1. Introduction
The diversity of biological functions, like ligand binding, conformational changes, or structural adaptability of proteins is used already since many years to engineer biosensors [
1]. In particular bacterial periplasmic binding proteins (PBPs) have been exploited to develop metabolite sensors [
2]. Typically, PBPs consist of two domains connected by a hinge region which includes a ligand binding site located at the interface of the two flanking domains. Depending on the ligand binding status the whole structure can adopt two different conformations: a ligand-free open form and a ligand-bound closed form, which interconvert through a bending and a swiveling twist motion about the hinge [
2,
3,
4,
5]. First approaches to make use of PBPs as biosensors were based on site-specific coupling of environmentally sensitive extrinsic fluorophores to the domain interface of the PBP. Ligand binding was reported as changes in fluorescence intensity, for example by local quenching effects. Often these quenching effects are related to large changes in solvent accessibility of the fluorophores between the ligand-free open state and the ligand-bound closed state [
5].
Later for in vivo applications the PBP-based sensors were equipped with two fluorescent proteins (FPs) that utilize Förster resonance energy transfer (FRET) to measure the ligand-induced conformational changes. For this purpose, variants of an appropriate FRET donor and acceptor pair, e.g., a cyan FP (donor) and a yellow FP (acceptor), were fused to the amino- and carboxyl-termini of the central ligand binding domain, respectively. Conformational changes of this sensing domain induce a change in the distance and/or the relative orientation of the FPs, which ultimately causes alterations of the FRET efficiency in a ligand concentration-dependent manner [
6,
7,
8]. These genetically encoded FRET biosensors can monitor steady-state levels of ions or metabolites in cells, for example to compare differences between mutants and wild types [
9,
10]. Therefore, genetically encoded fluorescence sensors have become essential tools in modern biological research and many recent advances have expanded the scope of applications [
1].
The design and optimization of genetically encoded FRET sensors is often a challenging task, since the change in the optical readout signal upon alteration of the ligand concentration needs to be sufficiently large [
6,
7,
10,
11,
12]. The major challenge for FRET-based biosensors, which make use of the Venus flytrap principle [
3], is given by the fact that a conformational change in the sensing domain must be translated into a significant variation of the relative distance and/or orientation of the attached FPs. At first glance the use of rather bulky FPs attached to the terminal ends of the sensing domain seems not to be a very promising approach to sense the conformational changes induced by the bound ligand. In practice though, the empirical screening of libraries of linker variants inserted between the sensing domain and the attached FPs have produced sensor constructs with dramatically improved signal changes [
6,
12]. However, this is often still a trial and error approach in which many constructs of various linker lengths, sensing domains and FPs need to be tested in order to obtain a sufficiently large response signal [
11].
An alternative approach makes use of sensing proteins (e.g., PBPs) to which a pair of small organic fluorescent dyes is attached at suitable positions in the protein sequence. These constructs are again employed as FRET sensors, in which the dye attachment is chemically performed at cysteine residues that were genetically introduced at the desired positions (see for example [
13,
14,
15]). However, the fluorescent dye-based approaches are generally hard to apply for in vivo measurements.
In this work we report on a comparison between a dye-labeled and a FP-based FRET biosensor and discuss advantages and disadvantages of both labeling schemes, as well as their potential for different applications. As a case study we compared a FP-equipped and dye-labeled FRET-based sensors for glucose [
6,
16] that use the same glucose/galactose binding protein (MglB) from
Escherichia coli. We employ single-molecule FRET (smFRET) analyses to compare the response of the respective sensors to glucose and crowding conditions.
