. 2017, 139, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Computational insight into the effect of natural compounds on the destabilization of preformed amyloid-β(1–40) fibrils. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Härd, T.; Lendel, C. Inhibition of amyloid formation. J. Mol. Biol. 2012, 421, 441–465. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AsA | AsA24h | AsA48h | DHA | DKG | |
|---|---|---|---|---|---|
| IC50 | 0.43 ± 0.07 | 0.29 ± 0.04 * | 0.20 ± 0.03 # | 0.17 ± 0.03 ## | 0.18 ± 0.03 ## |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.-F.; Zeng, C.-M. The Degradation Products of Ascorbic Acid Inhibit Amyloid Fibrillation of Insulin and Destabilize Preformed Fibrils. Molecules 2018, 23, 3122. https://doi.org/10.3390/molecules23123122
Yang L-F, Zeng C-M. The Degradation Products of Ascorbic Acid Inhibit Amyloid Fibrillation of Insulin and Destabilize Preformed Fibrils. Molecules. 2018; 23(12):3122. https://doi.org/10.3390/molecules23123122
Chicago/Turabian StyleYang, Lu-Fei, and Cheng-Ming Zeng. 2018. "The Degradation Products of Ascorbic Acid Inhibit Amyloid Fibrillation of Insulin and Destabilize Preformed Fibrils" Molecules 23, no. 12: 3122. https://doi.org/10.3390/molecules23123122
APA StyleYang, L.-F., & Zeng, C.-M. (2018). The Degradation Products of Ascorbic Acid Inhibit Amyloid Fibrillation of Insulin and Destabilize Preformed Fibrils. Molecules, 23(12), 3122. https://doi.org/10.3390/molecules23123122