3. Discussion
To the best of our knowledge we show here for the first time a direct comparative smFRET study of two optimized biosensors that are based on the same sensing protein equipped either with a pair of FPs or with a pair of fluorescent dyes. In particular, we compared an extensively optimized FP-equipped glucose sensor construct based on the glucose/galactose binding protein MglB with a dye-labeled MglB where the attachment positions were optimized to achieve a large FRET signal change. In contrast to ensemble data, single-molecule data provide detailed information on coexisting sensor subpopulations, which in addition permits the discrimination of heterogeneities arising from experimental artifacts or impaired sample preparation, including effects of incomplete FP chromophore maturation [
22,
24]. In this respect it is worth to mention that our smFRET approach allows the elimination of donor-only molecules which would otherwise bias the resulting FRET efficiencies drastically [
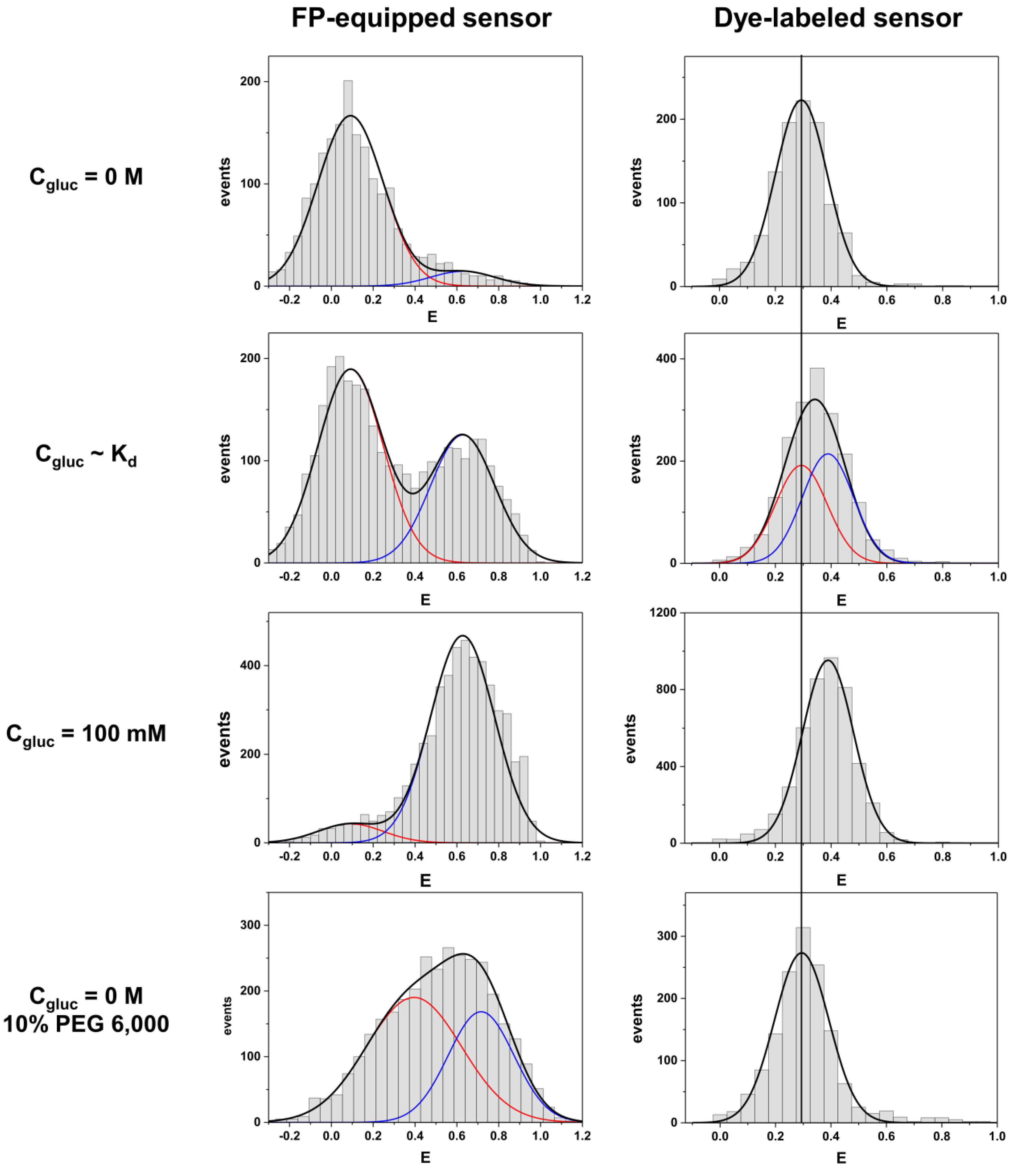
22]. Therefore, the smFRET approach is ideally suited to perform a rigorous comparison of both labeling schemes in order to judge the respective performances.
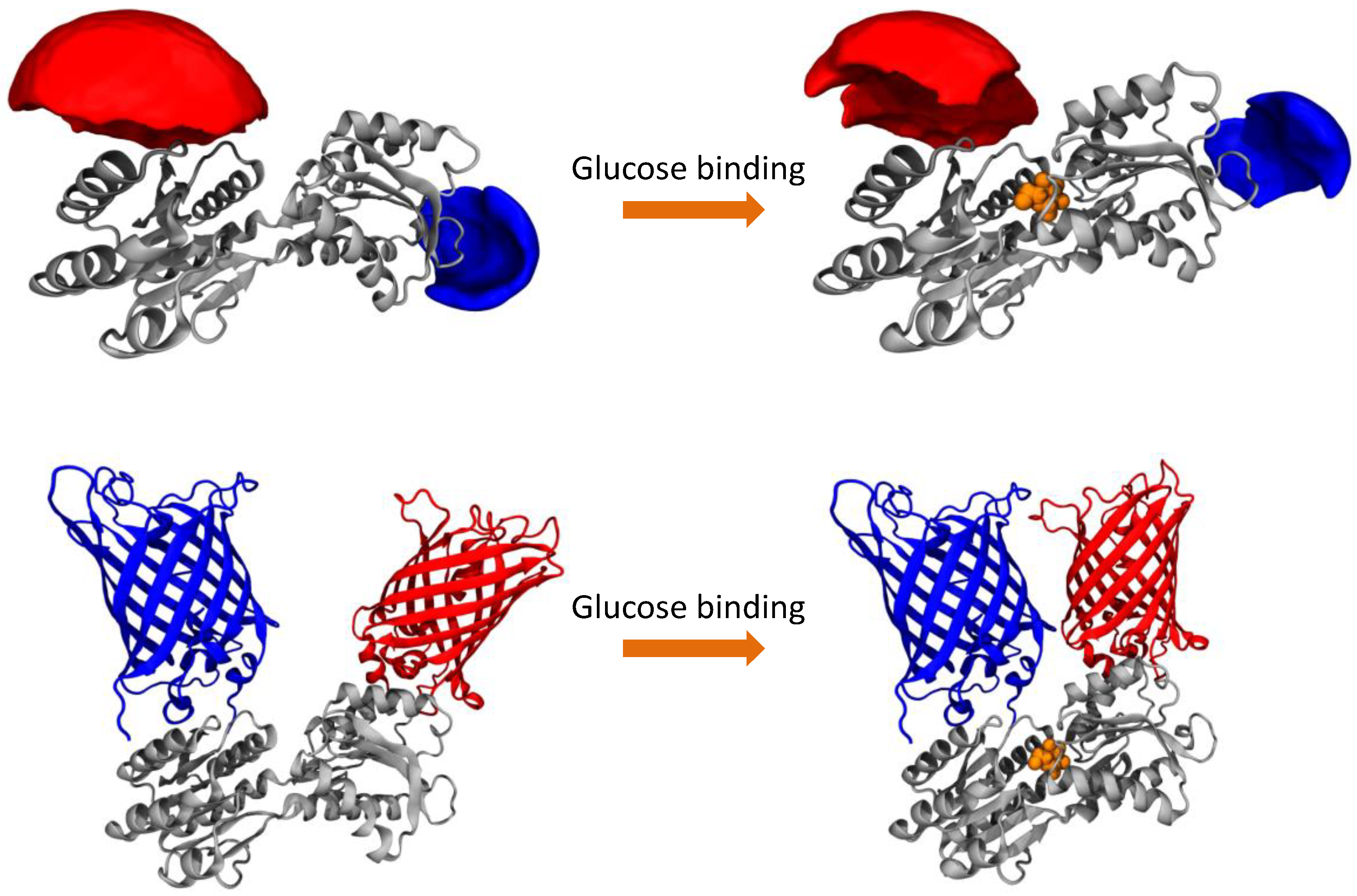
We compare both labeling schemes by means of FRET efficiency histograms because the energy transfer efficiency is the underlying physical parameter in contrast to derived parameters such as emission intensity ratios. The most obvious difference between the two labeling schemes is given by the fact that the dye-equipped sensor exhibits a much smaller energy transfer change between the ligand-free and the ligand-bound state as compared to the FP-equipped sensor. This is mainly caused by the fact that the dye-equipped sensor can only make use of changes in the inter-dye distance R
DA (see
Table 1). The extrinsic fluorophores are usually attached to the protein via a long and flexible linker, to avoid the interaction of the dyes with the surface of the protein, which could give rise to a quenching of the fluorescence, and to guarantee a fast and relatively free rotation of the dyes (see AVs in
Figure 1). As a consequence, the orientation factor that modulates the FRET efficiency can be approximated to a constant value of 2/3 and the only factor influencing the FRET efficiency is the inter-dye distance. The inter-dye distance change ΔR
DA is physically restricted by the absolute conformational change of the sensing proteins. For sensing proteins with sizes in the order of 30–35 kDa (like PBPs) or smaller, the conformational change is rather moderate, which remains an inherent problem. In addition, only attachment positions for which R
DA is close to the Förster radius R
0 can be used in order to be sensitive to distance changes with FRET. Although we could identify attachment points on the surface of the MglB protein which would result in larger ΔR
DA values, they are not favorable because their R
DA values are too small with respect to the actual R
0. However, even if we would consider the largest possible ΔR
DA value with the best matching R
0, the difference in transfer efficiency would still be significantly smaller than the value of the FP-equipped sensor.
In contrast, FP-equipped sensors are designed in a way that other factors rather than only the distance change ΔR
DA between both FPs contribute to the FRET signal change upon ligand binding. These approaches try to make use of a restriction of the spatial orientations occupied by the FPs. Thereby the orientation parameter κ
2 can be the dominant contribution to the FRET efficiency, which helps to increase the FRET signal change [
6,
7,
12]. In contrast to AV calculation for dyes, the problem for FP-based sensors is still to predict possible fluorophore orientations for a specific design, although there are some approaches reported in the literature [
25]. As a consequence, often many constructs with a variety of insertion positions of FPs and linker properties have to be tested. In most of these attempts at least a few of the tested constructs show a reasonable performance [
12,
22].
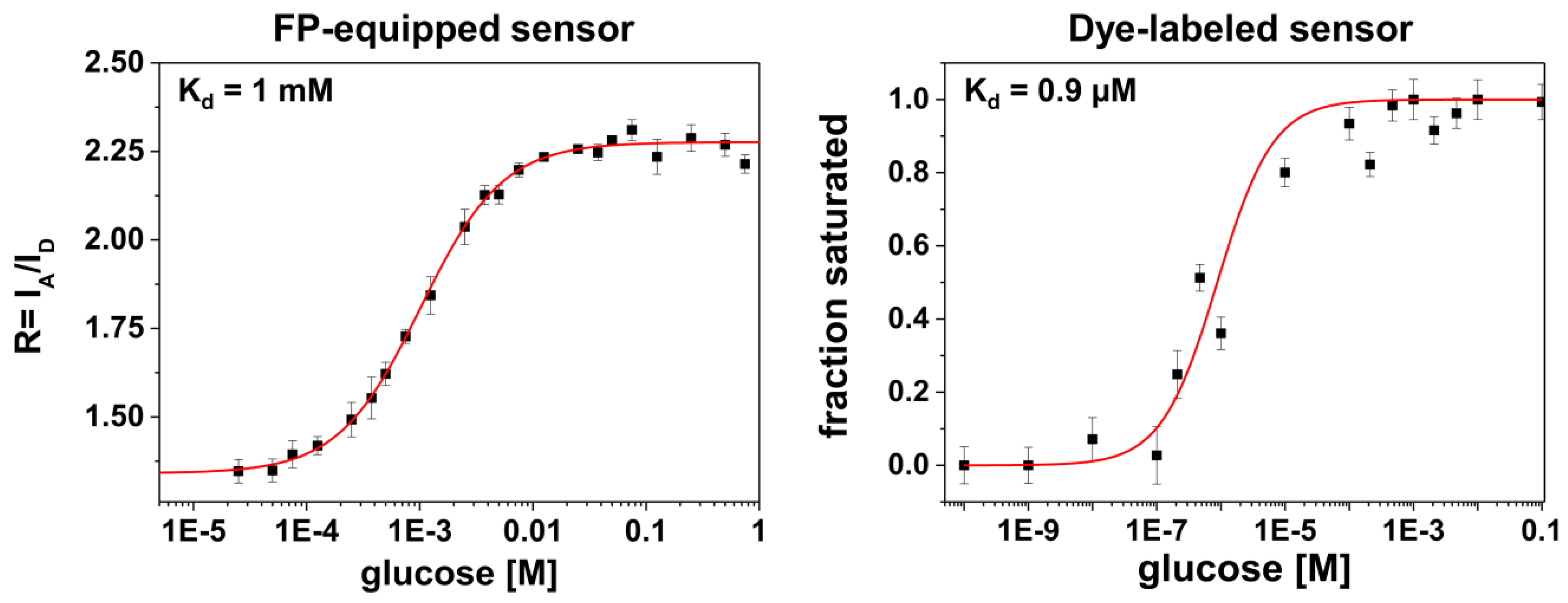
Another feature in the comparison between the different labeling schemes is given by rather different apparent binding affinities (in terms of K
d-values, see
Figure 3). It is known from previous studies that for FP-equipped as well as for dye-equipped sensors the binding affinity can be drastically reduced compared to the K
d of the isolated glucose/galactose binding protein (MglB) [
5,
26,
27]. On the one hand, we observe a K
d value of ~1 mM for the FP-equipped construct which is rather close to that of the starting construct (FLII
12Pglu600µ) [
6]. On the other hand, our dye-labeled sensor construct exhibits a K
d value of ~0.9 μM which is close to that of the wild-type MglB [
26]. Since for both sensor constructs almost identical MglB binding proteins were employed, the drastically reduced binding affinity of the FP-equipped sensor can be attributed to an impact predominantly originating from the FP attachment/insertion to/into the binding protein itself. In this respect the attachment of rather bulky FPs to the binding protein may alter the ligand accessibility of the binding site or modifies the well-balanced atomic structure of the binding site directly by FP induced strain.
The last pronounced difference between both sensors is related to their response to macromolecular crowding. Only the FP-equipped sensor exhibited a pronounced compaction in the presence of PEG 6000, while the dye-labeled species did not. This indicates that the volume expansion of the sensing protein alone is not much affected by this crowding agent. Interestingly, our observation somehow disagrees with recent findings which claim some PEG-induced compaction of the glucose binding protein [
28]. Most probably this disagreement is caused by the rather different experimental parameters which were measured in both studies. While smFRET measures the real physical dimension of the sensing protein, intrinsic protein fluorescence (emission wavelength shifts or altered emission intensities of intrinsic tryptophan residues) gives only nonspecific information about a change in the local environment of the tryptophan residues. Although for practical reasons genetically encoded FRET-based sensors are used to measure metabolite concentrations in cells, they would nevertheless require a specific calibration to consider crowding effects, which is a challenging task. In addition to ligand binding, also cellular crowding induces a structural compaction and thereby a FRET signal change which would bias the read out. An approach to analyze both effects separately is possible by comparing ordinary sensors to sensor variants with disabled ligand binding sites [
29].
Our comparison of two FRET-based glucose sensors reveals that the signal change of the FP-equipped sensor outperforms that of the dye-labeled analog. The attachment positions of the fluorophores on the surface of the glucose binding protein MglB vary for both sensors because both are optimized to reach a large signal change upon glucose binding. However, the choice of a sensor type may consider other aspects depending on the application of the sensor. The dye-labeled sensor may be beneficial if a high affinity is desired. Yet, the major advantage of the dye-labeled sensor is its insensitivity towards crowding. Thus, the application of dye-equipped sensors in living cells is a possible alternative using microinjection [
30], although this procedure is rather time-consuming. It can be likewise beneficial to use FP-equipped sensors for in vitro applications because of their large signal changes [
12,
16].
Finally, the transfer of our findings to other biosensors based on the family of periplasmic binding proteins (PBP) depends on two factors: (i) how extensively has the respective FP-equipped sensor already been optimized and (ii) are dye labeling positions of the respective PBP available that result in large distance changes in the range of the Förster radius. The latter can in principle be predicted ab initio, if high resolution crystal structures are known for the liganded and non-liganded state, as we have demonstrated here with the case study of MglB.
4. Materials and Methods
4.1. DNA Constructs
The gene encoding the glucose sensor FLII
12Pglu600µ, which consists of the glucose/galactose-binding protein MglB from
E. coli and the fluorescent proteins (FPs) ECFP and Citrine (EYFP with Q69M), was cloned between the NdeI and HindIII sites of the pRSET vector [
6]. The FPs were altered to the improved variants mTurquoise2 and Venus, respectively. mTurquoise2 was inserted after the 11 N-terminal amino acids of MglB to make it more rigid and Venus was fused to the C-terminal end as previously described [
22]. For sequence information see glucose sensor no. 2 in Supporting Information of ref. [
22]. For the dye-labeled sensor the MglB protein was amplified from the above mentioned construct and point mutations Q42C and K137C were introduced at the same time. The amino acid sequence of this construct is given in
Appendix B. The resulting PCR product was cloned between the NdeI and XhoI sites of the pRSET vector.
4.2. Production and Purification of MglB
The variant MglB Q42C K137C with C-terminal hexahistidine tag was produced in E. coli BL21 RIL cells for 3 h at 37 °C after induction with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density (O.D.) of 0.6. The cells were harvested and resuspended in buffer A containing one tablet of EDTA-free protease inhibitor (cOmplete Ultra, Merck, Darmstadt, Germany). Cells were lysed using a cell disruptor (Constant Systems Ltd., Daventry, UK). The cell lysate was filtered (pore size 0.2 µm) and applied to a Ni-NTA affinity column (Qiagen, Venlo, the Netherlands) equilibrated with buffer A (20 mM MOPS, 300 mM NaCl, pH 7.3). After washing with buffer A containing 10 mM and 20 mM imidazole, MglB was eluted with 250 mM imidazole. The protein was desalted in a Superdex 200 10/300 GL size exclusion column (GE Healthcare Life Sciences, Chicago, IL, USA) with 20 mM MOPS, pH 7.3, flash frozen and stored at −80 °C for further use.
4.3. Dye Labeling of the MglB Protein
The MglB protein was labeled on the two cysteine residues at positions 42 and 137 using approximately 5-fold excess of the acceptor dye Alexa Fluor 647 (AF647) and the donor dye Alexa Fluor 488 (AF488) in a 1.5:1 ratio. The excess of unbound fluorophores was removed by a Superdex 200 10/300 GL size-exclusion column. The double-labeled protein was purified via a MonoQ 5/50 GL ion-exchange column (GE Healthcare Life Sciences, Chicago, IL, USA) in an automated FPLC system (ÄKTAexplorer, GE Healthcare Life Sciences, Chicago, IL, USA) using a continuous NaCl gradient (0–500 mM NaCl) in 20 mM MOPS, pH 7.3. The different species were separated according to their charge due to the negative net charge of the bound fluorophores at pH 7.3. The obtained label stoichiometry is given by 43% donor only, 21% acceptor only, and 36% donor and acceptor labeled species. Since all smFRET measurements were performed using Pulsed Interleaved Excitation (PIE), we did not improve the label efficiency in this study.
4.4. smFRET Measurements with AF488 and AF647 Labeled MglB Sensors
A detailed description of smFRET measurements was published earlier [
15,
31]. We performed our studies with diffusing double labeled sensor constructs using a confocal inverted microscope MicroTime200 (PicoQuant, Berlin, Germany). Shortly, the fluorophores were excited using lasers with 485 nm and 640 nm (LDH-D-C 640B and LDH-D-C 485B from PicoQuant, Berlin, Germany), respectively. The excitation light was focused on the sample using a high numerical aperture water immersion objective (UPLSAPO 60x; Olympus, Hamburg, Germany); the fluorescence emitted was collected through the same objective and spatially filtered by a 75 μm pinhole in confocal configuration. The emission signal was separated by a dichroic mirror (T600lpxr, Chroma Technology, Olching, Germany) and filtered by a 535 nm band pass filter (535/55, Semrock, Rochester, NY, USA) for the donor channel and a 635 nm long-pass filter (635 LP, Semrock, Rochester, NY, USA) for the acceptor channel. Finally, photons were detected by single-photon avalanche diodes (τ-SPAD, PicoQuant, Berlin, Germany for the donor channel and SPCM-AQR-14, Perkin-Elmer Inc., Waltham, MA, USA for the acceptor channel). The arrival time of each photon was recorded with a time-correlated single-photon counting module (HydraHarp400, PicoQuant, Berlin, Germany). For smFRET, a PIE scheme was applied, in which excitation of the donor and acceptor is alternated in order to verify the presence of the acceptor by direct excitation and minimize the artifacts due to the presence of donor-only molecules. All measurements were made in presence of 0.001% Tween20 and a Trolox-cysteamine cocktail was used for photo-protection purposes.
4.5. Accessible Volume (AV)-Calculations and Data Analysis
The AVs for fluorescent dyes attached to MglB were determined using an algorithm that was previously described by Höfig et al. [
17]. In brief, this algorithm generates for each dye bound to the protein an AV cloud, where each point is sterically accessible for the fluorophore. This algorithm takes the length of the linker and the shape of the dyes into account as well as the fact that the dye is free to move around the anchor position. By averaging all the possible positions for both dyes, a mean donor-acceptor distance is calculated.
Analysis of smFRET data was performed using self-written Matlab routines (Mathworks, Natick, MA, USA), whose details are described in Gabba et al. [
15]. Briefly, the bursts (i.e., the photons emitted by the dyes when a labeled protein crosses the confocal volume) were identified from the raw data according to the inter-photon lag-time between two adjacent photons, which has to be below a certain threshold. This first selection was done on the photons emitted upon direct excitation of the acceptor, therefore ensuring the presence of this dye and in turn avoiding the selection of protein labeled only with the donor. To the bursts selected upon direct excitation of the acceptor, a further criterion was applied. Here, only the bursts having a total number of photons (donor + acceptor) emitted upon excitation of the donor (FRET photons) above a threshold value were considered for the analysis. According to established standard procedures, for each burst the number of photons emitted from the donor and from the acceptor were counted and the corresponding fluorescence intensities were calculated by correcting for the background photon counts, the crosstalk of the donor fluorescence into the acceptor channel, the detection efficiencies for the two channels, and for the different QY of the dyes [
24]. For each such selected burst, donor (D) and acceptor (A) photons after direct donor excitation were accumulated and corrected:
Here,
FD and
FA are the corrected intensities of donor and acceptor,
ID and
IA are the raw data of donor and acceptor counts, and
BGD and
BGA are the background counts of donor and acceptor obtained as the product of dwell time and average background count rate, respectively.
Lk represents the leakage of donor photons into the acceptor channel and
Dir gives the acceptor photons originating from direct excitation. For all bursts that fulfilled a threshold of
FD +
FA > 25, the energy transfer efficiency:
was calculated, where γ is a correction factor defined as:
Here, Φ
A and Φ
D are the fluorescence quantum yields while
gA and
gD are the transmission efficiencies of the acceptor and donor, respectively. For the calculation of expected energy transfer efficiency values from inter-dye distances R
DA known from the 3D protein structures, the relation:
is used, with a Förster radius of R
0 = 54.5 Å for the conditions used here.
4.6. Production, Measurements and Data Analysis of FP-Equipped MglB Constructs
All details about the design, production, and purification of FP-equipped sensor constructs are described in a recently published work [
22]. In this publication also all ensemble and smFRET measurements of the construct no. 2, the construct which we discuss in this work, are described in detail. With the help of two-color coincidence detection (TCCD) we also determined the label stoichiometry of the sensor construct. The sensor no. 2, for which data are presented here, is characterized by 31% donor only, 9% acceptor only, and 60% donor and acceptor labeled constructs (see ref. [
22]). These values are essentially determined by the chromophore maturation efficiency of the individual FPs within the given sensor construct.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}



